
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reductive cleavage of P4 by iron(I) centres: synthesis and structural characterisation of Fe2(P2)2 complexes with two bridging P22− ligands†
Shenglai
Yao
a,
Tibor
Szilvási
b,
Nils
Lindenmaier
a,
Yun
Xiong
a,
Shigeyoshi
Inoue
a,
Mario
Adelhardt
c,
Jörg
Sutter
c,
Karsten
Meyer
c and
Matthias
Driess
*a
aTechnische Universität Berlin, Department of Chemistry: Metalorganics and Inorganic Materials, Sekr. C2, Strasse des 17. Juni 135, D-10623 Berlin, Germany. E-mail: matthias.driess@tu-berlin.de; Fax: +49 (0)30-314-29732; Tel: +49 (0)30-314-29731
bDepartment of Inorganic and Analytical Chemistry, Budapest University of Technology and Economics, Szent Gellért tér 4, 1111 Budapest, Hungary
cInorganic Chemistry, Department of Chemistry and Pharmacy, Friedrich-Alexander-University Erlangen-Nürnberg (FAU), Egerlandstrasse 1, 91058 Erlangen, Germany
First published on 27th February 2015
Abstract
The selective transformation of white phosphorus with a β-diketiminato iron(I) toluene complex under mild reaction conditions is reported which furnishes a new dinuclear iron(III) Fe2(P2)2 complex with two bridging P22− ligands. Its reduction with potassium results in the formation of the first delocalised mixed-valent bis-diphosphido iron(II,III) complex which is isostructural with the neutral Fe2P4 precursor.
The exploration of low-valent transition metal (TM) complexes for coordination and subsequent reduction of white phosphorus (P4) is an important topic of current interest because metal-catalysed P4 derivatisation has been suggested to be an environmentally benign synthetic route for the synthesis of desirable organophosphorus compounds.1 During the past decades, several low-valent TM systems have been examined for transformation of P4. With a few exceptions,1,2 the approaches involve the use of cyclopentadienyl and/or carbonyl TM complexes under harsh conditions (thermal and/or photochemical) resulting in unpredictable P4 fragmentation. Thus new low-valent metal complex systems that are capable to transform P4 with high selectivity under mild conditions are highly desired.
Low-valent TM and main group element complexes supported by the β-diketiminato scaffold have been successfully utilised to activate a variety of small molecules, including P4.3 In 2004, Roesky et al. described the reduction of P4 with the monovalent aluminum MeLDippAl (MeLDipp = CH[CMeN(2,6-iPr2C6H3)]2), yielding [(MeLDipp)2Al2(μ:η2,η2-P4)] (I, Scheme 1) bearing the P44− ligand.3d Recently we communicated the coordination of P4 by a β-diketiminato ligand supported monovalent nickel species under very mild conditions, leading to the [(MeLDipp)2Ni2(μ2:η3,η3-P4)] complex (II).3g Applying a slightly modified β-diketiminato ligand, we developed the corresponding monovalent cobalt complex, which can perform facile transformation of P4 to afford [(LDippCo)2(μ2:η4,η4-P4)] (III, LDipp = CH[CHN(2,6-iPr2C6H3)]2) as the first complex featuring a neutral, rectangular-planar cyclo-P4 ligand.3h Interestingly, treating III with one molar equivalent of pottasium graphite transforms the rectangular cyclo-P4 moiety to the square-planar cyclo-P42− in IV as an anion. Similar cyclo-P42− and cyclo-P44− inverted sandwich β-diketiminato Nb and Ta complexes were more recently described by Bergman and Arnold et al.3i In this contribution, we report on the P4 transformation with a β-diketiminato ligand supported iron(I) species [LDippFe·toluene] (2, Scheme 2), affording selectively the dicuclear iron(III) complex [(LDippFe)2(μ2:η2,η2-P2)2] (3) with two P2 dianionic ligands. Remarkably, the reduction of 3 results in the delocalised mixed-valent iron(II,III) complex [(LDippFe)2(μ2:η2,η2-P2)2][K(thf)6] (4) comprising the same diphosphorus ligands of the oxidised species.
 | ||
| Scheme 1 Selected P4 transformaiton products I–IV mediated by low-valent β-diketiminato metals. | ||
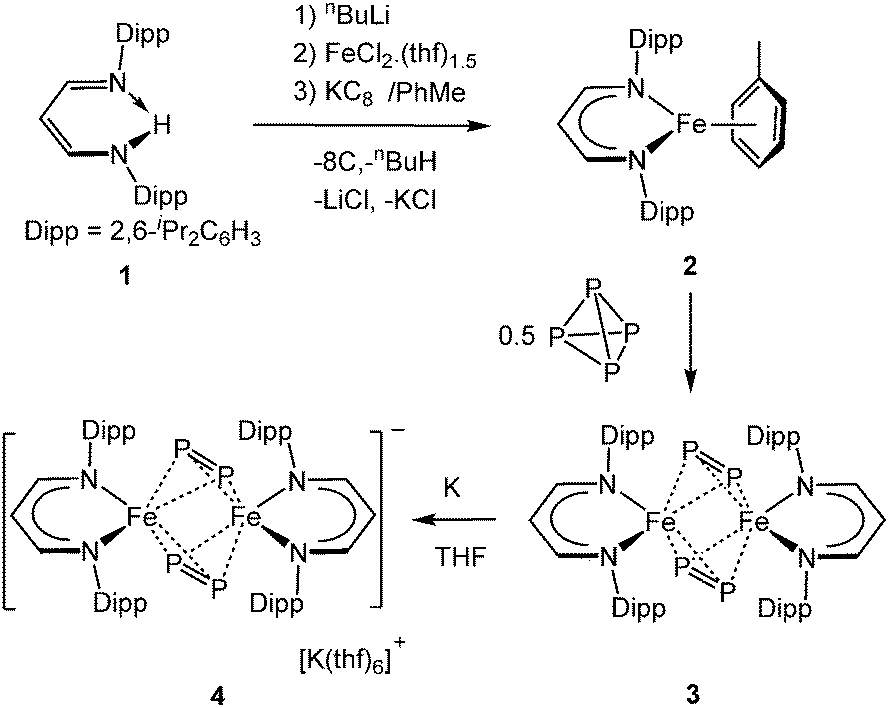 | ||
| Scheme 2 Synthesis of β-diketiminato iron(I) complex 2 and the transformation of P4 to iron complexes 3 and 4. | ||
The iron(I) precursor complex 2 can be readily prepared in a one-pot synthesis (Scheme 2). The lithiation of the β-diketiminto ligand [LDippH] (1)4a with nBuLi in THF, followed by metathesis with FeCl2(thf)1.5,4b leads to a yellow solid after removal of the solvent. Subsequent reduction of this solid dissolved in toluene with potassium graphite furnishes the desired iron(I) toluene complex 2 as dark green crystals in 75% isolated yield after work-up. Complex 2 is paramagnetic as evidenced by the room-temperature magnetic moment of 1.83 μB in solution. The single crystal X-ray diffraction study confirms its constitution (Fig. S1, ESI†). It is a mononuclear species with an η6-toluene ligand bound to the iron centre reminiscent to the low-spin β-diketiminato complex iron(I) [MeLDippFe(η6-benzene)], previously reported by Cundari, Rodgers, and Holland et al.5
Treatment of 2 in toluene solution with half an equivalent of P4 at room temperature affords a green precipitate of 3 (Scheme 2). After work-up, complex 3 could be isolated as a dark-green solid in approx. 90% yield. It is noteworthy that complex 3 is the only product of this reaction and results even with excess amounts of white phosphorus. Its composition was verified by elemental analysis and ESI mass spectrometry. Single crystals of 3 suitable for X-ray diffraction analysis could be obtained in benzene solutions.
The crystal structure of 3 revealed a centrosymmetric dinuclear iron complex, supported by two puckered β-diketiminato ligands (Fig. 1). The iron centres are bridged by two diphosphorus ligands in a μ2:η2,η2-coordination mode. A similar [(Fe)2(μ2:η2,η2-P2)2] core was previously proposed by Dahl and Barr6 for [(C5Me5)2(Fe)2(μ2:η2,η2-P2)2] (V) but – due to a severe disorder of the diphosphorus ligands – the solid state structure could not be fully refined. The Fe⋯Fe distance of 2.777 Å in 3 is significantly longer than that observed in V (2.585(6) Å) and rules out the presence of an Fe–Fe bond in the complex. The P⋯P distance (P2–P1′ and P2′–P1) between the two diphosphorus units (3.325 Å) also excludes any attractive P⋯P interaction between the two P2 fragments. This is in contrast to the β-diketiminato cobalt P4 complex III,3h for which two long P–P single bonds (2.298(1) Å) along with two short P–P bonds (2.124(1) Å) were observed. In fact, the P4 unit in 3 has been cleaved into two P2 moieties. The P–P distance of 3 (2.036(2) Å) within these P2 moieties are shorter than those typically observed for a P–P single bond (2.20–2.25 Å) and lie in the range of a P–P double bond (2.00–2.05 Å). The latter is slightly shorter than the P–P bond lengths of [(C5Me5)2(Co)2(μ2:η2,η2-P2)2] (2.053(4) and 2.058(4) Å).6 It is noteworthy that such P2 units represent an important type of transformation products resulting from TM-mediated degradation of P4.1 Generally, the P2 unit may behave as either a four or eight electron donor toward metal centres depending on the coordination environments.7
 | ||
| Fig. 1 Molecular structures of 3 (top) and the anion in 4·THF (bottom). Hydrogen atoms and solvents molecule are omitted for clarity. Thermal ellipsoids are drawn at 50% probability level. Symmetry transformations used to generate equivalent atoms (′) are −x + 1, −y + 2, −z (for 3); −x + 1, −y + 2, −z + 1 (for the anion in 4·THF). Selected distances (Å) and angles (°) for 3: Fe1–N1 2.023(3), Fe1–N2 2.025(3), Fe1–P1′ 2.344(1), Fe1–P2′ 2.356(1), Fe1–P1 2.373(1), Fe1–P2 2.377(1), P1–P2 2.036(2), N1–Fe1–N2 93.1(1), Fe1⋯Fe1′ 2.777(2); selected distances (Å) and angles (°) for the anion of 4: Fe1–N2 2.039(2), Fe1–N1 2.042(2), Fe1–P2′ 2.3560(5), Fe1–P1′ 2.3600(5), Fe1–P1 2.3669(5), Fe1–P2 2.3738(5), P1–P2 2.0353(8), N2–Fe1–N1 91.56(6), Fe1⋯Fe1′ 2.871(5). | ||
The cyclic voltammogram of 3, recorded in a THF solution containing 0.3 M nBu4NPF6 as electrolyte at 295 K, revealed a reversible redox event centred at E1/2 = −1.5 V (vs. Fc/Fc+) and additionally one quasi-reversible electron transfer, possibly coupled to a succeeding chemical reaction, at E1/2 = −2.8 V (for further details see ESI†). Accordingly, we performed the reduction of 3 with one equivalent of potassium in THF at room temperature (Scheme 2). A colour change from green to brown-red was observed during the period of reaction, leading to the formation of reduction product 4 that was isolated in 75% yield as a dark brown crystalline solid. Complex 4 crystallised in THF as a separated ion pair. The potassium cation is coordinated by six disordered THF molecules. Interestingly, the geometric parameters of the anion of 4 are almost identical to those observed for its Fe2P4 precursor 3 (Fig. 1 and Table 1). The apparent differences in the molecular structures of 3 and the anion of 4 are the P⋯P distance between the two (μ:η2,η2-P2) subunits (3.158 Å for 4vs. 3.3235 Å for 3) and the Fe⋯Fe distance (2.8713(5) Å for 4vs. 2.777(2) Å for 3).
| Complex | 3 | 4 | V | |
|---|---|---|---|---|
| P–P distance (short) | Exp. (Å) | 2.036(2) | 2.0353(8) | — |
| Calc. (Å) | 2.044 | 2.033 | 2.118 | |
| MBO | 1.53 | 1.63 | 1.39 | |
| P⋯P distance (long) | Exp. (Å) | 3.325 | 3.325 | — |
| Calc. (Å) | 3.243 | 3.318 | 3.185 | |
| MBO | 0.17 | 0.05 | 0.01 | |
The striking similarity of the two μ:η2,η2-P2 subunits in 3 and 4 prompted us to determine the oxidation states of the iron centres by 57Fe Mössbauer spectroscopy. The Mössbauer spectrum of 3, recorded at 77 K in a zero-field, exhibits a single quadrupole doublet with a quadrupole splitting (ΔEQ) of 1.15(1) mm s−1 and an isomer shift (δ) of 0.42(1) mm s−1, suggesting the presence of two equivalent high-spin iron(III) centres in 3 (Fig. 2). The Mössbauer spectrum of 4, recorded under the same conditions, to our surprise, also displayed a single quadrupole doublet with a slightly higher isomer shift and δ = 0.53(1) mm s−1 and a smaller quadrupole splitting ΔEQ = 0.74(1) mm s−1. Interestingly, the latter isomer shift is intermediate between the values expected for ferric and ferrous localised high-spin iron sites.8 On the time scale of Mössbauer spectroscopy (ca.10−7 s) no localised Fe(II)/Fe(III) state could be identified suggesting fast charge exchange. It is worth mentioning that such an unusual, delocalised system, evidenced by Mössbauer spectroscopy, is scarce and was previously reported only for [(LFe)2(μ-OH)3][(ClO4)2] (L = N,N′,N′′-trimethyl-l,4,7-triazacyclononane) and the porphyrin–phthalocyanine mixed-ligand [Fe2(μ-N)] complex.9
 | ||
| Fig. 2 Zero-field 57Fe Mössbauer spectra of 3 (left) and 4 (right) recorded at 77 K. The solid lines are fits of the experimental values with Lorentzian doublets exhibiting isomer shifts, δ, quadrupole splittings, ΔEQ, and linewidths, Γ: δ = 0.42(1) mm s−1, ΔEQ = 1.15(1) mm s−1, Γ = 0.30(1) mm s−1 (3); δ = 0.53(1) mm s−1, ΔEQ = 0.74(1) mm s−1, Γ = 0.48(1) mm s−1 (4). | ||
Both complexes 3 and 4 are paramagnetic in solution as well as in the solid state. Applying the Evans-method10 for the determination of the magnetic moment of 3 and 4, in THF-d8 solution revealed room temperature magnetic moments of 1.12 and 4.50 μB, respectively. Additionally, the solid-state magnetic susceptibility measurements in the temperature range from 300 to 2 K, carried out with an applied magnetic field B of 1.0 T, exhibited a temperature-dependent effective magnetic moment for both complexes (Fig. S9 and S10 in ESI†). For complex 3, the μeff varies slightly from 1.70 to 1.90 μB at temperatures between 20 and 300 K. In the case of 4, the effective magnetic moment is significantly more temperature dependent and increases from 2.0 to 3.9 μB as the temperature increases from 2 to 300 K.
The electronic nature of 3 and the the naion of 4 has been elucidated by broken-symmetry DFT calculations at the (B3LYP/6-31G(d)//BP86/6-31G(d) [P, Fe: aug-cc-pVTZ]) levels (see Table 1 and ESI†). The calculated metric parameters are in good agreement with those obtained by X-ray diffraction analyses. The Mayer Bond Order (MBO) suggests that the short P–P bonds have significant double bond character (MBO = 1.53 for 3 and 1.63 for 4 (Table 1)) as indicated also by NBO analysis (Fig. S11 and S12, ESI†). The negative charge on the P atoms in 3 (−0.16, Table S4, ESI†) confirms that the P2 moiety is best described as dianionic P22− (P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P dianion) while the NBO analysis indicated that the iron centres are in +3 oxidation state (Fig. S11 and S12, Table S4, ESI†). Interestingly, the one electron reduction of 3 does not change the electronic structure of the P22− moiety (charge: −0.19, Table S4, ESI†) but the iron centres since one Fe is reduced to +2 oxidation state which can be monitored from the charge (+0.16 compared to the previous +0.36) and net spin density (2.26 drops to 1.62).
P dianion) while the NBO analysis indicated that the iron centres are in +3 oxidation state (Fig. S11 and S12, Table S4, ESI†). Interestingly, the one electron reduction of 3 does not change the electronic structure of the P22− moiety (charge: −0.19, Table S4, ESI†) but the iron centres since one Fe is reduced to +2 oxidation state which can be monitored from the charge (+0.16 compared to the previous +0.36) and net spin density (2.26 drops to 1.62).
We also calculated the optmised geometry and the electronic structure of the previously proposed [{(C5Me5)Fe(μ2:η2,η2-P2)}2] V by Dahl and Barr.6 The short (2.118 Å) and long P–P distances (3.185 Å) are quite similar to that of 3 (Table 1), respectively. However, the Fe–Fe distance (2.552 Å) is significantly shorter than that in 3 (2.778 Å). We found that the P2 moiety can be regarded as a P22− unit because it also showes double bonding character as 3 and the P atoms bear partial negative charge (−0.17, Table S4, ESI†). The short Fe–Fe distance has, however, an important consequence, namely, the direct exchange interaction becomes dominant between the iron centres and thus the ground state adopts high spin character (Table S4, ESI†). NBO analysis even indicates a Fe–Fe bond which clearly distinguishes V from 3.
In summary, the β-diketiminato iron(I) complex 2 reacts readily with P4 to yield the diiron(III) complex 3 with two P22− ligands. One-electron reduction of 3 results in the formation of 4 by reducing one Fe(+3) centre to Fe(+2) without significantly changing the geometry of the [(Fe)2(μ2:η2,η2-P2)2] core. Remarkably, the mixed-valent iron(II,III) pair of 4 is delocalised as evidenced by Mössbauer spectroscopy and represents a rare case of that type. The facile and selective transformation of P4 by β-diketiminato low-valent TM species opens new possibilities to further functionalise the P2 units; respective investigations are currently pursued by our group.
Notes and references
- Selected reviews: (a) B. M. Cossairt, N. A. Piro and C. C. Cummins, Chem. Rev., 2010, 110, 4164 CrossRef CAS PubMed; (b) M. Caporali, L. Gonsalvi, A. Rossin and M. Peruzzini, Chem. Rev., 2010, 110, 4178 CrossRef CAS PubMed; (c) M. Scheer, G. Balázs and A. Seitz, Chem. Rev., 2010, 110, 4236 CrossRef CAS PubMed; (d) N. A. Giffin and J. D. Masuda, Coord. Chem. Rev., 2011, 255, 1342 CrossRef CAS PubMed; (e) S. Khan, S. S. Sen and H. W. Roesky, Chem. Commun., 2012, 48, 2169 RSC; (f) M. Serrano-Ruiz, A. Romerosa and P. Lorenzo-Luis, Eur. J. Inorg. Chem., 2014, 1587 CrossRef CAS; (g) M. Peruzzini, L. Gonsalvi and A. Romerosa, Chem. Soc. Rev., 2005, 34, 1038 RSC; (h) O. J. Scherer, Acc. Chem. Res., 1999, 32, 751 CrossRef CAS; (i) M. P. Ehses, A. Romerosa and M. Peruzzini, Top. Curr. Chem., 2002, 220, 107 CrossRef CAS; (j) M. Peruzzini, R. R. Abdreimova, Y. Budnikova, A. Romerosa, O. J. Scherer and H. Sitzmann, J. Organomet. Chem., 2004, 689, 4319 CrossRef CAS PubMed.
- See for examples: (a) J. S. Figueroa and C. C. Cummins, J. Am. Chem. Soc., 2003, 125, 4020 CrossRef CAS PubMed; (b) J. S. Figueroa and C. C. Cummins, Dalton Trans., 2006, 2161 RSC; (c) E. B. Hulley, P. T. Wolczanski and E. B. Lobkovsky, Chem. Commun., 2009, 6412 RSC; (d) M. Demange, X.-F. Le Goff, P. Le Floch and N. Mézailles, Chem. – Eur. J., 2010, 16, 12064 CrossRef CAS PubMed; (e) B. Zarzycki, T. Zell, D. Schmitdt and U. Radius, Eur. J. Inorg. Chem., 2013, 2051 CrossRef CAS.
- (a) L. Bourget-Merle, M. F. Lappert and J. R. Severn, Chem. Rev., 2002, 102, 3031 CrossRef CAS PubMed; (b) Y.-C. Tsai, Coord. Chem. Rev., 2012, 256, 722 CrossRef CAS PubMed; (c) S. Yao and M. Driess, Acc. Chem. Res., 2012, 45, 276 CrossRef CAS PubMed; (d) Y. Peng, H. Fan, H. Zhu, H. W. Roesky, J. Magull and C. E. Hughes, Angew. Chem., Int. Ed., 2004, 43, 3443 CrossRef CAS PubMed; (e) Y. Xiong, S. Yao and M. Driess, Angew. Chem., Int. Ed., 2007, 46, 4511 CrossRef CAS PubMed; (f) Y. Xiong, S. Yao, E. Bill and M. Driess, Inorg. Chem., 2009, 48, 7522 CrossRef CAS PubMed; (g) S. Yao, Y. Xiong, C. Milsmann, E. Bill, S. Pfirrmann, C. Limberg and M. Driess, Chem. – Eur. J., 2010, 16, 436 CrossRef CAS PubMed; (h) S. Yao, N. Lindenmaier, Y. Xiong, S. Inoue, T. Szilvási, M. Adelhardt, J. Sutter, K. Meyer and M. Driess, Angew. Chem., Int. Ed., 2015, 54, 1250 CrossRef CAS PubMed; (i) C. Camp, L. Maron, R. G. Bergman and J. Arnold, J. Am. Chem. Soc., 2014, 136, 17652 CrossRef CAS PubMed.
- (a) D. J. E. Spencer, A. M. Reynolds, P. L. Holland, B. A. Jazdzewski, C. Duboc-Toia, L. L. Pape, S. Yokota, Y. Tachi, S. Itoh and W. B. Tolman, Inorg. Chem., 2002, 47, 6307 CrossRef PubMed; (b) R. J. Kern, J. Inorg. Nucl. Chem., 1962, 24, 1105 CrossRef CAS.
- J. M. Smith, A. R. Sadique, T. R. Cundari, K. R. Rodgers, G. Lukat-Rodgers, R. J. Lachicotte, C. J. Flaschenriem, J. Vela and P. L. Holland, J. Am. Chem. Soc., 2006, 128, 756 CrossRef CAS PubMed.
- M. E. Barr and L. F. Dahl, Organometallics, 1991, 10, 3991 CrossRef CAS.
- M. Caporali, L. Gonsalvi, V. Mirabello, A. Ienco, G. Manca, F. Zanobini and M. Peruzzini, Eur. J. Inorg. Chem., 2014, 1652 CrossRef CAS , and cited references therein.
- See for examples: (a) P. K. Mascharak, G. C. Papaefthymiou, R. B. Frankel and R. H. Holm, J. Am. Chem. Soc., 1981, 103, 6110 CrossRef CAS; (b) C. Achim, M.-P. Golinelli, E. L. Bominaar, J. Meyer and E. Münck, J. Am. Chem. Soc., 1996, 118, 8168 CrossRef CAS; (c) C. Achim, E. L. Bominaar, J. Meyer, J. Peterson and E. Münck, J. Am. Chem. Soc., 1999, 121, 3704 CrossRef CAS.
- (a) S. Drüeke, P. Chaudhuri, K. Pohl, K. Wieghardt, X.-Q. Ding, E. Bill, A. Sawaryn, A. X. Trautwein, H. Winkler and S. J. Gurman, J. Chem. Soc., Chem. Commun., 1989, 59 RSC; (b) C. Ercolani, S. Hewage, R. Heucher and G. Rossi, Inorg. Chem., 1993, 32, 2975 CrossRef CAS.
- (a) D. F. Evans, J. Chem. Soc., 1959, 2003 RSC; (b) T. Ayers, R. Turk, C. Lane, J. Goins, D. Jameson and S. J. Slattery, Inorg. Chim. Acta, 2004, 357, 202 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The detailed synthesis, characterisation and crystallographic data of 2, 3, and 4 as well as the computational details of 3 and 4. CCDC 1042230–1042232. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc00147a |
| This journal is © The Royal Society of Chemistry 2015 |