
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiastereoselective synthesis of O symmetric heterometallic cubic cages†
Yang
Yang
a,
Jian-Hua
Jia
b,
Xiao-Li
Pei
a,
Hao
Zheng
a,
Zi-Ang
Nan
a and
Quan-Ming
Wang
*a
aState Key Lab of Physical Chemistry of Solid Surfaces, Collaborative Innovation Center of Chemistry for Energy Materials (iChEM), Department of Chemistry, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, Fujian, People's Republic of China. E-mail: qmwang@xmu.edu.cn
bSchool of Chemistry & Chemical Engineering, Sun Yat-Sen University, Guangzhou, Guangdong, People's Republic of China
First published on 27th January 2015
Abstract
Enantiopure chiral cubic cages have been diastereoselectively synthesized for the first time. Chiral amines lead to the isolation of O symmetric homochiral cubic cages, while an achiral amine gives a racemic mixture. CD enhancement is observed as a result of configuration rigidity.
Coordination cage compounds are of special interest due to their well-defined structures, and potential applications such as catalysis, encapsulation of intermediates, and storage of labile substances.1 Their formation is usually based on the self-assembly of metal centers and bridging ligands.2 Chirality is an important issue with cage compounds, as asymmetric arrangement of ligands around a metal center or spatial twisting of ligands could generate supramolecular chirality.3 The chiral environments enable guests to experience sensations of dissymmetry, facilitating chiral separation and/or stereoselective reactions.4,5
Chiral M4Ln (n = 4 or 6) coordination tetrahedral cages are well studied.5,6 In contrast, there is no report on homochiral M8Ln (n = 6 or 12) cubes, which have eight chiral vertices leading to much more possible stereoisomers.7 So far, all the structurally determined chiral cubic cages reported in the literature are racemic mixtures.8,9 Li et al. reported a spontaneously resolved chiral homometallic cage, unfortunately the absolute structure is not reliable due to an unsatisfactory Flack parameter.10 Therefore, definitive evidence has not been presented for resolving cubic cages or have they ever been diastereoselectively synthesized.
A cube has four C3 and three C4 axes, so the incorporation of C3 moieties into C4 units favors the formation of a coordination cubic cage. Previously we have reported a cubic cage assembled from C3 metalloligands and C4 symmetric palladium(II) ions.9a Nitschke et al. have prepared cubic cages by mixing C4 symmetric tetrakis-amine, 2-formylpyridines and octahedral metals (C3 symmetric).9,11 However, these examples are not homochiral. For efficient chirality control, we choose to diastereoselectively synthesize cubic cages with chiral ligands. Herein, we report a facile approach to synthesize homochiral cubic cages with the combination of C3 and C4 components. We designed a ligand precursor L1 consisting of a formylpyridyl and a pyridyl group, which can react with chiral amines to form chiral ditopic ligands containing both bidentate pyridylimine and monodentate pyridine donors. Two enantiomeric chiral ligands are facilely synthesized, which are available for integrating C3 octahedral metals and C4 square planar metals in one entity (Scheme 1).
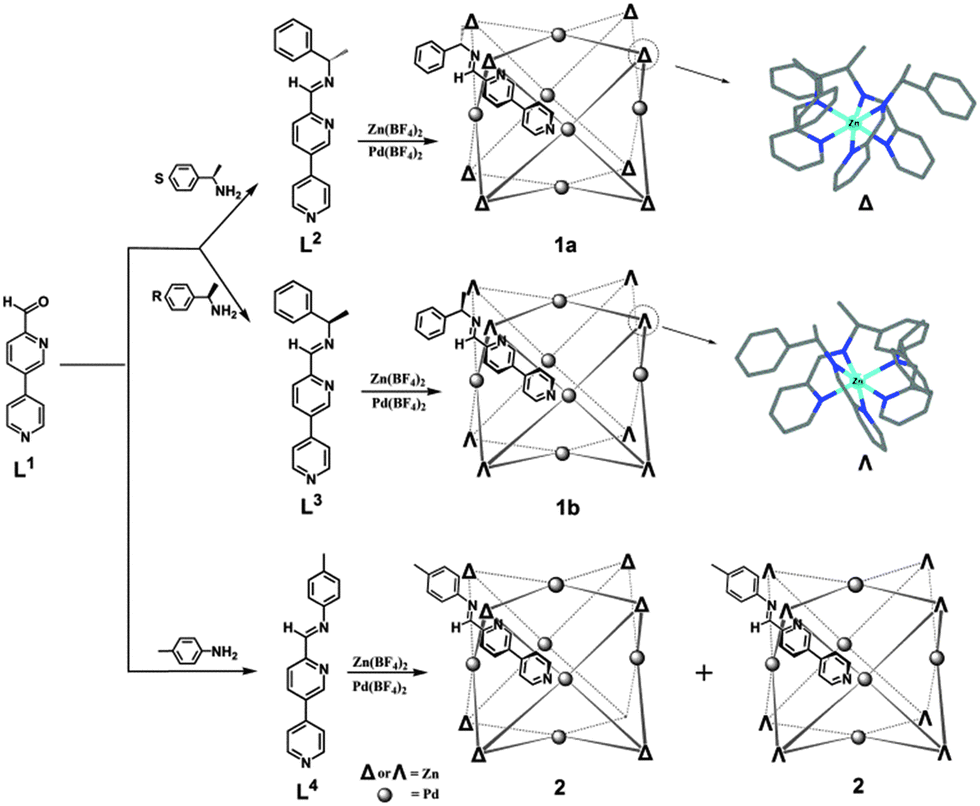 | ||
| Scheme 1 Synthetic routes of the ligands and the cubic cages. | ||
As shown in Scheme 1, the reaction between L1 and S-1-phenylethylamine, a chiral amine widely applied in controlling the absolute configuration,6e,12 gave a chiral heterotopic ligand L2. Then L2 was used in situ to ligate zinc(II) and finally palladium(II) ions in a ratio of 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8:6. This three-step synthesis afforded a cubic cage [Zn8Pd6(L2)24(BF4)28] (1a) in high yield.
:8:6. This three-step synthesis afforded a cubic cage [Zn8Pd6(L2)24(BF4)28] (1a) in high yield.
Crystalline 1a was grown by diffusion of diethylether into an acetonitrile solution of 1a, which was characterized by NMR, ESI-MS, IR and elemental analysis. Compound 1a is insoluble in common solvents such as methanol and dichloromethane, but it is highly soluble in acetonitrile. The solution behavior of 1a was studied in acetonitrile. The sharp and well resolvable 1H (Fig. 1), 13C NMR (Fig. S5, ESI†) signals of 1a in CD3CN revealed that the ligands were in an identical magnetically distinct environment, suggesting the cage's high symmetry. Compared with the free ligand, most of the signals were shifted indicating the coordination with metal ions. The full assignment was with the help of 2D COSY and NOESY spectra. In 1H diffusion ordered spectroscopy (DOSY), all the proton signals of 1a had the same diffusion coefficients, indicating that there was a single species in solution. According to the Stockes–Einstein equation,13 the dynamic radius of the species was calculated to be about 16 Å, which implies the self-assembly of a large sized cage.1e,14 The spectrum of the reaction mixture was similar to that prepared from the crystalline product, manifesting that the cage was formed in solution, not in the crystallization process.
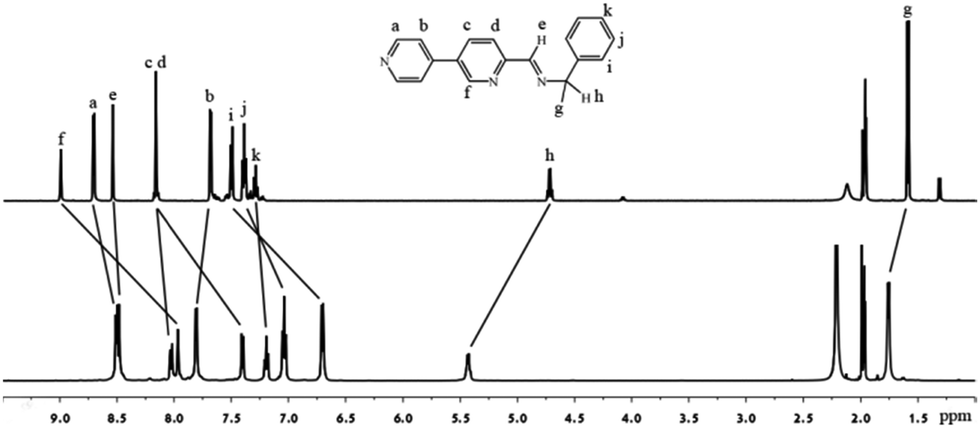 | ||
| Fig. 1 1H NMR (500.2 MHz) spectra of the free ligand L2 (top) and 1a (bottom) in CD3CN. | ||
Electrospray ionization mass spectrometry (ESI-MS) analysis further confirmed the formation of cage 1a, as shown in Fig. 2. A series of multi-charge peaks with m/z values at 962.0, 1078.6, 1224.3, 1411.6 and 1661.4 could be assigned to molecular ions of 1a from +10 to +6, respectively, which were derived from the loss of corresponding numbers of BF4− counterions. The assignment was verified by carefully matching the simulated data with experimental results. For example, the simulated isotopic patterns of [1a-9(BF4)]9+ and [1a-8(BF4)]8+ perfectly fit those experimental ones (Fig. 2b).
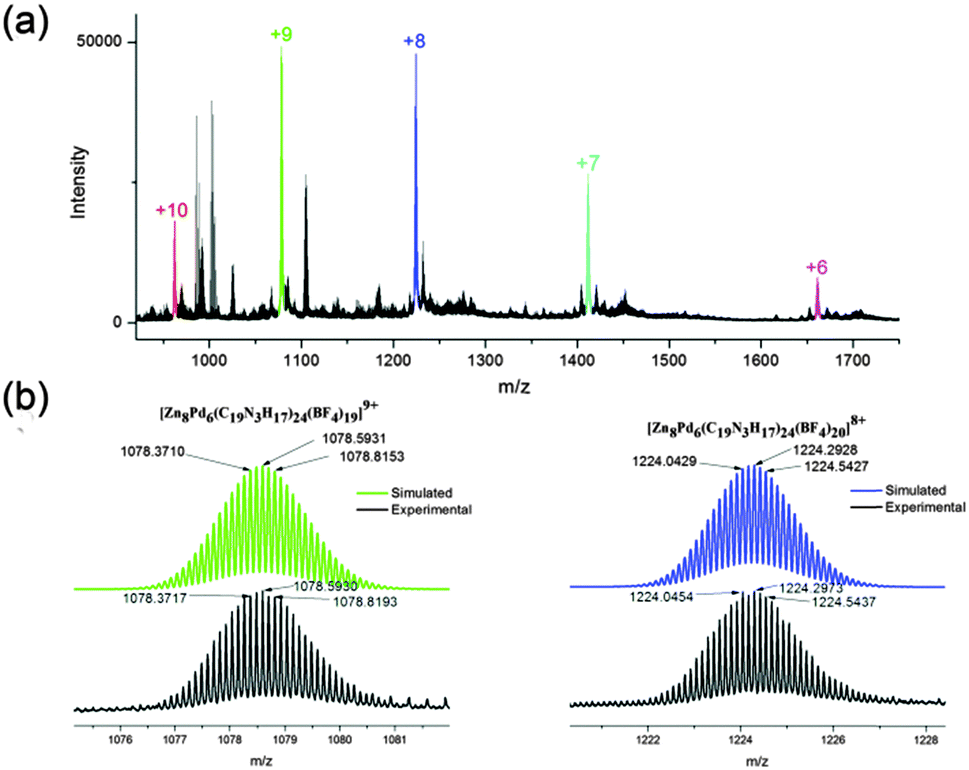 | ||
| Fig. 2 (a) ESI-MS spectrum of 1a in MeCN, molecular ion peaks with various multi-charges are shown in different colors. (b) The experimental (bottom trace) and simulated (top trace) isotopic patterns of the molecular ion peaks of [Zn8Pd6(L2)24(BF4)19]9+ (left) and [Zn8Pd6(L2)24(BF4)20]8+ (right). | ||
The cage structure was unambiguously revealed by single crystal X-ray structural analysis. 1a crystallizes in a chiral cubic P432 space group. There is only one L2 ligand in the asymmetric unit with the pyridylimine moiety chelating a zinc(II) cation and terminal pyridine coordinated to a palladium(II) cation. ZnII and PdII are located at crystallographically special positions. Cage 1a comprises 6 PdII, 8 ZnII and 24 L2 ligands (Fig. 3a). The eight vertices of the cube, each occupied by a C3-symmetric Zn-tris(pyridylimine) unit with identical Δ chiral configuration, and the six square faces, each capped by a C4-symmetric square-planar PdII coordinated to four pyridine units are observed. The arrangement style of the 24 ligands of 1a with O symmetry is analogous to that of the 24 protein subunits in ferritin.15 All cage molecules in crystal 1a are of the same handedness. This fact clearly demonstrates the successful diastereoselective synthesis of a chiral cubic cage. The phenyl groups of the stereogenic centers of the imine form face to face π–π interactions with the pyridyl ring of adjacent ligands, with a centroid to centroid distance of 3.64 Å. The distance between two PdII of two opposite square faces is about 16.18 Å. The longest Zn⋯Zn distance from diagonal vertices across the cubic center is about 24.42 Å. The diameter of the cube is approximately 33 Å, in accordance with the dynamic radius obtained from DOSY experiments. By switching the amine to its enantiomer R-1-phenylethylamine in the preparation (Scheme 1), we obtained [Zn8Pd6(L3)24(BF4)28] (1b). Structural determination confirmed that 1b and 1a are a pair of enantiomers (Fig. 3b). The ZnII centers at the eight corners of the cube are in Λ configurations. The Flack factors of both crystals are 0.02(2) for 1a and 0.035(17) for 1b, which verify the absolute configuration and enantiopurity. The crystals studied are representative of the entire sample since they showed the same solution behaviors with the bulk sample. Thus the chirality of the cage is dictated by the chiral amine used. In a control experiment, achiral 4-toluidine was used in place of S-1-phenylethylamine. Following the synthetic procedure of 1a, [Zn8Pd6(L4)24(BF4)28] (2) was formed. A yellow block single crystal of 2 was applied in X-ray structural analysis. Compound 2 crystallizes in a centrosymmetric space group P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) 1c, and it also has a cubic cage structure with 14 metal centers (Fig. 3d). Each individual cage is chiral with eight corners having either ΔΔΔΔΔΔΔΔ or ΛΛΛΛΛΛΛΛ configurations. However, both enantiomers are present equally giving a racemic mixture in contrast to enantiopure 1a and 1b.‡
1c, and it also has a cubic cage structure with 14 metal centers (Fig. 3d). Each individual cage is chiral with eight corners having either ΔΔΔΔΔΔΔΔ or ΛΛΛΛΛΛΛΛ configurations. However, both enantiomers are present equally giving a racemic mixture in contrast to enantiopure 1a and 1b.‡
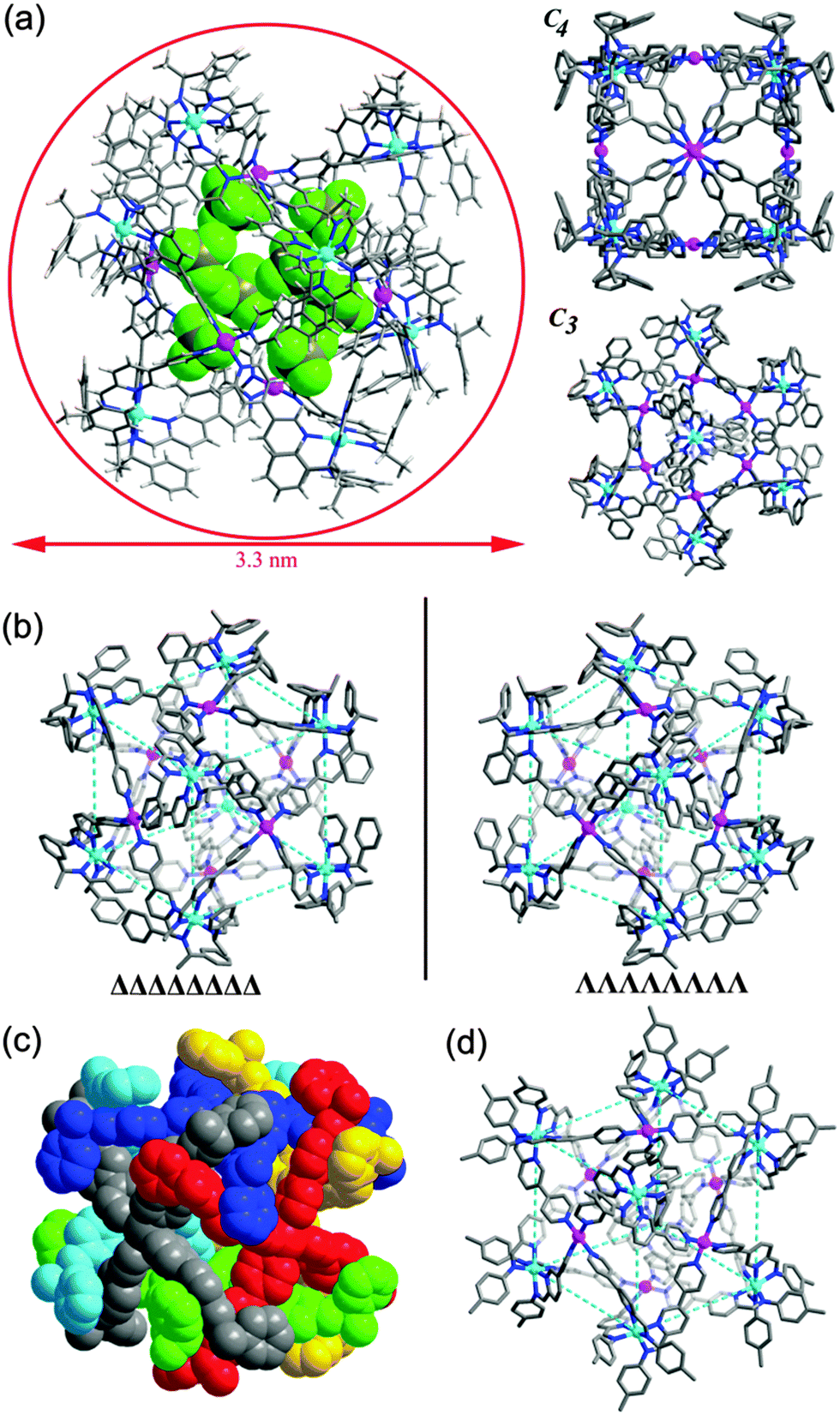 | ||
| Fig. 3 X-ray crystal structures of the cubic cages: (a) 1a with eight encapsulated tetrafluoroborate anions in space-filling mode, inset: view from one of the C4 axes (top) and view from one of the C3 axes (bottom); (b) ΔΔΔΔΔΔΔΔ-1a (left) and ΛΛΛΛΛΛΛΛ-1b (right); (c) space-filling mode of 1a with six faces in different colours; (d) ΛΛΛΛΛΛΛΛ-2. Hydrogen atoms and counter ions are omitted in the inset, (b), (c) and (d). Color legend: purple Pd; cyan Zn; green F; blue N; gray C; dark yellow B; white H. | ||
Eight counterions were enclosed inside each cage as observed in the X-ray crystal structure. The 19F NMR signal of the BF4− ions in cage 1a was shifted compared with free BF4− (Fig. S21, ESI†), suggesting rapid exchange of the anions with those inside the cage in solution. However the anion template effect for the cage formation was ruled out, because the same cage structure still formed with the use of counterions of different sizes and geometries (nitrate, trifluoromethanesulfonate, etc.) (Fig. S23, ESI†). We think that the binding angles between the donors of the ligand and the coordination preferences of the metals are crucial in the assembly of the cubic cages.
Circular dichroism (CD) spectroscopy was applied to examine the optical properties of the cubic cages (Fig. 4a). 1a and 1b in acetonitrile showed perfect mirror image signals, whereas 2 in solution was CD silent. Several solutions of 1a (or 1b), each prepared from only one single crystal, have the same profile, in line with bulk samples. This evidence rules out the formation of conglomerates. The CD spectra confirm that 1a and 1b are enantiomers and optically active, but 2 is a racemic mixture as found in crystal structures. Absorption bands at around 237–350 nm of 1 was assigned to ligand centered π–π* transitions.6e,16 Bisignate signals of the CD bands appear at this area, due to exciton coupling.12b,16 The positive cotton effect at a longer wavelength associated with Δ configuration was in agreement with the chirality at ZnII centers in the crystal structure of 1a and vice versa, a negative cotton effect at a longer wavelength was observed in 1b. It is worth noting that the CD signals of 1a were much more stronger than those of the same amount of free ligand L2. Simply mixing 24 equivalents of L2 with 8 equivalents of Zn(BF4)2 or 6 equivalents of Pd(BF4)2, respectively, did not improve the signals at all (Fig. 4b). The CD signals become intense only when the cage structure is formed. Such a CD enhancement is related to the fixed configuration after the cage structure is formed. Since 1a and 1b are the first examples of homochiral cubic cages, this is the first CD study of a chiral cubic cage structure integrating as much as eight chiral metal centers together.
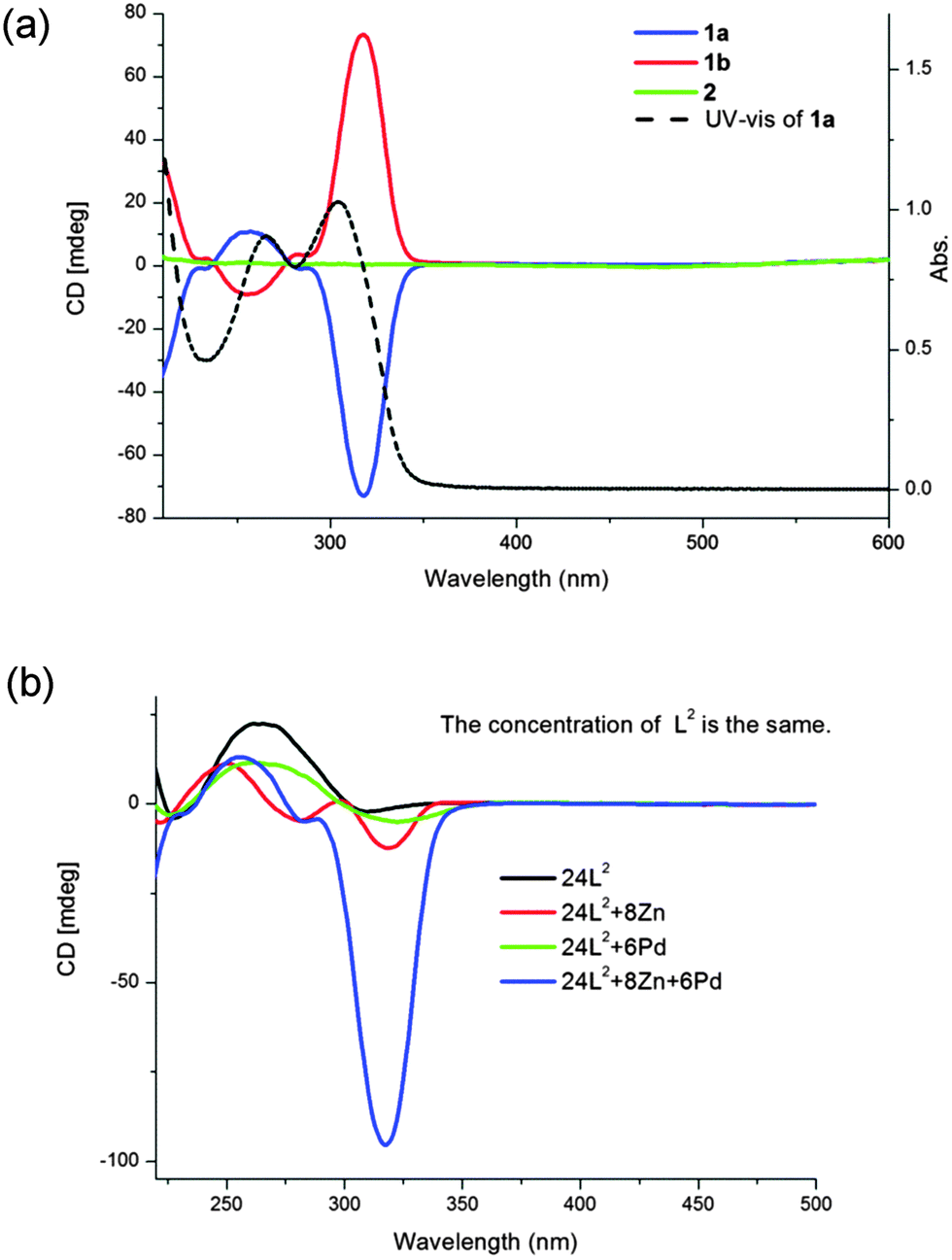 | ||
| Fig. 4 (a) CD spectra of 1a (blue line), 1b (red line), 2 (green line) and the UV-vis spectrum of 2a (black dashed line) in MeCN. (b) CD spectra of solutions containing: 24 equivalents of L2 (black); 24 equivalents of L2 and 8 equivalents of Zn(BF4)2 (red); 24 equivalents of L2 and 6 equivalents of Pd(BF4)2 (green); 24 equivalents of L2, 8 equivalents of Zn(BF4)2 and 6 equivalents of Pd(BF4)2 (blue). The concentration of L2 is the same (8.7 × 10−5 mol L−1) in all these solutions. | ||
No cage product 1a or 1b could be obtained starting from the racemic ligand indicating the lack of self-sorting of the chiral ligands. And once the cages formed, it is hard for them to exchange ligands. Adding large excess enantiomeric L2 into 1b could not invert the characteristic CD signals at around 300–350 nm of the cage, which would invert if all L3 were exchanged to L2. Partially exchanging ligands with opposite handedness will lower the symmetry of the cages resulting in a change in the NMR spectrum. Signals of 1b remained the same, which could be clearly distinguished from signals of free L2. And no obvious change has been observed in the NMR spectrum of a mixture of 1a and 1b after stirring overnight (Fig. S24–S26, ESI†).
The synthetic strategy can be applied in the preparation of a chiral cubic cage of larger size, with precursor ligand L5 having a longer backbone (Fig. 5). L5 was readily transformed into chiral L6 using S-1-phenylethylamine. A larger chiral cube [Zn8Pd6(L6)24(BF4)28] (3) was also obtained similar to the case of 1a. The formation of 3 has been confirmed by different NMR techniques. 1H NMR of 3 is in a similar pattern to 1a. And in comparison to 1a, a smaller diffusion factor for 3 was determined from DOSY experiments, which is indicative of a larger sized 3. Cage 3 also displays intense CD signals, much higher than the mixtures of L6 with Zn(BF4)2 or Pd(BF4)2, respectively (Fig. S27, ESI†). On the basis of the CD spectrum, 3 also has a ΔΔΔΔΔΔΔΔ configuration. Unfortunately, the crystals of 3 are too small for X-ray diffraction, and a molecular model of 3 is shown in Fig. 5 for reference.
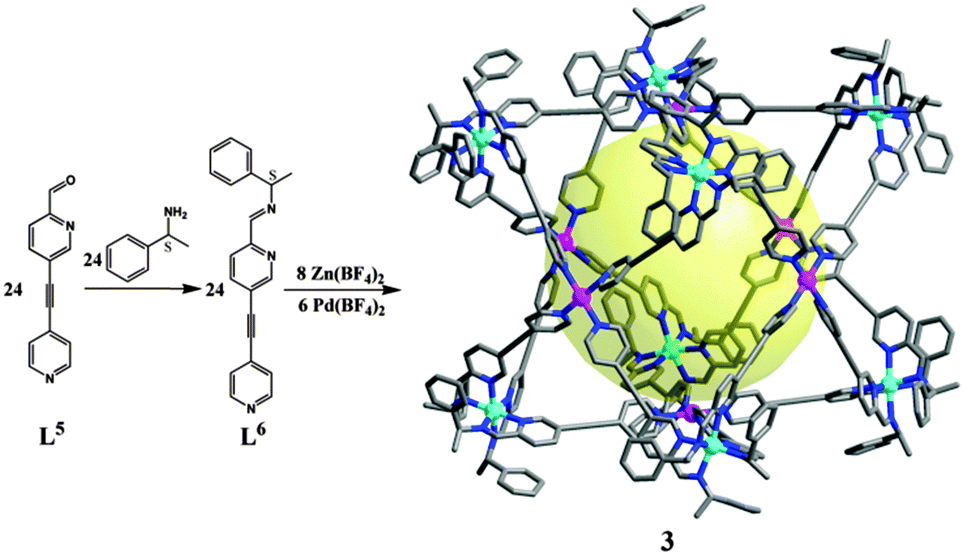 | ||
| Fig. 5 Synthetic path of 3 with a longer precursor L5; the large void inside the cube is shown as a yellow ball. Hydrogen atoms and counterions are omitted for clarity. Color legend: purple Pd; cyan Zn; blue N; gray C. | ||
In summary, we have successfully synthesized a series of new heterometallic chiral cubic cages by employing a heterotopic precursor ligand bearing a formyl group available for facile condensation with various amines. The structures and solution behavior of the cubic cages have been studied, and CD data confirm that homochiral cubic cages have been isolated. CD enhancement is observed with the formation of the cage structure. We demonstrate that a chiral amine is able to control the handedness of metal centers, leading to the diastereoselective formation of O symmetric cubic cages, i.e. the chirality of carbon centers of the chiral amine is transferred to the supramolecular entity. We are using this precursor ligand L1 to generate new ligands of different geometries and functionalities in the construction of various supramolecular coordination cages.
This work was supported by the 973 program (2014CB845603) and the National Natural Science Foundation of China (21125102, 21390390 and 21473139).
Notes and references
- (a) M. D. Pluth, R. G. Bergman and K. N. Raymond, Science, 2007, 316, 85 CrossRef CAS PubMed; (b) M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418 CrossRef CAS PubMed; (c) M. Yamashina, Y. Sei, M. Akita and M. Yoshizawa, Nat. Commun., 2014, 5, 4662 CAS; (d) P. Mal, B. Breiner, K. Rissanen and J. R. Nitschke, Science, 2009, 324, 1697 CrossRef CAS PubMed; (e) K. Li, L.-Y. Zhang, C. Yan, S.-C. Wei, M. Pan, L. Zhang and C.-Y. Su, J. Am. Chem. Soc., 2014, 136, 4456 CrossRef CAS PubMed.
- (a) M. M. J. Smulders, I. A. Riddell, C. Browne and J. R. Nitschke, Chem. Soc. Rev., 2013, 42, 1728 RSC; (b) R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810 CrossRef CAS PubMed; (c) S. Leininger, B. Olenyuk and P. J. Stang, Chem. Rev., 2000, 100, 853 CrossRef CAS PubMed; (d) S. R. Seidel and P. J. Stand, Acc. Chem. Res., 2002, 35, 972 CrossRef CAS PubMed.
- T. D. Hamilton and L. R. MacGillivray, Cryst. Growth Des., 2004, 4, 419 CAS.
- W. Xuan, M. Zhang, Y. Liu, Z. Chen and Y. Cui, J. Am. Chem. Soc., 2012, 134, 6904 CrossRef CAS PubMed.
- (a) T. Liu, Y. Liu, W. Xuan and Y. Cui, Angew. Chem., Int. Ed., 2010, 49, 4121 CrossRef CAS PubMed; (b) C. Zhao, Q.-F. Sun, W. M. Hart-Cooper, A. G. DiPasquale, F. D. Toste, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2013, 135, 18802 CrossRef CAS PubMed; (c) C. J. Brown, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2009, 131, 17530 CrossRef CAS PubMed.
- (a) S. Wan, L.-R. Lin, L. Zeng, Y. Lin and H. Zhang, Chem. Commun., 2014, 50, 15301 RSC; (b) A. J. Terpin, M. Ziegler, D. W. Johnson and K. N. Raymond, Angew. Chem., Int. Ed., 2001, 40, 157 CrossRef CAS; (c) S. P. Argent, T. Riis-Johannessen, J. C. Jeffery, L. P. Harding and M. D. Ward, Chem. Commun., 2005, 4647 RSC; (d) J. L. Bolliger, A. M. Belenguer and J. R. Nitschke, Angew. Chem., Int. Ed., 2013, 52, 7958 CrossRef CAS PubMed; (e) N. Ousaka, J. K. Clegg and J. R. Nitschke, Angew. Chem., Int. Ed., 2012, 51, 1464 CrossRef CAS PubMed.
- (a) C. Browne, S. Brenet, J. K. Clegg and J. R. Nitschke, Angew. Chem., Int. Ed., 2013, 52, 1944 CrossRef CAS PubMed; (b) I. S. Tidmarsh, T. B. Faust, H. Adams, L. P. Harding, L. Russo, W. Clegg and M. D. Ward, J. Am. Chem. Soc., 2008, 130, 15167 CrossRef CAS PubMed.
- (a) W. J. Ramsay, T. K. Ronson, J. K. Clegg and J. R. Nitschke, Angew. Chem., Int. Ed., 2013, 52, 13439 CrossRef CAS PubMed; (b) W. Meng, B. Breiner, K. Rissanen, J. D. Thoburn, J. K. Clegg and J. R. Nitschke, Angew. Chem., Int. Ed., 2011, 50, 3479 CrossRef CAS PubMed.
- (a) H.-B. Wu and Q.-M. Wang, Angew. Chem., Int. Ed., 2009, 48, 7343 CrossRef CAS PubMed; (b) M. B. Duriska, S. M. Neville, B. Moubaraki, J. D. Cashion, G. J. Halder, K. W. Chapman, C. Balde, J.-F. Létard, K. S. Murray, C. J. Kepert and S. R. Batten, Angew. Chem., Int. Ed., 2009, 48, 2549 CrossRef CAS PubMed; (c) M. B. Duriska, S. M. Neville, J. Lu, S. S. Iremonger, J. F. Boas, C. J. Kepert and S. R. Batten, Angew. Chem., Int. Ed., 2009, 48, 8949 Search PubMed; (d) F. Reichel, J. K. Clegg, K. Gloe, K. Gloe, J. J. Weigand, J. K. Reynolds, C.-G. Li, J. R. Aldrich-Wright, C. J. Kepert, L. F. Lindoy, H.-C. Yao and F. Li, Inorg. Chem., 2014, 53, 688 CrossRef CAS PubMed.
- X.-P. Zhou, J. Liu, S.-Z. Zhan, J.-R. Yang, D. Li, K.-M. Ng, R. W.-Y. Sun and C.-M. Che, J. Am. Chem. Soc., 2012, 134, 8042 CrossRef CAS PubMed.
- M. M. J. Smulders, A. Jiménez and J. R. Nitschke, Angew. Chem., Int. Ed., 2012, 51, 6681 CrossRef CAS PubMed.
- (a) S. E. Howson, L. E. N. Allan, N. P. Chmel, G. J. Clarkson, R. Gorkum and P. Scott, Chem. Commun., 2009, 1727 RSC; (b) J. M. Dragna, G. Pescitelli, L. Tran, V. M. Lynch, E. V. Anslyn and L. D. Bari, J. Am. Chem. Soc., 2012, 134, 4398 CrossRef CAS PubMed; (c) Y. Yang, X.-L. Pei and Q.-M. Wang, J. Am. Chem. Soc., 2013, 135, 16184 CrossRef CAS PubMed; (d) S. E. Howson, L. E. N. Allan, N. P. Chmel, G. J. Clarkson, R. J. Deeth, A. D. Faulkner, D. H. Simpson and P. Scott, Dalton Trans., 2011, 40, 10416 RSC.
- Y. Cohen, L. Avram and L. Frish, Angew. Chem., Int. Ed., 2005, 44, 520 CrossRef CAS PubMed.
- C. Gütz, R. Hovorka, C. Klein, Q.-Q. Jiang, C. Bannwarth, M. Engeser, C. Schmuck, W. Assenmacher, W. Mader, F. Topić, K. Rissanen, S. Grimme and A. Lützen, Angew. Chem., Int. Ed., 2014, 53, 1693 CrossRef PubMed.
- S. H. Banyard, D. K. Stammers and P. M. Harrison, Nature, 1978, 271, 282 CrossRef CAS PubMed.
- M. Ziegler and A. Zelewsky, Coord. Chem. Rev., 1998, 177, 257 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis, characterization details and X-ray crystallographic (CIF) data. CCDC 1036634 (1a), 1036635 (1b), 1036633 (2). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc00087d |
| ‡ (a) Crystal data for 1a: C456H408B28F112N72Pd6Zn8, a = b = c = 25.9101(3) Å, α = β = γ = 90.00°, V = 17394.3(3) Å3, cubic space group P432, Z = 1, T = 100(2) K, 15768 reflections measured, 5506 unique (Rint = 0.0661), final R1 = 0.0763, wR2 = 0.2291 for 3516 observed reflections [I > 2σ(I)]. Flack factor = 0.02(2). (b) Crystal data for 1b: C456H408B28F112N72Pd6Zn8, a = b = c = 25.9709(3) Å, α = β = γ = 90.00°, V = 17517.1(4) Å3, cubic space group P432, Z = 1, T = 100(2) K, 16722 reflections measured, 5830 unique (Rint = 0.0920), final R1 = 0.0736, wR2 = 0.1931 for 4406 observed reflections [I > 2σ(I)]. Flack factor = 0.035(17). (c) Crystal data for 2: C432H360B28F112N72Pd6Zn8, a = b = 31.0248(10), c = 42.9572(17) Å, α = β = 90.00°, γ = 120.00°, V = 35808(2) Å3, trigonal space group P1c, Z = 2, T = 100(2) K, 84155 reflections measured, 22914 unique (Rint = 0.0541), final R1 = 0.0986, wR2 = 0.2846 for 15153 observed reflections [I > 2σ(I)]. |
| This journal is © The Royal Society of Chemistry 2015 |