
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Isotope sensitive branching and kinetic isotope effects to analyse multiproduct terpenoid synthases from Zea mays†
Nathalie
Gatto
a,
Abith
Vattekkatte
a,
Tobias
Köllner
a,
Jörg
Degenhardt
b,
Jonathan
Gershenzon
a and
Wilhelm
Boland
*a
aMax Planck Institute for Chemical Ecology, Hans-Knöll-Strasse 8, D-07745 Jena, Germany. E-mail: boland@ice.mpg.deb
bInstitute for Pharmacy, University of Halle, Hoher Weg 8, D-06120 Halle, Germany
First published on 30th January 2015
Abstract
Multiproduct terpene synthases TPS4-B73 and TPS5-Delprim from Zea mays exhibit isotopically sensitive branching in the formation of mono- and sesquiterpene volatiles. The impact of the kinetic isotope effects and the stabilization of the reactive intermediates by hyperconjugation along with the shift of products from alkenes to alcohols are discussed.
Terpenes constitute the largest and most diverse class of plant natural products with more than 30
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 members.1,2 Volatile terpenes, which represent a major class among herbivore induced volatiles, are synthesized by specific terpene synthases. These enzymes have been extensively investigated in recent decades, and various cDNAs encoding plant terpene synthases involved in primary and secondary metabolism have been cloned and characterized.3,4 Terpene synthases are able to convert acyclic precursors such as geranyl diphosphate (GDP, C10), farnesyl diphosphate (FDP, C15) and geranylgeranyl diphosphates (GGDP, C20) into cyclic monoterpenes (C10), sesquiterpenes (C15) and diterpenes (C20), respectively.5 As the first reaction step, the unsaturated diphosphates dissociate into highly reactive carbocations and diphosphate anions. These cations interact with electron-rich double bonds in their vicinity resulting in intramolecular cyclizations. In addition, rearrangements, including hydride or methyl shifts, occur prior to stabilization by either deprotonation or reaction with a nucleophile.
000 members.1,2 Volatile terpenes, which represent a major class among herbivore induced volatiles, are synthesized by specific terpene synthases. These enzymes have been extensively investigated in recent decades, and various cDNAs encoding plant terpene synthases involved in primary and secondary metabolism have been cloned and characterized.3,4 Terpene synthases are able to convert acyclic precursors such as geranyl diphosphate (GDP, C10), farnesyl diphosphate (FDP, C15) and geranylgeranyl diphosphates (GGDP, C20) into cyclic monoterpenes (C10), sesquiterpenes (C15) and diterpenes (C20), respectively.5 As the first reaction step, the unsaturated diphosphates dissociate into highly reactive carbocations and diphosphate anions. These cations interact with electron-rich double bonds in their vicinity resulting in intramolecular cyclizations. In addition, rearrangements, including hydride or methyl shifts, occur prior to stabilization by either deprotonation or reaction with a nucleophile.
In addition to terpene synthases, which generate a single product, there are multiproduct terpene synthases, which generate a bouquet of acyclic and cyclic products from a single precursor.3 These enzymes have the advantage that a single mutation may generate a bouquet of new compounds improving the plant's defense.6 The δ-selinene synthase and γ-humulene synthase from Abies grandis hold the current record, producing 52 and 34 different sesquiterpenes, respectively.3 To better understand the mechanistic details of multiproduct terpenoid synthases and to determine whether the final deprotonation of cationic intermediates en route to mono- and sesquiterpenes is rate-limiting, deuterium kinetic isotope effects and product composition were investigated for TPS4 and TPS5 enzymes from B73 and Delprim maize varieties. The different terpene profiles were controlled by allelic variation of the closely related terpene synthase genes, TPS4 and TPS5.6 Although both enzymes showed the typical properties of sesquiterpene synthases, they not only accepted FDP (C15) but also GDP (C10) as a substrate. Both substrates were converted almost exclusively into two types of cyclic products, with cyclohexenyl- and bicyclo[3.1.0]hexyl moieties as structural elements (Scheme 1). To study the kinetics of the reaction cascade, we synthesized labeled substrates with deuterium atoms completely surrounding the cationic center at C(3) of the key intermediates (Scheme 1). Depending on their stability and ease with which deprotonation reactions, different reaction channels may be favoured.
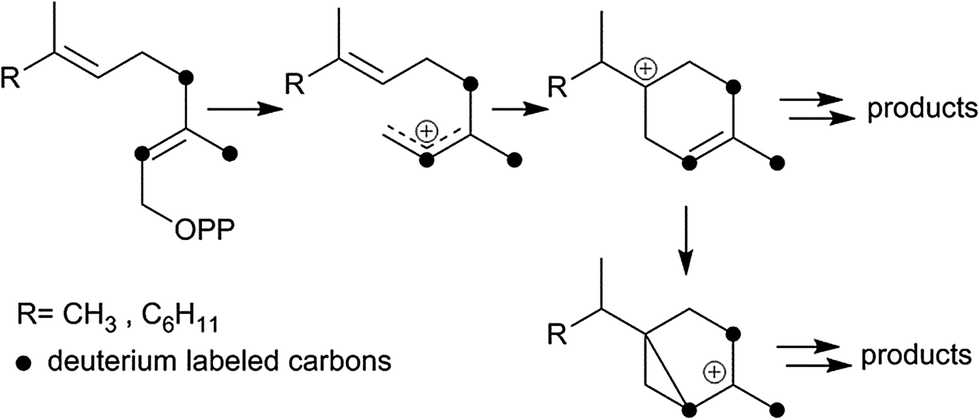 | ||
| Scheme 1 Isotope sensitive branching strategy. | ||
[2-2H]- and [2,4,4,9,9,9-2H6]-GDP as well as [2-2H]- and [2,4,4,13,13,13-2H6]-FDP were prepared according to the protocol of Arigoni et al. (Scheme 2).7 For hexadeuterated analogues, [1,1,1,3,3-2H5] ketones 1b and 1d were prepared by proton-deuterium exchange reaction in D2O in presence of potassium carbonate. The subsequent Peterson olefination of the ketones 1a–d with [2,2-2H2]-trimethylsilylacetate 2 afforded the [2-2H]- or [2,4,4,9,9,9-2H6]-carboxylic acids. The corresponding methyl esters were converted to the mono- and hexadeuterated alcohols 4a–d using diisopropyl aluminum hydride. Labeled diphosphates were prepared according to Woodside et al.8 and yielded the corresponding trisammonium geranyl diphosphates 5a–b and trisammonium farnesyl diphosphates 5c–d.
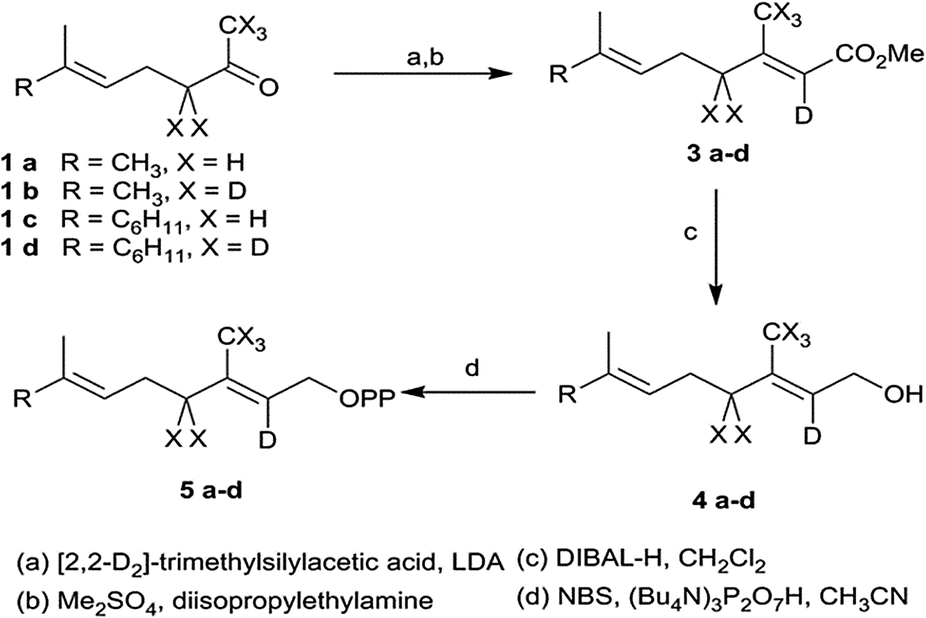 | ||
| Scheme 2 Synthesis of deuterated GDP and FDP. | ||
The synthesized deuterium labelled GDP and FDP substrates were incubated with TPS4 and TPS5. The resulting terpenoid profiles were quantified by GC-FID and showed predominantly cyclic substances with a terpinan/sabinan (monoterpenes) and a bisabolane/sesquisabinane (sesquiterpenes) skeleton, respectiveely.9 Acyclic products were also present, including β-myrcene, and linalool from GDP and two sesquiterpenoids, (E)-β-farnesene and (3R)-(E)-nerolidol from FDP. The acyclic terpenes result from the deprotonation or water-capture of the first carbocation formed upon cleavage of the diphosphate. Comparing product ratios from labeled versus unlabeled precursors, we found that both cyclases exhibited a constant 1:2 ratio (acyclic/cyclic monoterpenes) for the GDP substrates as well as constant ratios of 1:5 for TPS4 and 1:10 for TPS5 for the FDP substrates. These results suggest that the labeling with stable isotopes did not influence the kinetics of the first ring closure, although the delocalized positive charge is partly surrounded by deuterium.
The relative overall rates of mono- and sesquiterpene formation from incubation of deuterated GDP and FDP with TPS4 and TPS5 and the apparent isotope effects kH/kD are given in Table 1. Incubation of monodeuterated [2H1]-GDP 5a and [2H1]-FDP 5c with both cyclases resulted in relative overall rates that were almost identical to those obtained with unlabeled substrates. The isotope effects for monodeuterated [2H1]-GDP 5a and [2H1]-FDP 5c are close to unity. However, the incubation of hexadeuterated substrates noticeably, reduced the rate of product formation. The relative total rate was reduced by 15% (TPS4) or 25% (TPS5) upon incubation with [2H6]-GDP 5b. The rate of suppression corresponded to an apparent isotope effect kH/kD of 1.17 and 1.33, respectively. Similarly, enzymatic incubation with [2H6]-FDP 5d decreased the relative amount of sesquiterpenes (19% and 21% with TPS4 and TPS5, respectively) equivalent to kH/kD of 1.23 and 1.25, respectively. The observed overall rate reductions upon incubation with hexadeuterated GDP and FDP result from primary isotope effects (loss of 2H+). In contrast, the isotope labels in the monodeuterated substrates have no direct influence on the reaction mechanism, since the C(2)–D bond is not cleaved during the whole cyclization cascade. Moreover, the variations in the product profiles and the overall rates were a result of apparent secondary isotope effects. The difference in the rate of volatile formation was even more pronounced in the case of sesquiterpenes. The production of all cyclic products requires an (E/Z)-isomerization step of the C(2)–C(3) double bond of the (E)-configured substrates, which is achieved through tertiary allylic phosphate intermediates, and linalyl- and nerolidyl diphosphate, respectively. This phenomenon has been reported for the product formation of maize sesquiterpene synthases TPS6, TPS10, TPS11,3,10 and other terpene synthases.11
| Substrate | TPS4-B73 | TPS5-Delprim | ||
|---|---|---|---|---|
| Relative ratea (%) | k H/kD | Relative ratea (%) | k H/kD | |
| a Relative overall rates compared to those of incubation with unlabeled GDP or FDP substrates (set at 100). Mean values from six replicates, ±1 SD. b Apparent total rate isotope effects compared to those of incubation with unlabeled GDP or FDP substrates. Note: oxygenated cyclic volatiles not considered. | ||||
| [2H1]-GDP 5a | 101.09 ± 0.45 | ∼1b | 99.98 ± 0.84 | ∼1b |
| [2H6]-GDP 5b | 85.06 ± 0.84 | 1.17b | 75.02 ± 0.78 | 1.33b |
| [2H1]-FDP 5c | 106.21 ± 6.95 | ∼1b | 101.30 ± 1.11 | ∼1b |
| [2H6]-FDP 5d | 80.95 ± 7.55 | 1.23b | 79.65 ± 1.94 | 1.25b |
As mentioned above, the oxygenated cyclic volatiles were not considered in the present data. To estimate the weight of this approximation, quantitative kinetic measurements were carried out. For [2H6]-FDP 5d with TPS4 with kH/kD = 1.15, 13% fewer sesquiterpene formation (relative to FDP). Similar results were obtained when the oxygenated cyclic volatiles were not considered (a 19% decrease in the volatile production corresponded to a kH/kD = 1.23) and justify the approximation made above.
Both cyclases showed minor changes in the formation of limonene (4), α-terpinolene (6) and linalool (7) (Fig. 1). In contrast a significant decrease of α-thujene (1) and sabinene (2) along with a corresponding increase in sabinene hydrate (5) was observed when TPS4 and TPS5 were incubated with [2H1]-GDP 5a or [2H6]-GDP 5b when compared to incubation with GDP. The observed kinetic isotope effects (KIE)s, corresponding to a difference of 5 to 6 deuterium atoms between the substrates, are in the range of kH/kD = 2.91–5.68. These values are in agreement with those observed for terminating deprotonation reactions of other monoterpene cyclases.12,13 In case of substrates differing by only one deuterium atom, the observed KIEs were much smaller (within a range of kH/kD = 1.10–1.18).
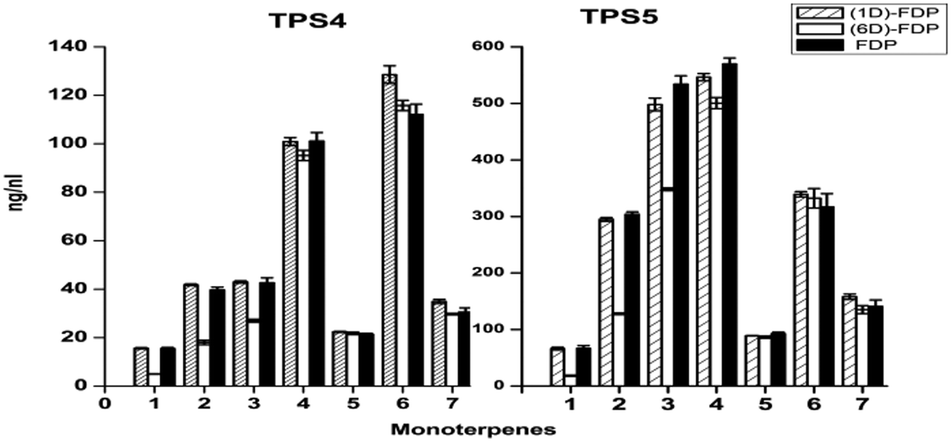 | ||
| Fig. 1 Product distribution of main monoterpenes from incubations of deuterated GDP with TPS4 and TPS5. (1) α-Thujene, (2) sabinene, (3) β-myrcene, (4) limonene, (5) sabinene hydrate, (6) α-terpinolene, (7) linalool. | ||
The first set of sesquiterpenes, comprising (S)-β-bisabolene (11), (E)-γ-bisabolene (12) and zingiberene (9), (Fig. 2), were not affected by the isotopically sensitive branching experiments, and only minor changes in product composition were observed with hexadeuterated substrates (relative to unlabeled or monodeuterated analogues). A significant decrease in 7-epi-sesquithujene (1), sesquithujene (2), sesquisabinenes A (5) and B (6) and (E)-α-bergamotene isomers (3, 4) was observed after incubating [2H6]-FDP 5d with TPS4 (10–64%) and with TPS5 (26–61%), when compared to natural FDP (Fig. 2).
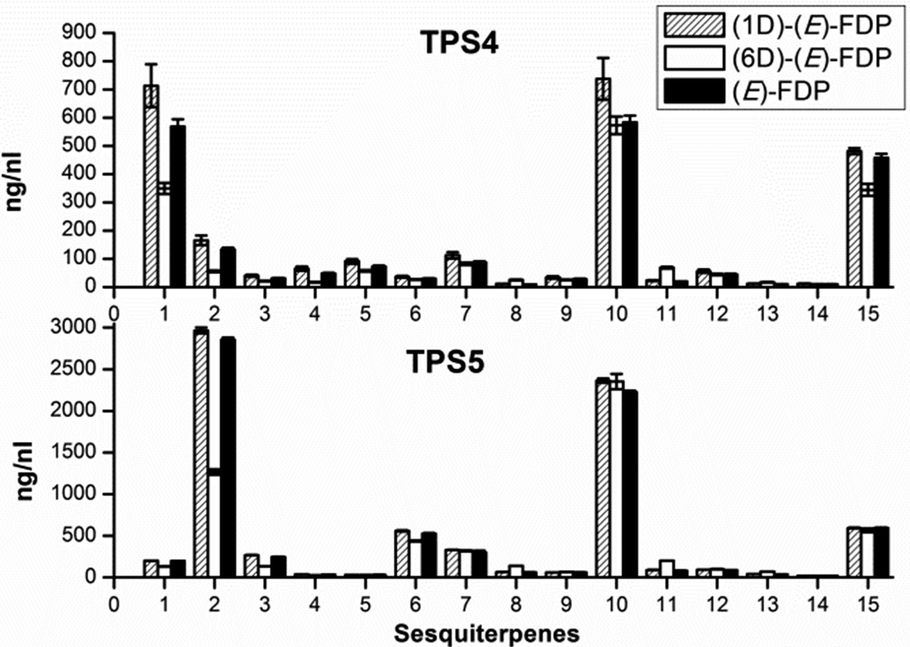 | ||
| Fig. 2 Product distribution of main sesquiterpenes from incubations of deuterated FDP with TPS4 and TPS5. (1) 7-epi-Sesquithujene, (2) sesquithujene, (3) (Z)-α-bergamotene, (4) (E)-α-bergamotene, (5) sesquisabinene A, (6) sesquisabinene B, (7) (E)-β-farnesene, (8) γ-curcumene, (9) zingiberene, (10) (S)-β-bisabolene, (11) β-curcumene, (12) (E)-γ-bisabolene, (13) 7-epi-sesquithujene hydrate**, (14) sesquithujene hydrate**, (15) (3R)-(E)-nerolidol. ** Tentatively assigned structure. | ||
This rate suppression was coupled with a corresponding increase in the formation of γ- and β-curcumene isomers, sesquithujene hydrate and 7-epi-sesquithujene hydrate by 175–400% for TPS4 and 193–255% for TPS5. Because the biosynthesis of sesquiterpene volatiles is mechanistically much more complex than that of monoterpenes volatiles, only the observed KIEs for the deprotonation reactions leading to 7-epi-sesquithujene and sesquithujene are presented. Thus, the observed KIEs were in the range of kH/kD = 3.38–4.08 for the terminating deprotonation reaction, leading to 7-epi-sesquithujene, and in the range of kH/kD = 4.17–6.24 for the terminating deprotonation reaction leading to sesquithujene. These values are in agreement with those obtained with the monoterpene series and with previous studies on other monoterpene cyclases.12,13 The corresponding KIEs were close to unity, demonstrating that the monodeuterated labelling at C(3) had almost no influence on the reaction cascade.
We previously proposed6 a reaction mechanism for the formation of mono- and sesquiterpene products by TPS4 and TPS5. The stabilization of the carbocationic intermediates is partly facilitated by interactions (e.g. π–cation interactions) with the hydrophobic, aromatic-rich environment and the DDxxD motif of the active-site of the enzyme.9 Nevertheless, hyperconjugation is an important factor for the stability of carbocation intermediates. Hyperconjugation is considered to be the interaction of the vacant p-type orbital on the cationic center with adjacent C–H or C–C σ-bonds. Because a C–D bond is stronger than a C–H bond, a C–H hyperconjugation stabilizes an adjacent positive charge more than a C–D hyperconjugation. Accordingly, reactions involving breaking C–D are slowed down. Such hyperconjugative weakening in reaction intermediates due to isotopes induces secondary KIEs. In the present study, all KIEs lead to the alteration of product distributions after isotopically sensitive branching. To illustrate the effects of hyperconjugation, the secondary KIEs of the cyclisation reactions of [2H6]-FDP 5d (Fig. 3) and [2H6]-GDP 5b (Fig. 4) are discussed.
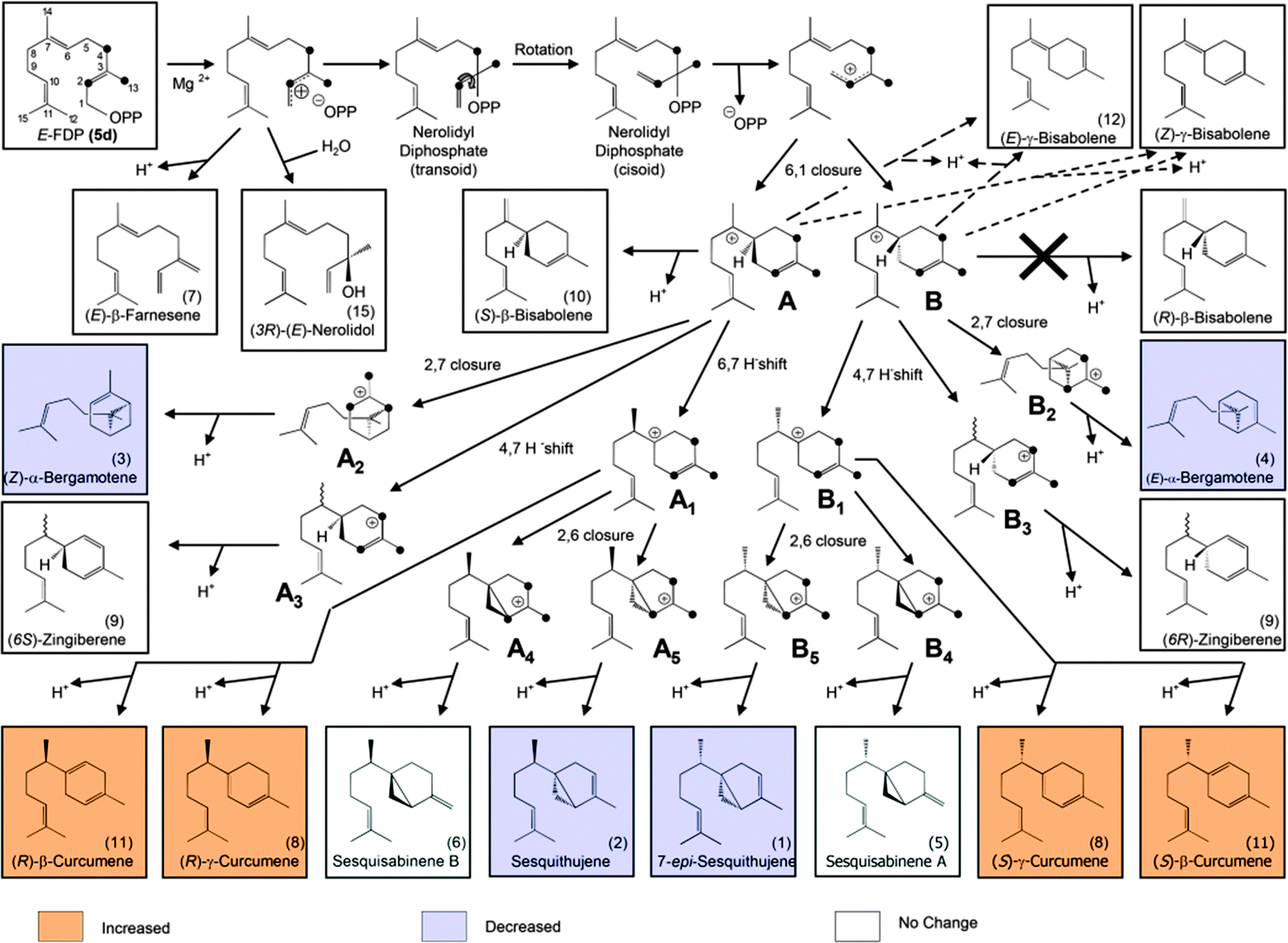 | ||
| Fig. 3 Proposed mechanism for sesquiterpene formation from [2H6]-FDP. The black dots represent deuterated carbons.6 | ||
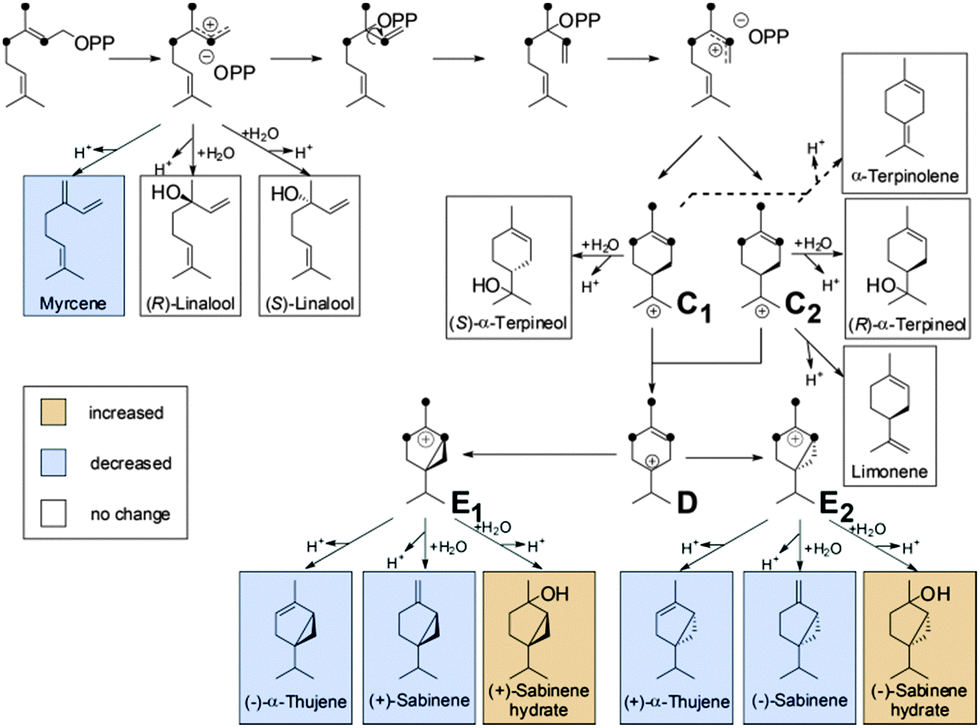 | ||
| Fig. 4 Proposed reaction mechanism for the formation of monoterpenes by TPS4 and TPS5 from (E)-[2H6]-GDP [7b]. | ||
From [2H6]-FDP 5d, after the initial ionization–isomerization–ionization sequence, the cyclization cascade is initiated by the formation of (S)- and (R)-bisabolyl cations (A and B). These first carbocations can be directly deprotonated to produce (S)-β-bisabolene without noticeable KIEs (the positive charge being located far from the deuterated center). Tertiary carbocations A1 and B1 are almost as stable as A and B because the positive charge is distant from the deuterium labeled carbon center and, hence, can be stabilized by C–H hyperconjugation. Secondary carbocations A3 and B3, which are energetically less favorable than tertiary ones (e.g. A4 and B4), are relatively stable due to surrounding hydrogen atoms. Carbocations A4, A5, B4 and B5 are the least stable ones, since the positive charge is fully surrounded by deuterium atoms and hence, cannot be delocalized and stabilized by C–D hyperconjugation. The stability of terminal carbocations results in the production of the corresponding cyclic terpene. Taken together, these observations are consistent with the product distribution obtained by isotopically sensitive branching experiments. Deuterium isotope effects are less pronounced in the monodeuterated analogues (Table 1). Nevertheless, strong KIEs for the formation of sesquithujene, 7-epi-sesquithujene or sesquisabinenes A and B were observed after incubation of both enzymes with hexadeuterated substrates (Fig. 4). In case of the bisabolyl carbocations A and B, (Fig. 3), minor KIEs were observed for β- or γ-bisabolene formation since the reactive carbocation intermediates are located in an exclusive [1H]-environment. Similarly, only minor KIEs were observed for zingiberene isomers since the final deprotonation involves only the loss of a hydrogen atom (no primary KIE). Deuterium isotope effects on the monoterpene product distribution can also be rationalized in terms of hyperconjugation. As shown in Fig. 4, the first step of the cyclization cascade is the C(1)–C(6) ring closure of the linaloyl diphosphate, resulting in the formation of (S)- and (R)-terpinyl carbocations C1 and C2. As for bisabolyl intermediates A and B in the biosynthesis of sesquiterpenes, the positive charge is positioned far away from the influence of the deuterated carbons. Hence terminating steps via deprotonation or water capture leading to (S)-(−)-limonene and α-terpineol or α-terpinolene, can occur spontaneously and are not affected by kinetic isotope effects. As discussed below, minor variations in product distribution were observed when unlabeled or [2H1]-GDP were used as substrates. These minor KIEs observed for limonene, α-terpinolene and α-terpineol reflect the absence of destabilizing effects in the two α-terpinyl carbocations. From C1 and C2, the cyclization cascade proceeded to carbocation D which was subsequently rearranged to the tertiary carbocations E1 and E2. Because the positive charge fully surrounded by deuterium atoms is less efficiently stabilized as by C–H hyperconjugation, strong deuterium isotope effects on the formation of sabinene, sabinene hydrate and α-thujene were observed. Obviously, E1 and E2 preferentially stabilize by reacting with water as a nucleophile rather than losing a positively charged hydrogen isotope.
Multiproduct terpene synthases TPS4 and TPS5 from Zea mays show isotopically sensitive branching in the reaction cascade of prenyl diphosphates en route to mono- and sesquiterpene volatiles via a common carbocationic intermediate along a branched reaction sequence. The primary kinetic isotope effects of deuterium atoms on terminating deprotonations and effects resulting from lower stabilization of the reactive intermediates by hyperconjugation direct the reaction to an enhanced formation of alcohols instead of olefinic products. Accordingly, the extensive deuterium labeling of intermediary cations is a valuable tool to identify branching points in complex cyclization sequences of multiproduct terpenoid synthases.
Notes and references
- J. D. Connolly and R. A. Hill, Dictionary of Terpenoids, Chapman & Hall, New York, 1992 Search PubMed.
- J. C. Sacchettini and C. D. Poulter, Science, 1997, 277, 1788–1789 CrossRef CAS.
- J. Degenhardt, T. G. Köllner and J. Gershenzon, Phytochemistry, 2009, 70, 1621–1637 CrossRef CAS PubMed.
- D. J. Schenk, C. M. Starks, K. R. Manna, J. Chappell, J. P. Noel and R. M. Coates, Arch. Biochem. Biophys., 2006, 448, 31–44 CrossRef CAS PubMed.
- E. M. Davis and R. Croteau, Biosynthesis: Aromatic Polyketides, Isoprenoids, Alkaloids, Springer-Verlag, Berlin, 2000, pp. 53–95 Search PubMed.
- T. G. Köllner, C. Schnee, J. Gershenzon and J. Degenhardt, Plant Cell, 2004, 16, 1115–1131 CrossRef PubMed.
- D. Arigoni, D. E. Cane, J. H. Shim, R. Croteau and K. A. Wagschal, Phytochemistry, 1993, 32, 623–631 CrossRef CAS.
- A. B. Woodside, Z. Huang and C. D. Poulter, Org. Synth., 1993, 8, 616–623 Search PubMed.
- T. G. Köllner, P. E. O'Maille, N. Gatto, W. Boland, J. Gershenzon and J. Degenhardt, Arch. Biochem. Biophys., 2006, 448, 83–92 CrossRef PubMed.
- T. G. Köllner, C. Schnee, S. Li, A. Svatos, B. Schneider, J. Gershenzon and J. Degenhardt, J. Biol. Chem., 2008, 283, 20779–20788 CrossRef PubMed.
- D. E. Cane, H. T. Chiu, P. H. Liang and K. S. Anderson, Biochemistry, 1997, 36, 8332–8339 CrossRef CAS PubMed.
- H. J. Pyun, R. M. Coates, K. C. Wagschal, P. McGeady and R. B. Croteau, J. Org. Chem., 1993, 58, 3998–4009 CrossRef CAS.
- K. C. Wagschal, H. J. Pyun, R. M. Coates and R. Croteau, Arch. Biochem. Biophys., 1994, 308, 477 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cc10395e |
| This journal is © The Royal Society of Chemistry 2015 |