
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMesostructured zeolites: bridging the gap between zeolites and MCM-41†
Teerawit
Prasomsri
a,
Wenqian
Jiao
bc,
Steve Z.
Weng
a and
Javier
Garcia Martinez
*ac
aRive Technology, Inc., 1 Deer Park Drive, Monmouth Junction, NJ 08852, USA. E-mail: j.garcia@ua.es
bShanghai Key Lab of Green Chemistry and Chemical Processes, Department of Chemistry, East China Normal University, Shanghai, China
cMolecular Nanotechnology Lab, Department of Inorganic Chemistry, University of Alicante, Campus de San Vicente, 03690 Alicante, Spain
First published on 1st April 2015
Abstract
Surfactant-templating is one of the most versatile and useful techniques to implement mesoporous systems into solid materials. Various strategies based on various interactions between surfactants and solid precursors have been explored to produce new structures. Zeolites are invaluable as size- and shape-selective solid acid catalysts. Nevertheless, their micropores impose limitations on the mass transport of bulky feed and/or product molecules. Many studies have attempted to address this by utilizing surfactant-assisting technology to alleviate the diffusion constraints. However, most efforts have failed due to micro/mesopore phase separation. Recently, a new technique combining the uses of cationic surfactants and mild basic solutions was introduced to synthesise mesostructured zeolites. These materials sustain the unique characteristics of zeolites (i.e., strong acidity, crystallinity, microporosity, and hydrothermal stability), including tunable mesopore sizes and degrees of mesoporosity. The mesostructured zeolites are now commercially available through Rive Technology, and show superior performance in VGO cracking. This feature article provides an overview of recent explorations in the introduction of mesoporosity into zeolites using surfactant-templating techniques. Various porous materials, preparation methods, physical and catalytic properties of mesostructured zeolites will be discussed.
Introduction
Zeolites are a family of aluminosilicate materials with crystalline framework structures constructed by TO4 tetrahedral (T = Si, Al) units. Each TO4 tetrahedron covalently bonds with four neighboring TO4 tetrahedrons forming three-dimensional zeolite frameworks which include cavities and channels of 0.2–1.3 nm in size.1–23 The tetrahedrons link to one another in a variety of ways leading to the emerging of diverse zeolite topological structures. So far, 218 distinct zeolite framework topologies have been recognized by the Structure Commission of the International Zeolite Association (IZA-SC).4 Due to the continued development of novel synthesis strategies, the number of topological structures is increasing rapidly.5By virtue of the flexible framework structures, controllable framework compositions and uniform porosities,6 zeolites exhibit unique physical/chemical properties. In comparison with amorphous materials, they exhibit better ion exchange abilities, stronger acidities and higher thermal/hydrothermal stabilities. Based on these characteristics, zeolites have found a variety of applications in many areas including the kitchen and space, as well as, industries,7,8 such as purification, adsorption, separation, and catalysis in petroleum refining, as well as petrochemical and fine chemical processing. The intrinsic pore structures of zeolites, i.e., the pore opening dimensions, channel size, shape and inter-connectivity, may affect their practical applications as molecules need to enter the channels to either be stored there or react and transform into other compounds.8 The typically small sized channels (<0.8 nm) and cavities (< 1.3 nm) of zeolites can offer size/shape selectivity and play a role as molecular sieves. On the other hand, they can impose diffusion limitations, which may hinder the large-sized reactant/product molecules from approaching/leaving the active sites in the pores, leading to lowered conversion or further reactions which prompt coke deposition and catalyst deactivation.9–14
Mass transfer limitations may greatly affect reaction processes in the industrial use of zeolites.6,15,16 Thus, much effort has been made focussing on alleviating the diffusional constraints imposed by zeolitic microporous structures. One method involves the preparation of nano-sized zeolite particles to increase the external surface area and shorten the diffusion path lengths.17–21 Nevertheless, zeolite nano-particles with crystal size below 100 nm not only may cause filtration problems due to their colloidal nature, but also a decrease of micropore volume and a reduction of thermal/hydrothermal stability corresponding to the poor crystalline structures.6 Another procedure usually applied to improve the diffusion of large-sized molecules into the host porous materials is the synthesis of extra-large pore zeolites with channel windows circumvented by more than 12 T atoms.22–26 In 1988, Davis and co-workers reported the first successful synthesis of extra-large microporous molecular sieves with pore openings consisting of 18 T atoms in the presence of amine structure directing agents.27–29 These molecular sieves, coined VPI-5, exhibited higher adsorption efficiency of triisopropyl benzene in comparison with conventional faujasite (FAU) zeolite, which further confirmed the presence of large-sized pores in the new materials.27 Consequently, some other extra-large microporous zeolites were also synthesized, such as Cloverite,30,31 JDF-2032 and ULM-5,33 which have phosphate-based frameworks, as well as UTD-134 and CIT-5,23,35 which have pure silica or aluminosilicate frameworks. Due to the modest increase of the pore size, the extra-large microporous molecular sieves can, to some extent, circumvent the diffusion limitations and improve the mass transfer in the reaction processes. However, in most cases, their complicated synthesis, the need of costly organic structure directing agents, and their inferior acidity and thermal/hydrothermal stability caused by the framework structural features of extra large microporous molecular sieves exclude their practical industrial use.6
Expansion of the pore size using surfactants
Synthesis of mesoporous materials
Besides the large amount of work dedicated to widening the channels' window sizes or reducing the particle sizes of microporous zeolites, sizeable attention has also been directed toward the synthesis of molecular sieves with even larger pore sizes on the mesoscale (2 nm < d < 50 nm) to solve the diffusion problems. In the 90's, a new family of ordered mesoporous silica and aluminosilicate materials, M41S's, was developed by the Mobil Oil Company in a soft-templating method using quaternary ammonium surfactant micellar aggregates, rather than molecular species as templates.36,37 MCM-41 with its hexagonal arrangement of uniform mesopores is a typical phase produced. The pore size of the materials can be adjusted from 1.5 to larger than 10 nm and the specific surface area reached 700 m2 g−1 with pore volumes increased to 0.7 cc g−1 by hydrocarbon sorption. To illustrate the formation process of these materials, a liquid crystal templating (LCT) mechanism was proposed: the surfactant liquid crystal structures (surfactant micelles) serve as organic templates and the properties of the surfactant, such as surfactant chain length and solution chemistry, may greatly affect the materials' textural and structural properties.In the LCT mechanism (Fig. 1), mesostructured organic–inorganic hybrids formed either in a “true” liquid-crystal templating procedure or in a cooperative self-assembly pathway.9,38,39 Either way, the interactions between organic template molecules and the inorganic silicon species were immensely important to direct the occurrence of mesoporous structures. Six different synthesis pathways, namely S+I−, S−I+, S−M+I−, S+X−I+, S0I0 and N0I0, have been employed to synthesize mesoporous molecular sieves and explain the assembling process of the mesostructures, depending on the utility of different silica sources, the addition of varied surfactant compounds and tuned synthesis conditions.38–41 In the pathways, S represents the surfactants, where S+ and S− are the cationic and anionic templates, respectively. I represents the inorganic species, while M+ symbolizes mediating cations and X− is the mediating anionic species. I0 is hydrated neutral inorganic oligomer; S0 and N0 are the neutral amine and the neutral surfactant, respectively. Many procedures in which cationic surfactants were used as templates for the preparation of mesostructured silica materials have been reported. As the isoelectric point of silica lies at a pH value of ∼2,42 silicon species in alkaline solutions are negatively charged which can be linked and stabilized by cationic surfactants (S+) through strong electrostatic Coulomb forces in the S+I− pathway, leading to the formation of ordered mesostructured silica materials such as M41S,36,37 FDU-2,43 FDU-11 and FDU-13,44 SBA-2,45 SBA-646 and SBA-8,47etc. Likewise, cationic surfactants can also be used as templates to prepare silica mesostructures in strong acidic solutions (pH < 2) where the silicon species are positively charged. In this case, the cationic surfactant molecules cannot be directly connected with silicon species due to the strong repulsion force. Therefore, mesostructured materials can only be prepared through S+X−I+ reactions in strong acidic solutions.48
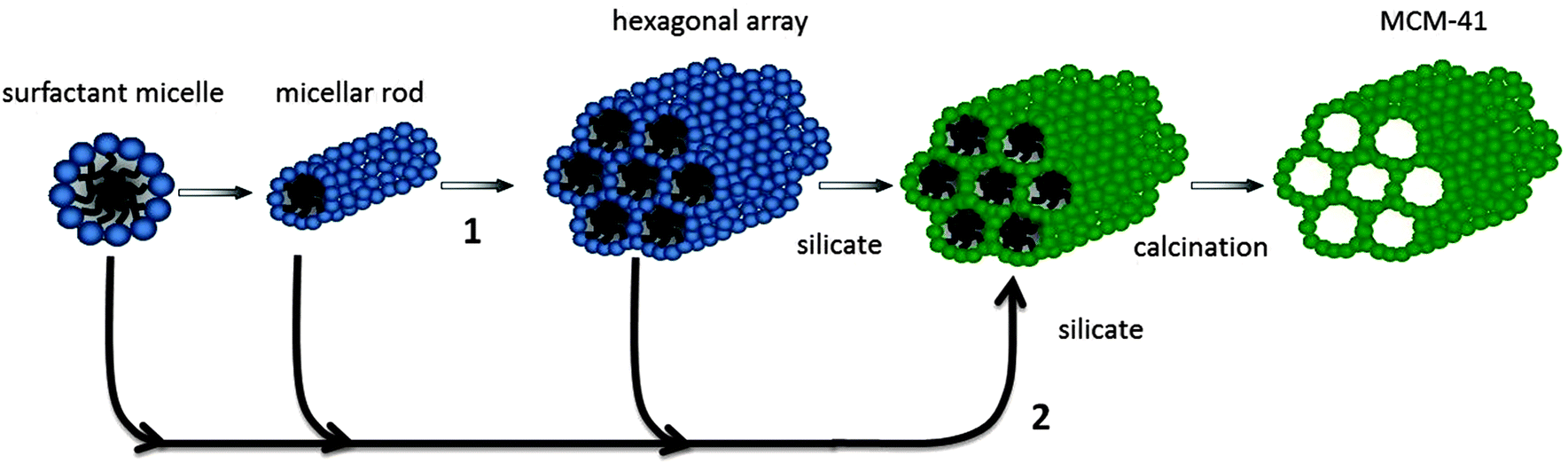 | ||
| Fig. 1 Schematic illustration for the liquid-crystal templating mechanism: (1) preformation of surfactant micellar rods, and (2) formation of surfactant– silicate rods on the basis of organic–inorganic interaction. Adapted with permission from ref. 39. Copyright 1992 American Chemical Society. | ||
Anionic surfactants, due to their highly potent detergency, low toxicity and low cost of manufacture, are much more promising as templates for the fabrication of ordered mesoporous materials.49 However, mesoporous silica materials were rarely synthesized when anionic surfactants were introduced as templates, in either the S−I+ or the S−X+I− pathway. Instead some mesoporous metal oxides, such as Al2O3, W and Mo oxides, were successfully prepared.48,50 As an exception, Che and co-workers once synthesized a family of mesoporous silica materials (AMS-n) with anionic surfactants as structure directing agents (SDAs). 3-Aminopropyltrimethoxysilane (APS) or N-trimethoxysilylpropyl-N,N,N-trimethylammonium (TMAPS) was added as a co-structure-directing agent (CSDA).49,51–53 The APS or TMAPS molecules interacting with the anionic surfactant molecules can act as a silicon co-precursor to co-condense with inorganic silica species throughout the alkoxysilane site (Fig. 2). Therefore, mesostructured silica materials formed more likely in an S−N+–I− pathway, instead of an S−M+I− pathway, where N+ are cationic amino groups of organoalkoxysilanes.38,39
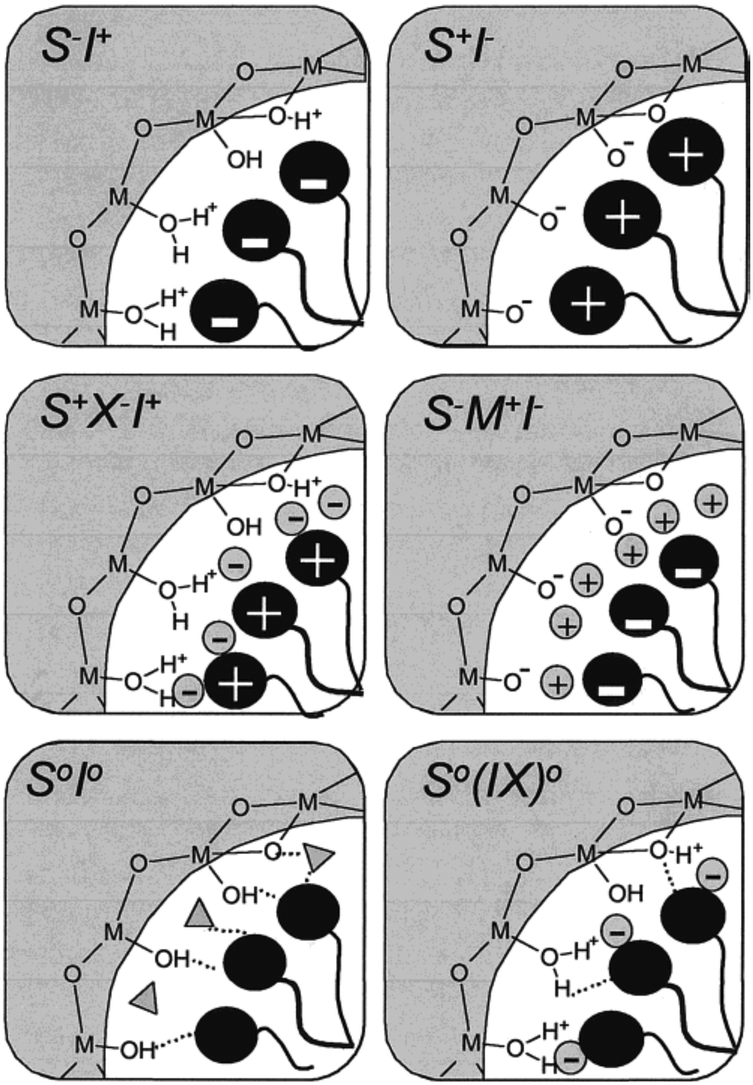 | ||
| Fig. 2 Schematic illustration of the different types of silica–surfactant interfaces. Reprinted with permission from ref. 9. Copyright 2002 American Chemical Society. | ||
Besides ionic surfactants, neutral amines and PEO-derived non-ionic surfactants were also used as templates for the synthesis of mesoporous silica materials. A series of HMS (hexagonal mesoporous silica)54,55 and MSU (Michigan State University)56 silica molecular sieves were prepared by Pinnavaia and co-workers under neutral conditions using neutral amine micelles (dodecylamine, hexadecylamine, N,N-dimethyl dodecylamine, N,N-dimethyl hexadodecylamine, etc., S0) and poly(ethylene oxide) (PEO) surfactants (N0) as templates, respectively. The amine micelles or poly(ethylene oxide) surfactants interact with silicate oligomers (I0) derived from tetraethyl orthosilicate (TEOS) through hydrogen bonding interactions to direct the assembling of the organic–inorganic composites. Thus, the N0I0 and S0I0 pathways were proposed to illustrate the material formation processes. The silicate oligomers (I0) formed in neutral solutions (pH > 2) were negatively charged. Therefore, the neutral templates were assumed to be partially protonated or positively charged to neutralize the negative charges of silicon species and form organic–inorganic hybrids. Stucky and co-workers in Santa Barbara57,58 spread the applications of non-ionic surfactants (S0) in preparations of mesoporous silica materials in strong acidic solutions. Well-ordered hexagonal mesoporous silica structures (SBA-15) with uniform pore sizes of up to approximately 30 nm were prepared by using amphiphilic triblock copolymers as templates. The obtained materials exhibited high hydrothermal stability thanks to the large silica wall thickness. Meanwhile, Ryoo and co-workers59–61 from the Korea Advanced Institute of Science and Technology also used non-ionic triblock copolymers as templates to synthesize a series of mesoporous silica (KIT-n) materials in aqueous HCl solutions with pore sizes ranging from 3–12 nm. The structures of the KIT-n materials can be facilely tuned from disordered wormlike channels to cubic Ia3d or cubic Fm3m symmetry by varying the synthesis recipe or by adding or removing additional salts or organic additives. The mesopore diameters and apertures of the KIT-n materials can also be tailored in the range of 4–12 nm by changing the hydrothermal treatment temperatures. In order to understand the assembly process of the organic–inorganic composites occurring in the acidic environment, a new pathway, S0H+X−I−, was proposed based on the electrostatic Coulomb force and double layer hydrogen interactions.38,39
A soft-templating method has been one of the most successful and versatile strategies to produce mesoporous solids to date. A huge number of mesostructured silica or aluminosilicate materials have been produced and used in adsorption, separation and catalysis processes due to their large pore size and high specific surface areas.10,62–65 Nevertheless, the nature of amorphous frameworks and thin pore walls of this sort of materials may result in poorer hydrothermal stabilities and weaker acidities than that of zeolites, which would inhibit their large-scale manufactures or industrial applications.6,66
Several additional techniques have been applied to improve aluminosilicate mesostructures. It has been reported that the assembly of nanoclustered microporous zeolite precursors (seeds) within hexagonal aluminosilicate mesostructures (Al-MSU) imparts improved hydrothermal stability and acidity.67,68 The XRD low angle data suggest that the composite remains amorphous (see Fig. 3). Though the XRD patterns suggest a lack of crystallinity, the 27Al NMR spectra imply the presence of zeolite-like connectivities in Al-MSU. The mesostructure of materials prepared using the seeding technique can be maintained after steaming (800 °C, 5 h). The inclusion of zeolite seeds within the MSU mesopores (i.e., 10%Al-MSU) shows improved retention of the catalytic capability, over even that of an ultrastable form of 14%Al-MCM-41 prepared using the grafting technique. In addition, there have been efforts to alleviate the diffusion limitations through a synthesis of extra-large-pore molecular sieves (e.g., ITQ-21, ITQ-37 and ITQ-40) using organic structure directing agents (SDAs).24,69,70 Corma et al. exhibited that the synthesized materials can contain large pore openings (i.e., up to 16-member rings) or large cavities (i.e., 1.8 nm-wide). The results from catalytic cracking of a vacuum gas oil show that the activity of ITQ-21 is higher than that of the USY zeolite, but it produces a lower gasoline yield.24
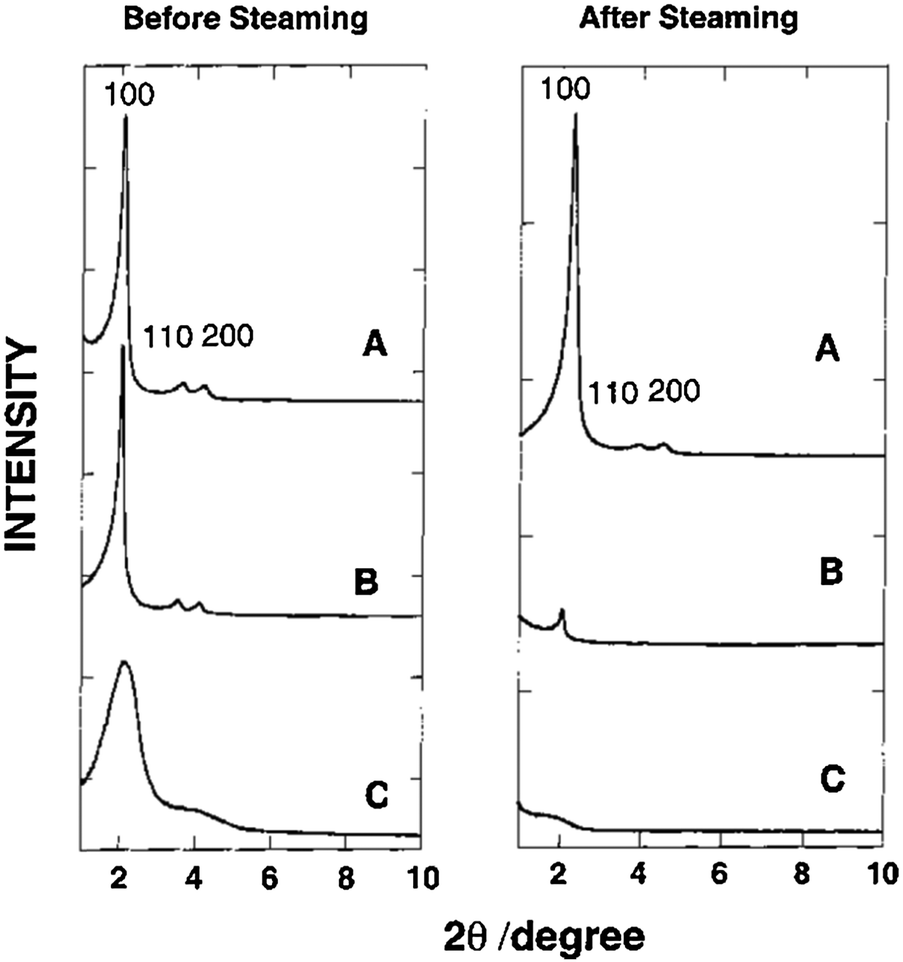 | ||
| Fig. 3 XRD patterns of mesoporous aluminosilicate materials before and after steaming: (A) hexagonal 10%Al-MSU prepared from zeolite Y seeds; (B) “ultrastable” hexagonal 14%Al-MCM-41 prepared by grafting; (C) disordered 10%Al-MCM-41 prepared by direct synthesis. Reprinted with permission from ref. 67. Copyright 2000 American Chemical Society. | ||
Creating mesopores in zeolite crystals increases the external surface area thereby improving micropore accessibility. The aforementioned properties can be attained through a breakthrough zeolite synthesis pioneered by Ryoo and co-workers.71,72 By using diquaternary ammonium surfactants, having a long aliphatic chain and two quaternary ammonium groups spaced by a short alkyl linkage, or gemini-type polyquaternary ammonium surfactants, phase segregation was avoided and zeolite nanosheets were obtained.71,72 The use of multiammonium surfactants inhibits Ostwald ripening, a thermodynamic process that minimizes the surface free energy of crystals and results in the aggregation of smaller crystals into larger crystals. The long aliphatic chain induces the formation of mesoscale spacing, while the multiammonium group serves as a structure-directing agent for nano-thick zeolites (see Fig. 4). The concentration of Na+ ions affects the synthesis of the multilamellar or unilamellar nanosheets. The mesoporosity is created by (i) the retention of mesostructure due to the intergrown crystals preventing the complete collapse, and (ii) the slight crystal orientation deviation which prevented the complete condensation of the nanosheet layers. It is remarkable that the nanosheet thickness and framework topology (i.e., MFI-like or Beta zeolite) can be manipulated by varying gemini like head groups, and the size of the mesopores can be adjusted according to the hydrophobic tail length. The evolution of framework transformation during hydrothermal syntheses of the mesostructured zeolites over time scales was comprehensively investigated using NMR, XRD and TEM analyses.73 As shown in Fig. 5, surfactant-directed mesoporous material with an amorphous framework was initially formed after 1 day. Over the period of one week, it gradually transformed into intermediate nanolayers that eventually crystallized into full MFI nanosheets after 12 days. These materials possess numerous characteristics which are beneficial to catalytic conversion, namely minimizing diffusion path lengths resulting in reduced coke deposition and more available acidic sites at the external surface. It has been demonstrated that the catalytic conversion of bulky molecules by the zeolite nanosheets was higher than that of a conventional zeolite.71,72 In addition, the MFI nanosheets showed an increased catalyst lifetime in methanol-to-gasoline conversion, and exhibited not only less coke deposition, but also that most of the coke formation occurred at the external surface.71
 | ||
| Fig. 4 SEM image of MFI nanosheets synthesized in a flake-like morphology (left), and TEM image of the cross-section of the flake (right). Adapted with permission from ref. 71. Copyright 2009 Macmillan Publishers Limited. | ||
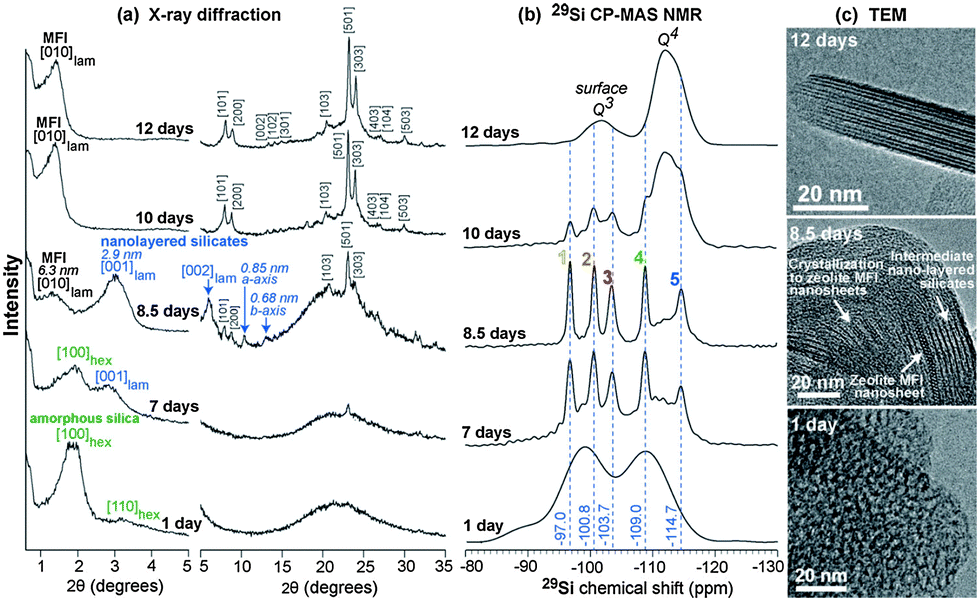 | ||
| Fig. 5 (a) Powder XRD patterns, (b) solid-state 29Si CP-MAS NMR spectra, and (c) TEM images of mesostructured zeolite MFI nanosheets obtained during hydrothermal synthesis. Reprinted with permission from ref. 73. Copyright 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
In addition, Che and co-workers reported a similar approach for the synthesis of single-crystalline mesostructured zeolite nanosheets using a single quaternary ammonium template with strong ordered self-assembling ability through the π–π interaction of aromatic groups in the amphiphilic molecules.74 Generally, the aggregation of surfactants with one-head quaternary ammonium groups is less energetically favourable than that of multiple quaternary head molecules, and therefore the formation of a laminar structure directed from one-head quaternary molecules is unlikely to occur. However, introducing at least two benzene rings (e.g., biphenyl and naphthyl) into the hydrophobic alkyl tails of one-head quaternary ammonium surfactants leads to the formation of more stable bilayer assemblies, as confirmed by a molecular dynamic simulation study, and makes the synthesis of mesostructured zeolite nanosheets plausible. The special configuration of molecules and the strong stacking give rise to a new type of mesoporous zeolite nanosheet joined with a 90° rotational boundary (see Fig. 6).
 | ||
| Fig. 6 HRTEM images of as-made (a, b) and calcined (c, d) single-crystalline mesostructured zeolite nanosheets. The scale bars in a, b, c, d represent 200 nm, 10 nm, 50 nm and 10 nm, respectively. Reprinted with permission from ref. 74. Copyright 2014 Macmillan Publishers Limited. | ||
Preparations of micro–mesoporous composites
Many studies have been devoted to expand the use of surfactants to introduce mesoporosity in zeolites as to combine the advantages of crystalline microporous zeolites with amorphous mesoporous materials. Through creative use of multiple surfactants, several research groups have attempted to induce mesoporosity into microporous zeolites. The “bottom-up” approach uses surfactants and templates at the same time (i.e., the so-called dual-templating) to create micro- and meso-porous structures simultaneously. Beck et al.75 investigated the ability of alkyltrimethylammonium surfactants with various chain lengths (i.e., CnH2n+1(CH3)3NBr, n = 6, 8, 10, 12, 14, and 16) to form micro- and meso-porous molecular sieve structures under various synthesis conditions, and exhibited the tunability of MFI–MCM-41 composite compositions. It was found that the reaction temperatures play an important role during synthesis. The aggregation of the cationic surfactants to form micelles is more likely at lower temperatures, giving rise to the formation of the amorphous mesostructures. While at higher temperatures, the micelle structures are destabilized yielding single molecule templating, leading to the formation of zeolitic materials. It is clear that the variation of the alkyl chain length of surfactants can be either mesopore (20–500 Angstrom) or micropore (<20 Angstrom) promoting. In the 100–200 °C synthesis temperature range, the shortest chain length (n = 6), with medium to high temperatures, gives rise to primarily microporous materials, while the longer chains (n = 8 to 12) are both mesopore and micropore directing. The longest chains (n = 14 and 16) promote the formation of MCM-41 at lower and intermediate synthesis temperatures.Karlsson et al. demonstrated the formation of the micro- and mesoporous composites of MFI-type/MCM-41 using a dual-templating syntheses gel system (i.e., mixtures of C6H13(CH3)3NBr and C14H29(CH3)3NBr).76 The compositions of the final products were tailored by adjusting the ratio of the template surfactants and the synthesis temperatures. As shown in Fig. 7, the sample derived from the C14 surfactant templating alone shows the sharp diffraction peaks corresponding to the amorphous MCM-41 structure (i.e., low angle region peaks). Upon the addition of the C6 surfactant co-templates, the low angle diffraction peaks representing the mesostructures became less pronounced, suggesting a higher yield of the microporous MFI-type structure in the final products. The presence of the mesostructures is confirmed by the N2 adsorption isotherms of the zeolite samples with varying ratios of C6 and C14 cationic surfactants, which reveal a sharp uptake in the curve at P/P0 = ca. 0.33 due to pore condensation typical for a mesoporous sample (see Fig. 8). The synthesis temperatures have a direct effect on the composition of the MFI–MCM-41 composite. At a fixed C6 and C14 ratio, the proportion of the MFI-type phase significantly increases when a higher synthesis temperature was used. This behavior can be explained by the micelle destabilization effect proposed by Beck and co-workers.75 Some of the additional structures exhibit partial MFI- or MCM-41 morphologies. This would suggest that the templates worked independently and were competing. Due to the seemingly amorphous nature of MCM-41 when analyzed with XRD, the characterization of these additional structures remains elusive.
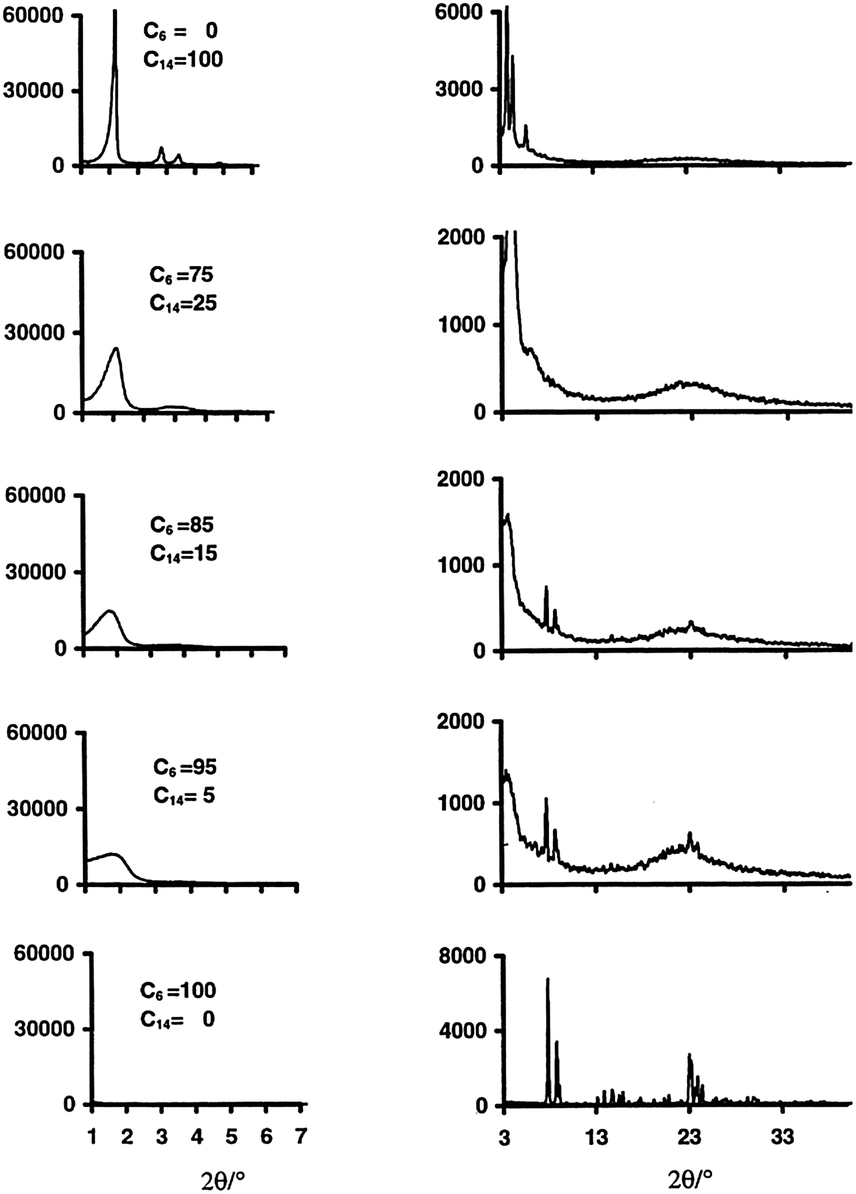 | ||
| Fig. 7 XRD patterns representing mesoporous (left) and microporous (right) structures of the composite samples obtained using various combinations of C6 and C14 ammonium cations at a synthesis temperature of 150 °C. Reprinted with permission from ref. 76. Copyright 1999 Elsevier Science B.V. | ||
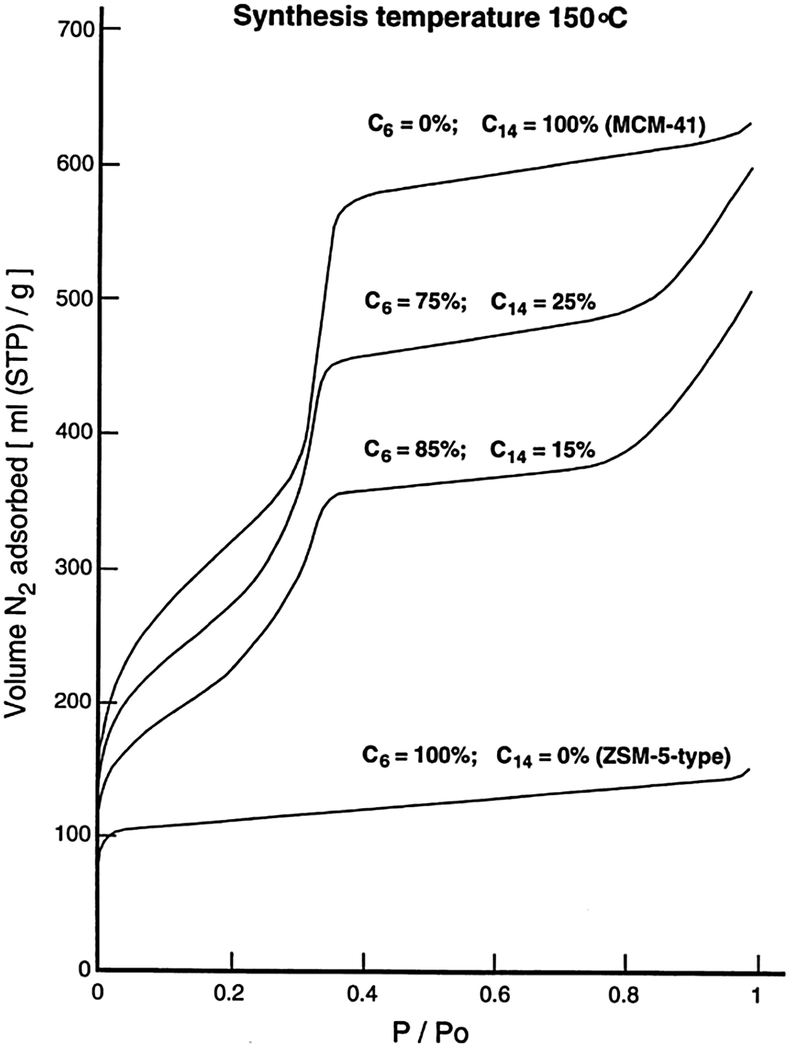 | ||
| Fig. 8 N2-adsorption isotherms of the composite samples obtained using various combinations of C6 and C14 ammonium cations at a synthesis temperature of 150 °C. Reprinted with permission from ref. 76. Copyright 1999 Elsevier Science B.V. | ||
Kloetstra et al. added mesoporosity to faujasite (FAU) through the deposition of MCM-41, resulting in MCM-41–FAU composite materials.77 The use of cetyltrimethylammonium bromide (CTAB) as a template led to a separated MCM-41 phase as well as an outgrowth of MCM-41 on the FAU-type zeolite. While the surfactant was expected to bind perpendicularly to the crystal, the MCM-41 self-assembled independently, suggesting a lack of nucleation at the surfactant–Y zeolite surface. The use of steamed ultrastable H–Y (USY) led to an overgrowth of MCM-41 after pH adjustment due to the inherent rough crystal surface of USY. When tested in the cracking of vacuum gasoil, the MCM-41 coated USY showed a 10% lower conversion, as compared to the original USY. However, the selectivities toward the valuable products were improved (i.e., comparing at the same conversion level, 3% higher gasoline, 1.5% higher light cycle oil, 1.5% lower bottoms, and 2% lower coke).
Zeolitic recrystallization is another technique to introduce mesoporosity into zeolite crystals. The process is considered as a “top-down” approach consisting of two steps: (i) the partial dissolution of a zeolite with a basic solution, followed by (ii) a hydrothermal treatment with CTAB. Ivanova et al., reported the synthesis of composite micro/mesoporous mordenite zeolite (MOR) using the “2-step” process.78 They found that the addition of more severe caustic treatments led to a decrease in the tetrahedral coordinated Al species while conversely an increase in the octahedral Al species.11,78–80 In contrast, the second step in the presence of CTAB led to the reduction of tetrahedrally coordinated Al species, while the appearance of the octahedral Al species gradually increased with the degree of recrystallization.80 The presence of octahedral aluminum is indicative of the presence of amorphous mesoporous materials. The “2-step” recrystallization approach for making mesoporous molecular sieve MCM-41 materials with zeolitic pore walls can be achieved using dissolved MOR81 or Beta82 gels and CTAB in alkaline solutions. It has been proposed that the good catalytic activity of these composite materials came from zeolite building units in the mesopore walls. In addition, zeolitic pore walls in amorphous mesoporous materials can be obtained through the partial recrystallization of MCM-41 in the presence of a tetrapropylammonium template for the ZSM-5 phase.83
Surfactant-templated zeolites
It has long been a goal of researchers to develop a hierarchically structured zeolite, where the mesopore walls are composed of a crystalline zeolitic framework. In 2005, Garcia-Martinez et al.84–88 introduced a unique post-synthesis technique to make fully crystalline zeolites (i.e., Y, MOR, and ZSM-5) with well controlled mesoporosity using surfactants. The sizes of the introduced mesopores are directed by the sizes of the surfactants used, and have very narrow pore-size distributions compared to the ones obtained from a conventional desilication process. This approach involves a “single-step” treatment based on the utilization of cationic surfactants (such as cetyltrimethylammonium bromide (CTAB) and chloride (CTAC)) in dilute basic solutions (such as NaOH, NH3OH, Na2CO3 or tetrapropylammonium hydroxide). The templates are removed through calcination or extraction to expose the introduced mesoporosity. The mesostructuring treatment leads to a significant improvement of physical properties, while maintaining the key properties of the zeolite such as crystallinity, acidity, and hydrothermal stability.84–88 For example, in the case of using CBV720 as a starting material with a C16 surfactant, the mesopore volume (i.e., pore diameter 20–300 Å) increases from 0.16 cc g−1 to 0.5 cc g−1. This corresponds to the increase of the “external” surface area by ca. 530 m2 g−1, as compared to the starting material.86 However this increase in mesopore volume does not sacrifice acidity. Temperature programmed ammonium desorption (TPAD) measurements revealed that the acidity of the mesostructured and conventional USY (CBV500) was comparable (i.e., 1.18 vs. 1.25 mmolH+ g−1).86 This feature is crucial in the development of a successful catalyst.One of the benefits of the surfactant-directing approach is that the mesopore diameter can be simply manipulated by using different cationic surfactants with various chain lengths. Fig. 9 shows the corresponding results derived from a series of mesostructured NH4–Y zeolites that were prepared using alkyltrimethylammonium bromide surfactants with various chain lengths (i.e., CnH2n+1(CH3)3NBr, n = 8–18) in NaOH aqueous solutions. The uniform size distribution of mesopores in the mesostructured Y-zeolite increased from ca. 15 Å to 45 Å as the surfactant alkyl chain lengths increased from C8 to C18. It is noteworthy that a linear correlation was found between the modal mesopore diameters and the carbon chain lengths.
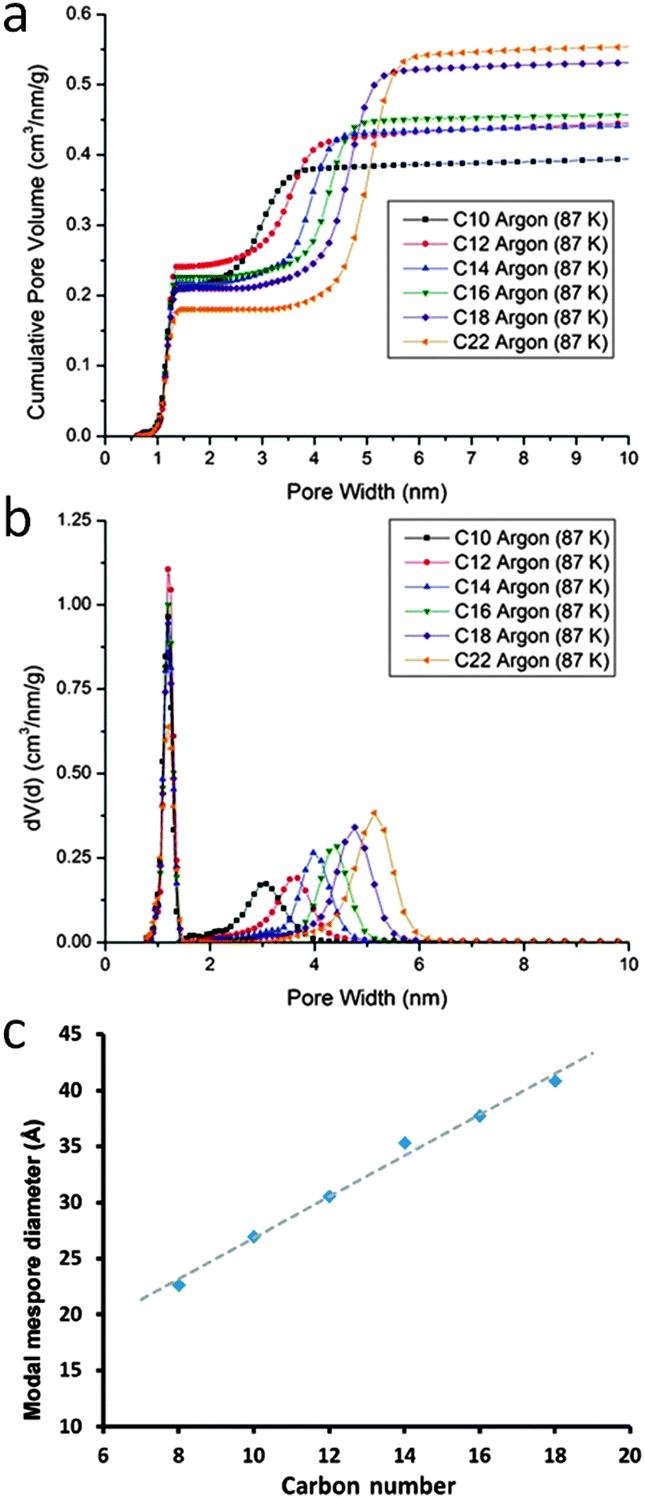 | ||
| Fig. 9 (a) Cumulative pore volumes, (b) nonlocal density functional theory (NLDFT) pore size distribution curves calculated from Ar (87 K) isotherms, and (c) a relationship between the surfactant carbon chain lengths and the modal mesopore diameters. Adapted with permission from ref. 86 and 87. Copyright 2012 and 2014 Royal Society of Chemistry, respectively. | ||
The proposed mechanism of the surfactant-assisted crystal rearrangement is depicted in Fig. 10. The crystalline Si–O–Si bonds of the original zeolite frameworks are open and consequently yield negatively charged Si–O− species. The diffusion of the cationic surfactants into the zeolite crystals is driven by the electrostatic interaction between the negatively charged sites and the positively charged surfactants. When the local concentration of the surfactant is high enough, it spontaneously assembles to form micelles within the zeolite crystals. As a result, the crystal structure rearranging to form mesopores around the micelles is driven by (i) the electrostatic interaction, and (ii) the hydrophobic effect responsible for the micelle formation, causing the crystal expansion as has been determined by SEM analysis (see Fig. S1, ESI†). Nevertheless, the true synthesis mechanism at the molecular level is still unclear.
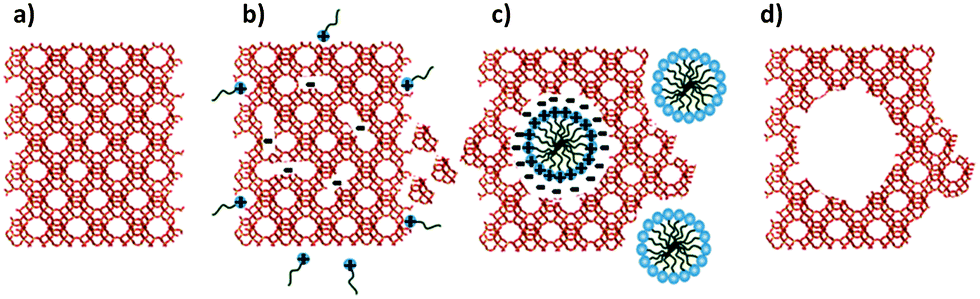 | ||
| Fig. 10 Proposed mechanism of the surfactant-templated mesostructuring process in zeolites: (a) pristine Y-zeolite; (b) Si–O–Si bond-opening/reconstruction in basic media; (c) crystal rearrangement to accommodate the surfactant micelles; and (d) removal of the template to expose the introduced mesoporosity. Reprinted with permission from ref. 86. Copyright 2012 Royal Society of Chemistry. | ||
For the zeolite Y containing a higher aluminum content (i.e., Si/Al ratio < 3), an additional acid wash pre-treatment is required prior to the surfactant/base solution treatment.89,90 The Si–O–Al bonds are not as labile as the Si–O–Si bonds under basic conditions. Therefore, the zeolites containing higher framework aluminum contents are not able to restructure to generate the micelle-directed mesoporosity. The process of pretreating the zeolite with a dilute acid weakens the crystalline structure. The pretreatment partially breaks the Si–O–Al bonds without collapsing the zeolite structure. The cleavage of the framework O–Al bonds and the formation of the “hydroxyl nest” defect sites are evident in the peak broadening and shifting from the X-ray diffraction (see Fig. 11).86
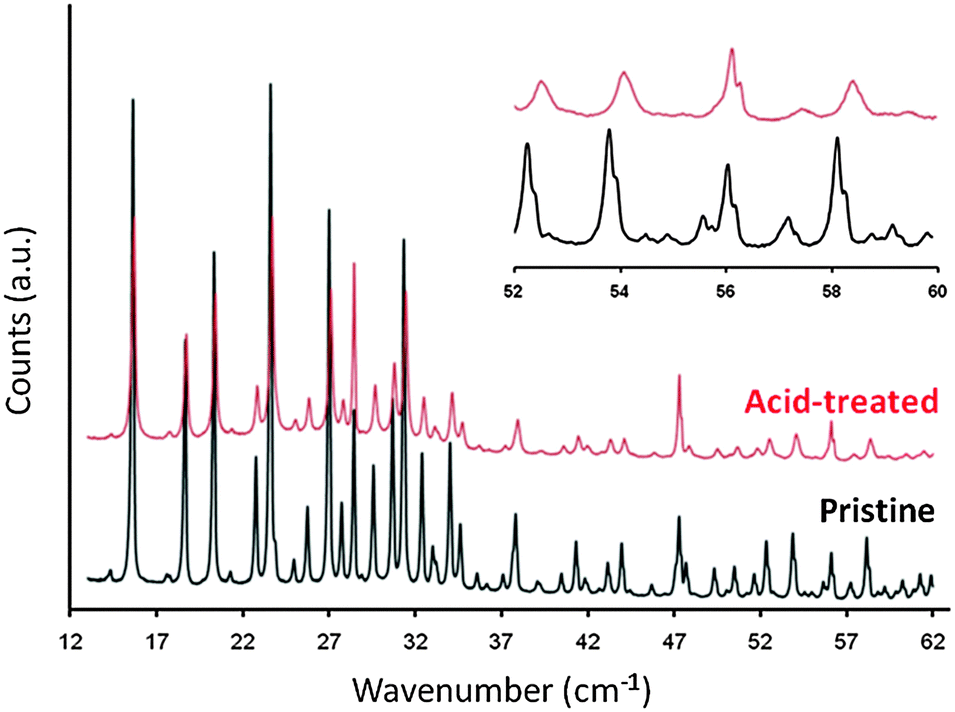 | ||
| Fig. 11 X-ray diffraction patterns of pristine (bottom) and acid-treated (top) Y zeolites. Adapted with permission from ref. 86. Copyright 2012 Royal Society of Chemistry. | ||
Moreover, the evidence of the hydroxyl nests was also observed from FT-IR analysis. In Y-zeolites, the vibrational frequencies of strong Brønsted acid O–H bonds and silanol O–H bonds are typically assigned at ca. 3640 cm−1 and 3740 cm−1, respectively. Fig. 12 shows the evolution of the O–H stretch signals of Y-zeolite samples along the treatment process. A significant increase in the intensity of the peak at 3740 cm−1 was observed after the acid treatment while the intensity of the peak at 3640 cm−1 decreased. This behaviour suggests that the framework Al representing the Brønsted acid sites was abstracted during the acid treatment and the terminal silanol groups (i.e., hydroxyl nests) were subsequently formed. The significant decrease of the peak at 3640 cm−1 after the CTAB/NaOH treatment is due to the ion exchange between H+ and Na+, however the intensity corresponding to the Brønsted acid sites was recovered after the calcination and NH4+ exchange.
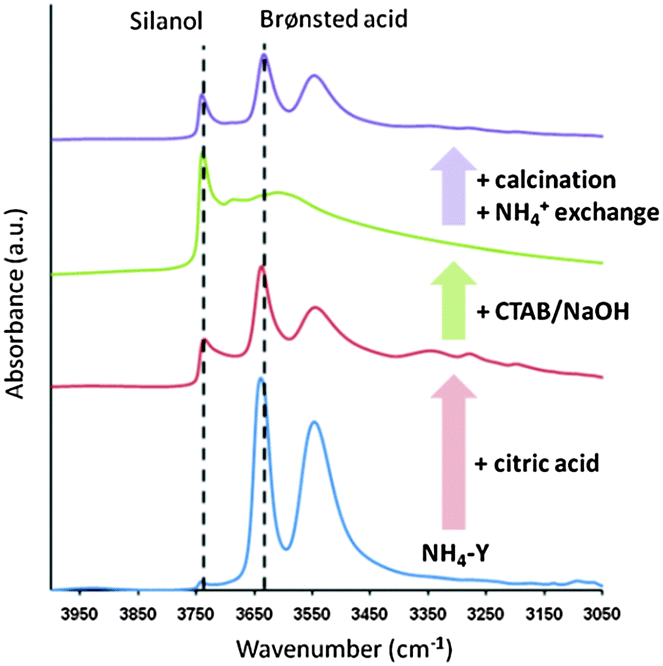 | ||
| Fig. 12 The FT-IR (O–H stretch region) of various Y zeolite samples along the synthesis process. From bottom to top, the starting NH4–Y (CBV300), after citric acid pre-treatment, after treatment with CTAB in NaOH solution, after removal of template by calcination and NH4+ exchange. Reprinted with permission from ref. 86. Copyright 2012 Royal Society of Chemistry. | ||
Recently, the modification of Si and Al species during the surfactant-assisted crystal rearrangement process was investigated through solid 29Si and 27Al MAS NMR and reported by Jiao and co-workers (see Fig. 13).91 It is notable that after the acid (i.e., lactic acid) treatment, the peaks of Si(nAl, n = 2–4) species decrease along with the increase of the Si(nAl, n = 0 and 1) peaks, suggesting the occurrence of dealumination in the Al-rich framework of the zeolite. The extracted Al ions are more likely to form aluminum lactate species which dissolve into the solution instead of remaining as extra-framework Al species in the zeolite structures. As a result, the 27Al NMR peak with a chemical shift of ca. 60 ppm was observed, as well as a trace amount of extra-framework Al species (i.e., a chemical shift of 0 ppm). After the treatment with CTAB in NH4OH or NaOH solutions, the corresponding 29Si NMR spectrum reveals a different pattern. The changes in the intensities of the Si(nAl, n = 1 and 3) peaks result from the partial desilication which is more severe in NaOH solution. The lack of chemical shift at 0 ppm indicates no extra-framework Al was present in the sample. There may have been no extra-framework Al formed upon the treatment in aqueous NaOH solutions.86 On the other hand, the silicon dissolution could have given rise to the creation of extra-framework Al species remained in the zeolite structures after the NH4OH treatment, as shown in Fig. 13II-c. However, when a NaOH solution was used, the created extra-framework Al species were removed, leading to the disappearance of the 27Al NMR peak with a chemical shift of ca. 0 ppm (see Fig. 13II-d).91
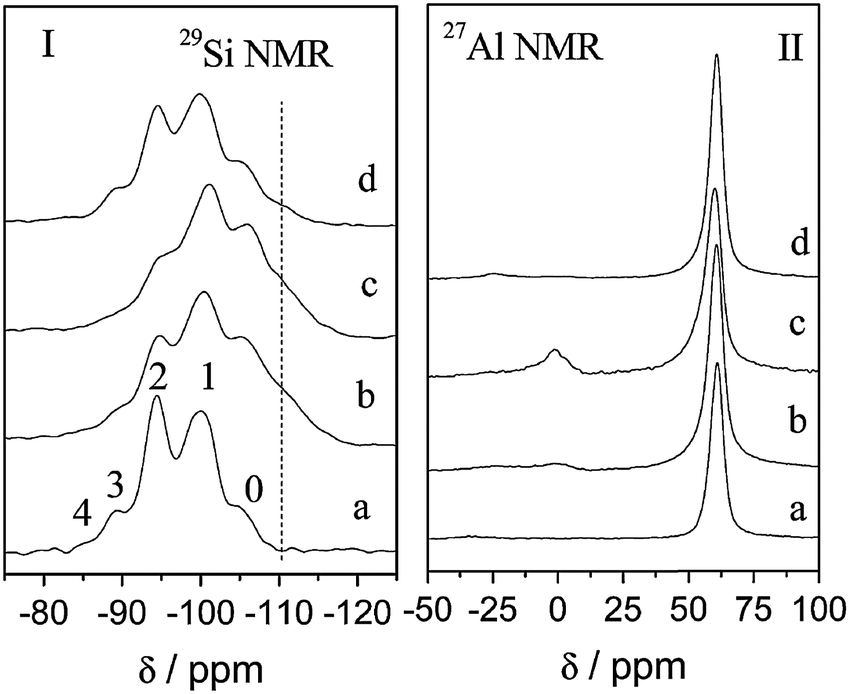 | ||
| Fig. 13 29Si and 27Al MAS-NMR of various Y zeolite samples along the synthesis process. From bottom to top, (a) the starting NaY, (b) after lactic acid pre-treatment, (c) after treatment with CTAB in NH4OH solution, and (d) after treatment with CTAB in NaOH solution. Reprinted with permission from ref. 91. Copyright 2014 Royal Society of Chemistry. | ||
The “single-step” treatment creates intracrystalline mesopores connected to micropores. The Si/Al ratio of the treated zeolites remains unchanged and the product recovery yield close to 100%, suggesting that no significant desilication took place during the synthesis process when the dilute basic solution was used. The use of cationic surfactants and mild basic conditions allowed for the structural rearrangement that was necessary for the formation of mesoporosity without severe desilication. The co-existence of intracrystalline mesostructures within the zeolite crystal can be exhibited by TEM (Fig. 14) and SEM analyses (Fig. 15). The TEM in Fig. 14 reveals that the uniformly sized and distributed smaller mesopores were created to replace the larger channel-like mesopores in the starting commercial zeolite Y (CBV720) while maintaining the original crystal shapes after the mesostructuring treatment. This evidence supports the previously described crystal rearrangement mechanism for the mesopore formation.86
 | ||
| Fig. 14 TEM images of (a) the starting zeolite Y (CBV720), (b–d) mesostructured Y zeolites demonstrating the co-existence of the intramesopores and the crystal lattice fringe lines. Reprinted with permission from ref. 87. Copyright 2014 Royal Society of Chemistry. | ||
 | ||
| Fig. 15 FE-SEM images of the untreated NaY zeolite (top) and the mesostructured Y-zeolite crystals (bottom). Reprinted with permission from ref. 86. Copyright 2014 Royal Society of Chemistry. | ||
In addition, the 3-dimentional distribution and connectivity of the intracrystallinemesopores were studied using advanced gas sorption, electron tomography (ET), and rotation electron diffraction (RED) techniques.87Fig. 16 clearly shows that the mesopores generated by the “single-step” treatment method are well-connected and form a network of mesopore-channels distributed throughout the crystal. With evidence reported by Chal et al., the pseudomorphic transformation zeolite crystals lead to interconnected micro- and mesopores, and have a minimal effect on the zeolite coherence domain (crystallite) size.92 In addition, the analysis of power X-ray diffraction patterns suggested that an intense peak at low angle corresponds to a pore–pore correlation distance of 45.8 Å, ascribing to a vermicular mesopore structure.
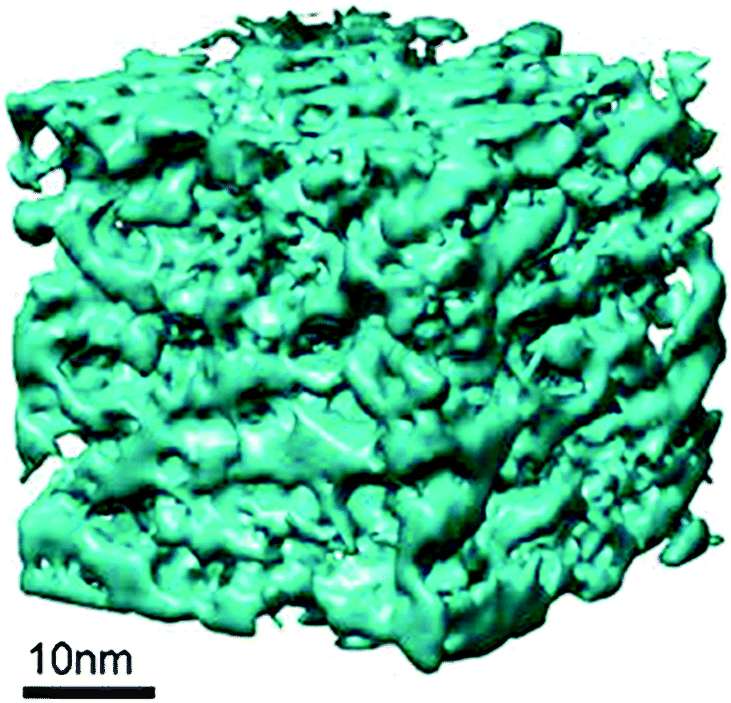 | ||
| Fig. 16 The tomogram of the mesostructured Y-zeolite crystal prepared using C16 surfactants. Reprinted with permission from ref. 87. Copyright 2014 Royal Society of Chemistry. | ||
As previously mentioned, another surfactant-templated post-synthetic method involves a “two-step” process: (1) partial or full dissolution of zeolites (i.e., ZSM-5 and MOR) in basic solution, followed by (2) hydrothermal treatment in the presence of the cationic surfactant (i.e., CTAB).11,78,80 Unlike the “single-step” process, the NaOH solution was added before the CTAB in the “two-step” process, and the obtained materials can be significantly different as depicted in Fig. 17. During the NaOH treatment in the first step, some mesoporosity was generated within the zeolite crystals by the typical desilication process. At the same time, partial dissolution of the crystals also took place. Upon the addition of the surfactants in the second step, the dissolved zeolite species from the dissolution in the first step are re-assembled around the micelle surface to form the second mesoporous silica phase and eventually deposited back onto the external surface of zeolite crystals. The material generated is considered a composite as its compositions of zeolite/mesoporous materials can be varied depending on the severity of the zeolite dissolution in the first step. The two types of the mesoporous structures can be observed through the adsorption isotherm at two different nitrogen uptakes at P/P0ca. 0.35 and 0.95, corresponding to the well-controlled mesopores covering the crystal (i.e., ca. 4 nm mesopores of the surfactant-templated amorphous mesoporous material) and the broader mesopores (i.e., created by desilication) residing inside the zeolite crystals, respectively.78 From the TEM images shown in Fig. 17, it is clearly seen that the mesoporosity (lighter colored spots) created from the “single-step” and the “two-step” approaches is significantly different. For the single-phase mesostructured materials derived from the “single-step” approach, evenly distributed non-order mesopores were observed throughout the crystal (as confirmed in ultramicrotomed samples).86 In contrast, two-phase materials (i.e., well-ordered mesochannel MCM-41–microporous zeolite composites) were observed in the samples prepared using the “two-step” approach.91
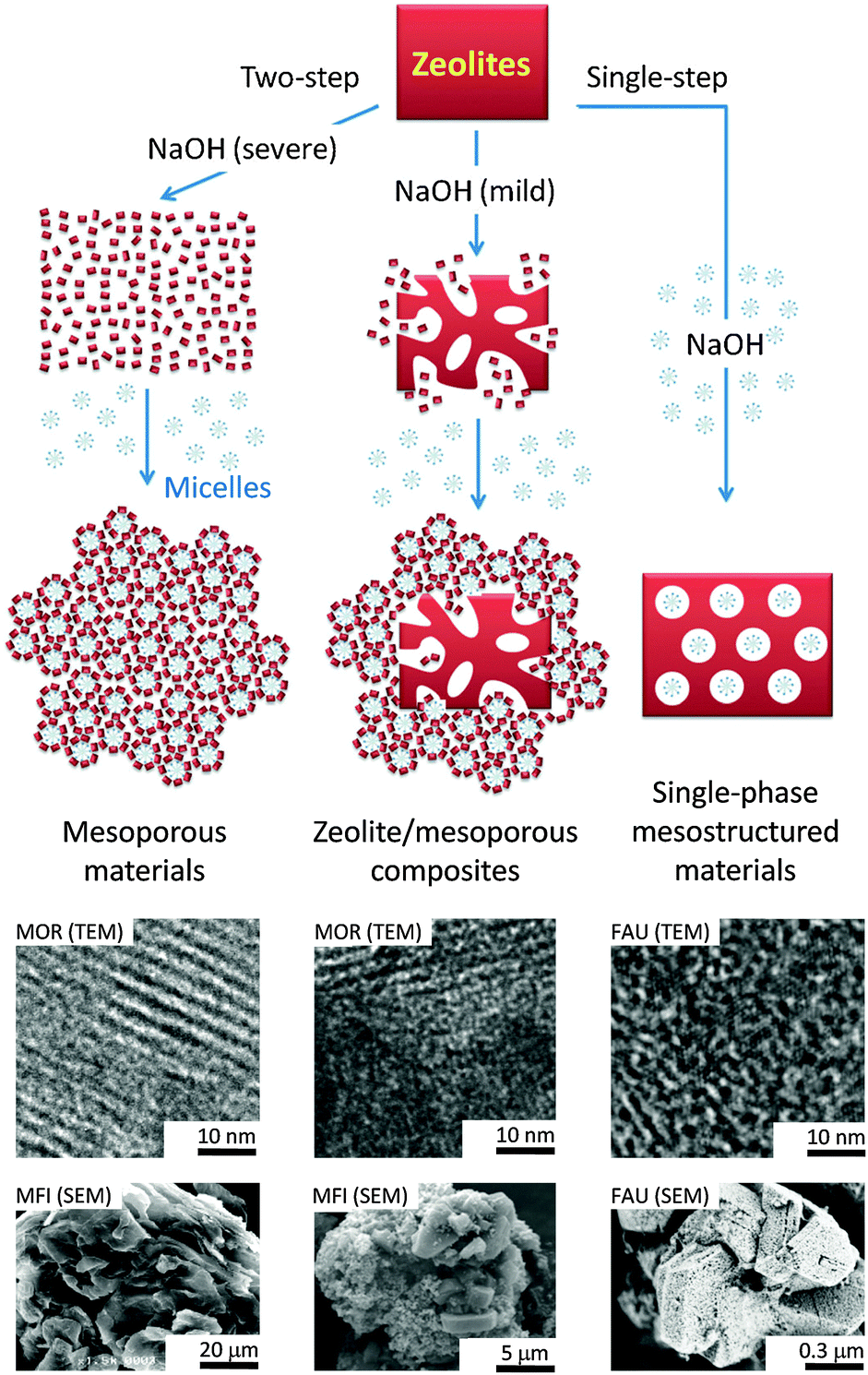 | ||
| Fig. 17 Schematic diagram illustrating the “single-step” and the “two-step” approaches, and examples of micrographs of the corresponding materials obtained from the processes. Reprinted with permission from ref. 80. Copyright 2014 Royal Society of Chemistry. | ||
Y-zeolites have been extensively used as catalysts for hydrocarbon processing in refineries. It is the key active component of fluid catalytic cracking (FCC) catalysts due to several unique properties including strong Brønsted acidity, high surface area, and high hydrothermal stability. However, the relatively large micropores (ca. 7.4 Å in diameter) of the Y-zeolite are sterically limited when performing catalytic cracking of heavy feeds such as vacuum gas oils (VGO). The introduction of mesoporosity into the zeolites could alleviate the diffusion limitation appearing in the conventional materials and, therefore, enhance the catalytic performance (i.e., maximize activity and desirable product selectivity). Minimizing the diffusion limitation of the Y-zeolite in FCC catalysts would help reduce bottoms by improving accessibility of larger hydrocarbons in the feed to active sites and therefore cracking into smaller products. At the same time, the selectivity of valuable products such as gasoline, LCO and LPG can be enhanced by allowing them to diffuse out before they over-crack into undesirable coke and dry gases.93
The catalyst evaluations of pure steam-deactivated mesostructured derived from the “single-step” process and conventional zeolite samples were performed in a Micro Activity Test unit (MAT) and an Advanced Catalyst Evaluation (ACE) test unit. The abovementioned VGO cracking benefits were exhibited in Fig. 18. The mesostructured materials showed outstanding hydrothermal stability as confirmed both at the lab scale (at 788 °C and 100% for 4 h) and the refinery scale. Since 2013, a FCC catalyst containing mesoporous zeolite Y prepared by surfactant-templated post-synthetic modification (i.e., “single-step” approach) has been commercially supplied by Rive Technology. The result from a commercial trial suggests the estimated economic uplift over $2.50 per bbl of the FCC feed when the refinery replaced the incumbent catalyst with the FCC catalyst containing the mesostructured zeolite.94 Unlike the materials obtained from the “single-step” process, the composite materials containing a non-zeolitic mesoporous component have exhibited inferior hydrothermal stability and catalytic activity, and caused them to fail as suitable FCC catalysts.
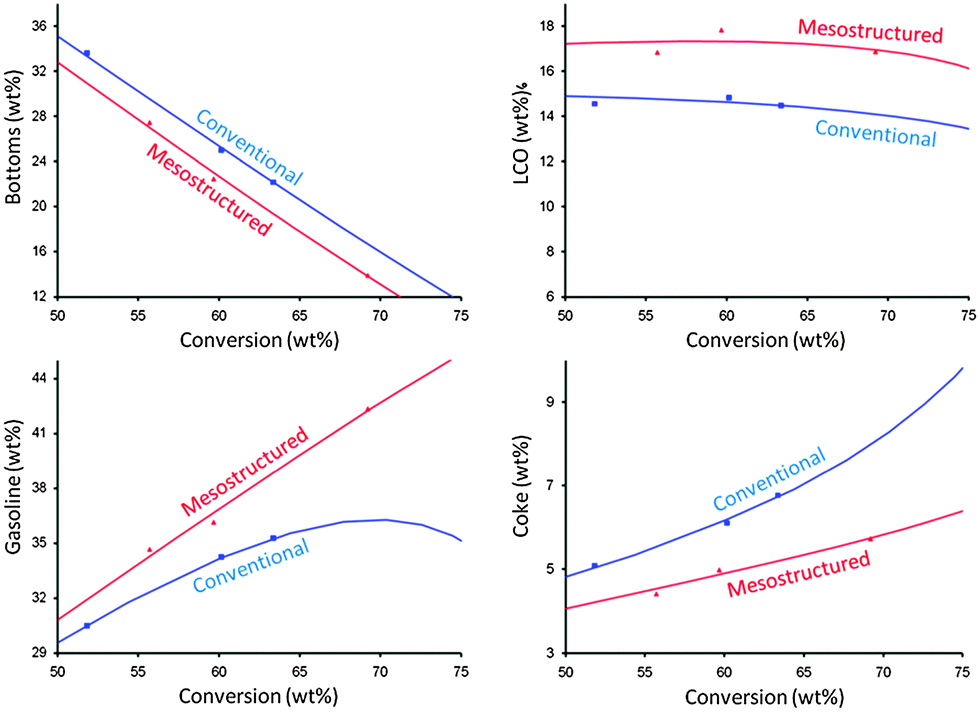 | ||
| Fig. 18 The MAT test results of (red) mesostructured zeolite USY and (blue) conventional zeolite USY. The zeolites were ultrastabilized, and then deactivated at 788 °C in 100% steam for 4 h before being tested. The curves were fitted by a kinetic lump model. Reprinted with permission from ref. 86. Copyright 2012 Royal Society of Chemistry. | ||
Conclusions
A novel area of research emerging from the relatively mature field of zeolite focuses on incorporating mesoporosity into microporous zeolites. Surfactant-templating is a useful and versatile technique to implement mesopore systems. The major goals are to enable the processing of bulky molecules and, at the same time, improve the diffusion into and away from the catalytic active sites in order to enhance the catalytic activity and avoid undesirable side reactions. Various surfactant-templating strategies have been explored and studied to produce new materials. The synthesis using the “dual-templating” approach can create micro- and mesoporous structures simultaneously; however, the materials prepared by this method mainly consist of a composite of amorphous and zeolite particles (i.e., MCM-41–zeolite composites). The presence of the amorphous phase results in poorer hydrothermal stabilities and weaker acidities than that of crystalline zeolites, which would hinder their catalytic performance. Recently, post-synthetic modification approaches combining the uses of cationic surfactants and mild basic solutions were introduced to synthesis mesostructured zeolites. It should be noted that this mesostructuring process undergoes structural re-arrangement which allows the formation of one phase materials containing mesoporosity within zeolite crystals while the microporous structures are well preserved. In contrast, the “two-step” process creates the two phases (i.e., MCM-41–zeolite composites) through the dissolution–precipitation mechanism. By contrast, surfactant-templated zeolites are one phase mesoporous zeolites, while maintaining key properties such as crystallinity, acidity and hydrothermal stability. Steam-deactivated mesostructured Y zeolites have been extensively tested in ACE (Advanced Catalytic Evaluation) units and several refinery trials, exhibiting superior performance in heavy oil (VGO) processing (i.e., lower bottoms, coke, and dry gases, and higher gasoline, LCO and LPG). They represent the bridge between zeolites and surfactant-templated materials and the successful commercial realization of the promising mesoporous materials in those processes where zeolites are diffusion limited.Acknowledgements
The authors would like to acknowledge the contribution of the employees, management, investors, and partners of Rive Technology to the great advancement of the mesostructured zeolite technology. We thank K. Li (Rive Technology, Inc.) for support and helpful discussions.References
- D. W. Breck, Zeolite molecular sieves: structure, chemistry, and use, R.E. Krieger, 1984.
- J. Cejka, H. van Bekkum, A. Corma and F. Schueth, Introduction to Zeolite Molecular Sieves, Elsevier Science, 2007 Search PubMed.
- J. Weitkamp and L. Puppe, Catalysis and Zeolites: Fundamentals and Applications, Springer, 1999 Search PubMed.
- C. Baerlocher and L. B. McCusker, Database of Zeolite Structures, http://www.iza-structure.org/databases/.
- Y. Li and J. Yu, Chem. Rev., 2014, 114, 7268–7316 CrossRef CAS PubMed.
- Y. Tao, H. Kanoh, L. Abrams and K. Kaneko, Chem. Rev., 2006, 106, 896–910 CrossRef CAS PubMed.
- H. Ghobarkar, O. Schäf and U. Guth, Prog. Solid State Chem., 1999, 27, 29–73 CrossRef CAS.
- B. Louis, G. Laugel, P. Pale and M. M. Pereira, ChemCatChem, 2011, 3, 1263–1272 CrossRef CAS PubMed.
- G. J. d. A. A. Soler-Illia, C. Sanchez, B. Lebeau and J. Patarin, Chem. Rev., 2002, 102, 4093–4138 CrossRef PubMed.
- A. Corma, Chem. Rev., 1997, 97, 2373–2420 CrossRef CAS PubMed.
- I. I. Ivanova and E. E. Knyazeva, Chem. Soc. Rev., 2013, 42, 3671–3688 RSC.
- J. Perez-Ramirez, C. H. Christensen, K. Egeblad, C. H. Christensen and J. C. Groen, Chem. Soc. Rev., 2008, 37, 2530–2542 RSC.
- D. P. Serrano, J. M. Escola and P. Pizarro, Chem. Soc. Rev., 2013, 42, 4004–4035 RSC.
- K. Moller and T. Bein, Chem. Soc. Rev., 2013, 42, 3689–3707 RSC.
- N. Chen, T. Degnan and C. M. Smith, Molecular transport and reaction in zeolites, 1994 Search PubMed.
- N. S. Nesterenko, F. Thibault-Starzyk, V. Montouillout, V. V. Yuschenko, C. Fernandez, J. P. Gilson, F. Fajula and I. I. Ivanova, Microporous Mesoporous Mater., 2004, 71, 157–166 CrossRef CAS PubMed.
- L. Tosheva and V. P. Valtchev, Chem. Mater., 2005, 17, 2494–2513 CrossRef CAS.
- B. A. Holmberg, H. Wang, J. M. Norbeck and Y. Yan, Microporous Mesoporous Mater., 2003, 59, 13–28 CrossRef CAS.
- S. Mintova, N. H. Olson and T. Bein, Angew. Chem., Int. Ed., 1999, 38, 3201–3204 CrossRef CAS.
- W. Song, V. H. Grassian and S. C. Larsen, Chem. Commun., 2005, 2951–2953 RSC.
- G. Zhu, S. Qiu, J. Yu, Y. Sakamoto, F. Xiao, R. Xu and O. Terasaki, Chem. Mater., 1998, 10, 1483–1486 CrossRef CAS.
- C. C. Freyhardt, M. Tsapatsis, R. F. Lobo, K. J. Balkus and M. E. Davis, Nature, 1996, 381, 295–298 CrossRef CAS.
- M. Yoshikawa, P. Wagner, M. Lovallo, K. Tsuji, T. Takewaki, C.-Y. Chen, L. W. Beck, C. Jones, M. Tsapatsis, S. I. Zones and M. E. Davis, J. Phys. Chem. B, 1998, 102, 7139–7147 CrossRef CAS.
- A. Corma, M. J. Diaz-Cabanas, J. Martinez-Triguero, F. Rey and J. Rius, Nature, 2002, 418, 514–517 CrossRef CAS PubMed.
- A. Corma, Chem. Rev., 1995, 95, 559–614 CrossRef CAS.
- M. E. Davis, Nature, 2002, 417, 813–821 CrossRef CAS PubMed.
- M. E. Davis, C. Saldarriaga, C. Montes, J. Garces and C. Crowdert, Nature, 1988, 331, 698–699 CrossRef CAS.
- M. E. Davis, C. Montes, P. E. Hathaway, J. P. Arhancet, D. L. Hasha and J. M. Garces, J. Am. Chem. Soc., 1989, 111, 3919–3924 CrossRef CAS.
- M. E. Davis, C. Saldarriaga and C. Montes, Zeolites, 1988, 8, 362–366 CrossRef CAS.
- A. Merrouche, J. Patarin, H. Kessler, M. Soulard, L. Delmotte, J. L. Guth and J. F. Joly, Zeolites, 1992, 12, 226–232 CrossRef CAS.
- T. L. Barr, J. Klinowski, H. He, K. Albert, G. Muller and J. A. Lercher, Nature, 1993, 365, 429–431 CrossRef CAS.
- Q. Huo, R. Xu, S. Li, Z. Ma, J. M. Thomas, R. H. Jones and A. M. Chippindale, J. Chem. Soc., Chem. Commun., 1992, 875–876 RSC.
- M. Estermann, L. B. McCusker, C. Baerlocher, A. Merrouche and H. Kessler, Nature, 1991, 352, 320–323 CrossRef CAS.
- R. F. Lobo, M. Tsapatsis, C. C. Freyhardt, S. Khodabandeh, P. Wagner, C.-Y. Chen, K. J. Balkus, S. I. Zones and M. E. Davis, J. Am. Chem. Soc., 1997, 119, 8474–8484 CrossRef CAS.
- P. Wagner, M. Yoshikawa, M. Lovallo, K. Tsuji, T. Michael and M. E. Davis, Chem. Commun., 1997, 2179–2180 RSC.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T. W. Chu, D. H. Olson and E. W. Sheppard, J. Am. Chem. Soc., 1992, 114, 10834–10843 CrossRef CAS.
- Y. Wan, Zhao, Chem. Rev., 2007, 107, 2821–2860 CrossRef CAS PubMed.
- D. Zhao, Y. Wan and W. Zhou, Ordered Mesoporous Materials, Wiley, 2012 Search PubMed.
- N. Pal and A. Bhaumik, Adv. Colloid Interface Sci., 2013, 189–190, 21–41 CrossRef CAS PubMed.
- T. Linssen, K. Cassiers, P. Cool and E. F. Vansant, Adv. Colloid Interface Sci., 2003, 103, 121–147 CrossRef CAS.
- G. A. Parks, Chem. Rev., 1965, 65, 177–198 CrossRef CAS.
- S. Shen, Y. Li, Z. Zhang, J. Fan, B. Tu, W. Zhou and D. Zhao, Chem. Commun., 2002, 2212–2213 RSC.
- S. Shen, A. E. Garcia-Bennett, Z. Liu, Q. Lu, Y. Shi, Y. Yan, C. Yu, W. Liu, Y. Cai, O. Terasaki and D. Zhao, J. Am. Chem. Soc., 2005, 127, 6780–6787 CrossRef CAS PubMed.
- Q. Huo, D. I. Margolese and G. D. Stucky, Chem. Mater., 1996, 8, 1147–1160 CrossRef CAS.
- Y. Sakamoto, M. Kaneda, O. Terasaki, D. Y. Zhao, J. M. Kim, G. Stucky, H. J. Shin and R. Ryoo, Nature, 2000, 408, 449–453 CrossRef CAS PubMed.
- D. Zhao, Q. Huo, J. Feng, J. Kim, Y. Han and G. D. Stucky, Chem. Mater., 1999, 11, 2668–2672 CrossRef CAS.
- Q. Huo, D. I. Margolese, U. Ciesla, P. Feng, T. E. Gier, P. Sieger, R. Leon, P. M. Petroff, F. Schuth and G. D. Stucky, Nature, 1994, 368, 317–321 CrossRef CAS.
- S. Che, A. E. Garcia-Bennett, T. Yokoi, K. Sakamoto, H. Kunieda, O. Terasaki and T. Tatsumi, Nat. Mater., 2003, 2, 801–805 CrossRef CAS PubMed.
- J. Y. Ying, C. P. Mehnert and M. S. Wong, Angew. Chem., Int. Ed., 1999, 38, 56–77 CrossRef CAS.
- S. Che, Z. Liu, T. Ohsuna, K. Sakamoto, O. Terasaki and T. Tatsumi, Nature, 2004, 429, 281–284 CrossRef CAS PubMed.
- A. E. Garcia-Bennett, N. Kupferschmidt, Y. Sakamoto, S. Che and O. Terasaki, Angew. Chem., Int. Ed., 2005, 44, 5317–5322 CrossRef CAS PubMed.
- C. Gao, Y. Sakamoto, K. Sakamoto, O. Terasaki and S. Che, Angew. Chem., Int. Ed., 2006, 45, 4295–4298 CrossRef CAS PubMed.
- P. T. Tanev and T. J. Pinnavaia, Science, 1995, 267, 865–867 CrossRef CAS PubMed.
- P. T. Tanev, M. Chibwe and T. J. Pinnavaia, Nature, 1994, 368, 321–323 CrossRef CAS PubMed.
- S. A. Bagshaw, E. Prouzet and T. J. Pinnavaia, Science, 1995, 269, 1242–1244 CrossRef PubMed.
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548–552 CrossRef CAS.
- D. Zhao, Q. Huo, J. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024–6036 CrossRef CAS.
- R. Ryoo, J. M. Kim, C. H. Ko and C. H. Shin, J. Phys. Chem., 1996, 100, 17718–17721 CrossRef CAS.
- F. Kleitz, D. Liu, G. M. Anilkumar, I.-S. Park, L. A. Solovyov, A. N. Shmakov and R. Ryoo, J. Phys. Chem. B, 2003, 107, 14296–14300 CrossRef CAS.
- F. Kleitz, S. Hei Choi and R. Ryoo, Chem. Commun., 2003, 2136–2137 RSC.
- L. T. Gibson, Chem. Soc. Rev., 2014, 43, 5163–5172 RSC.
- L. T. Gibson, Chem. Soc. Rev., 2014, 43, 5173–5182 RSC.
- A. Corma, M. S. Grande, V. Gonzalez-Alfaro and A. V. Orchilles, J. Catal., 1996, 159, 375–382 CrossRef CAS.
- Z. Wang, Y. Jiang, R. Rachwalik, Z. Liu, J. Shi, M. Hunger and J. Huang, ChemCatChem, 2013, 5, 3889–3896 CrossRef CAS PubMed.
- C. Perego and R. Millini, Chem. Soc. Rev., 2013, 42, 3956–3976 RSC.
- Y. Liu, W. Zhang and T. J. Pinnavaia, J. Am. Chem. Soc., 2000, 122, 8791–8792 CrossRef CAS.
- Y. Liu and T. J. Pinnavaia, J. Mater. Chem., 2002, 12, 3179–3190 RSC.
- J. Jiang, J. Yu and A. Corma, Angew. Chem., Int. Ed., 2010, 49, 3120–3145 CrossRef CAS PubMed.
- A. Corma, M. J. DíazCabañas, J. Jiang, M. Afeworki, D. L. Dorset and A. K.G. S. S. L. Soled, PNAS, 2010, 107(32), 13997–14002 CrossRef CAS PubMed.
- M. Choi, K. Na, J. Kim, Y. Sakamoto, O. Terasaki and R. Ryoo, Nature, 2009, 461, 246–249 CrossRef CAS PubMed.
- K. Na, C. Jo, J. Kim, K. Cho, J. Jung, Y. Seo, R. J. Messinger, B. F. Chmelka and R. Ryoo, Science, 2011, 333, 328–332 CrossRef CAS PubMed.
- R. J. Messinger, K. Na, Y. Seo, R. Ryoo and B. F. Chmelka, Angew. Chem., Int. Ed., 2015, 54, 1521–3773 CrossRef PubMed.
- D. Xu, Y. Ma, Z. Jing, L. Han, B. Singh, J. Feng, X. Shen, F. Cao, P. Oleynikov, H. Sun, O. Terasaki and S. Che, Nat. Commun., 2014, 5, Article No. 4262 Search PubMed.
- J. S. Beck, J. C. Vartuli, G. J. Kennedy, C. T. Kresge, W. J. Roth and S. E. Schramm, Chem. Mater., 1994, 6, 1816–1821 CrossRef CAS.
- A. Karlsson, M. Stöcker and R. Schmidt, Microporous Mesoporous Mater., 1999, 27, 181–192 CrossRef CAS.
- K. R. Kloetstra, H. W. Zandbergen, J. C. Jansen and H. van Bekkum, Microporous Mater., 1996, 6, 287–293 CrossRef CAS.
- I. I. Ivanova, A. S. Kuznetsov, V. V. Yuschenko and E. E. Knyazeva, Pure Appl. Chem., 2004, 76, 1647–1657 CrossRef CAS.
- I. I. Ivanova, I. A. Kasyanov, A. A. Maerle and V. I. Zaikovskii, Microporous Mesoporous Mater., 2014, 189, 163–172 CrossRef CAS PubMed.
- I. I. Ivanova, A. S. Kuznetsov, O. A. Ponomareva, V. V. Yuschenko and E. E. Knyazeva, in Stud. Surf. Sci. Catal., ed. N. Ž. J. Čejka and P. Nachtigall, Elsevier, 2005, vol. 158, Part A, pp. 121–128 Search PubMed.
- S. Wang, T. Dou, Y. Li, Y. Zhang, X. Li and Z. Yan, J. Solid State Chem., 2004, 177, 4800–4805 CrossRef CAS PubMed.
- W. P. Guo, C. R. Xiong, L. M. Huang and Q. Z. Li, J. Mater. Chem., 2001, 11, 1886–1890 RSC.
- K. R. Kloetstra and J. C. Jansen, Chem. Commun., 1997, 2281–2282 RSC.
- J. Garcia-Martinez, K. Li and G. Krishnaiah, Chem. Commun., 2012, 48, 11841–11843 RSC.
- K. Li, J. Valla and J. Garcia-Martinez, ChemCatChem, 2014, 6, 46–66 CrossRef CAS PubMed.
- J. Garcia-Martinez, M. Johnson, J. Valla, K. Li and J. Y. Ying, Catal. Sci. Technol., 2012, 2, 987–994 CAS.
- J. Garcia-Martinez, C. Xiao, K. A. Cychosz, K. Li, W. Wan, X. Zou and M. Thommes, ChemCatChem, 2014, 6, 3110–3115 CrossRef CAS PubMed.
- J. Ying and J. Garcia-Martinez, US pat., 20050239634, 2005 Search PubMed.
- J. Garcia-Martinez, M. M. Johnson and l. Valla, US pat., 20100196263, 2010 Search PubMed.
- J. Garcia-Martinez, M. M. Johnson and L. Valla, EP pat., 2379449, 2011 Search PubMed.
- W. Q. Jiao, W. H. Fu, X. M. Liang, Y. M. Wang and M.-Y. He, RSC Adv., 2014, 4, 58596–58607 RSC.
- R. Chal, T. Cacciaguerra, S. van Donk and C. Gerardin, Chem. Commun., 2010, 46, 7840–7842 RSC.
- B. Speronello, J. Garcia-Martinez, A. Hansen and R. Hu, Refinery Operations, 2011, 2, 1–6 Search PubMed.
- G. Krishnaiah, B. Speronello, A. Hansen and J. Crosby, American Fuels and Petrochemical Manufacturers Annual Meeting, San Antonio, 2013 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cc10391b |
| This journal is © The Royal Society of Chemistry 2015 |