
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic epoxidation by perrhenate through the formation of organic-phase supramolecular ion pairs†
Mirza
Cokoja
*a,
Iulius I. E.
Markovits‡
a,
Michael H.
Anthofer‡
a,
Saner
Poplata
a,
Alexander
Pöthig
a,
Danny S.
Morris
b,
Peter A.
Tasker
b,
Wolfgang A.
Herrmann
a,
Fritz E.
Kühn
*a and
Jason B.
Love
*b
aCatalysis Research Center and Department of Chemistry, Technische Universität München, Lichtenbergstraße 4, 85747 Garching bei München, Germany. E-mail: mirza.cokoja@tum.de; fritz.kuehn@ch.tum.de
bEaStCHEM School of Chemistry, University of Edinburgh, David Brewster Road, Edinburgh EH9 3FJ, UK. E-mail: jason.love@ed.ac.uk
First published on 26th January 2015
Abstract
Organic-phase supramolecular ion pair (SIP) host–guest assemblies of perrhenate anions (ReO4−) with ammonium amide receptor cations are reported. These compounds act as catalysts for the epoxidation of alkenes by aqueous hydrogen peroxide under biphasic conditions and can be recycled several times with no loss in activity.
The transfer of an anion into an organic phase can have enormous impact on its reactivity as the resulting hydrophobic environment accentuates supramolecular interactions between the anion, reactants, and potential substrates.1 For example, it is well known that the phase transfer of ion pairs enhances reactions of organic compounds with nucleophiles (e.g. halides, alkoxides).2 Furthermore, permanganate acts as a catalyst for the oxidation of olefins to alcohols or acids in the organic phase,3 and polyoxometallates catalyse oxidation reactions such as epoxidation under biphasic conditions.4 Unlike permanganate, the Group VII congener perrhenate is generally considered inactive in oxidation catalysis, and perrhenic acid5 or immobilized perrhenates6 act as poor epoxidation catalysts, with the nature of the active species unclear. Notably however, imidazolium perrhenates mediate the stoichiometric epoxidation of simple alkenes through the activation of H2O2 by hydrogen bonding to [ReO4]− in a hydrophobic ionic liquid.7 Furthermore, the activity of molecular catalysts such as methyltrioxorhenium (MTO) in the biphasic epoxidation of alkenes strongly depends on the nature of the reaction medium, being highest in hydrophobic ionic liquids.8–10 Given that the transfer of [ReO4]− into an organic phase has been established,11 we considered it likely that the amido-ammonium and -pyridinium receptors L1–L3 (Scheme 1), used recently in the solvent extraction of halidometalates,12,13 could be exploited to transfer [ReO4]− into a hydrophobic environment; this would potentially enhance its activity in oxidation catalysis by favouring supramolecular interactions with substrates such as H2O2. Herein, we report the synthesis and characterization of supramolecular ion pairs (SIPs) constructed from perrhenate and the organic receptor cations [HL1]+–[HL3]+, and their application in biphasic epoxidation catalysis.
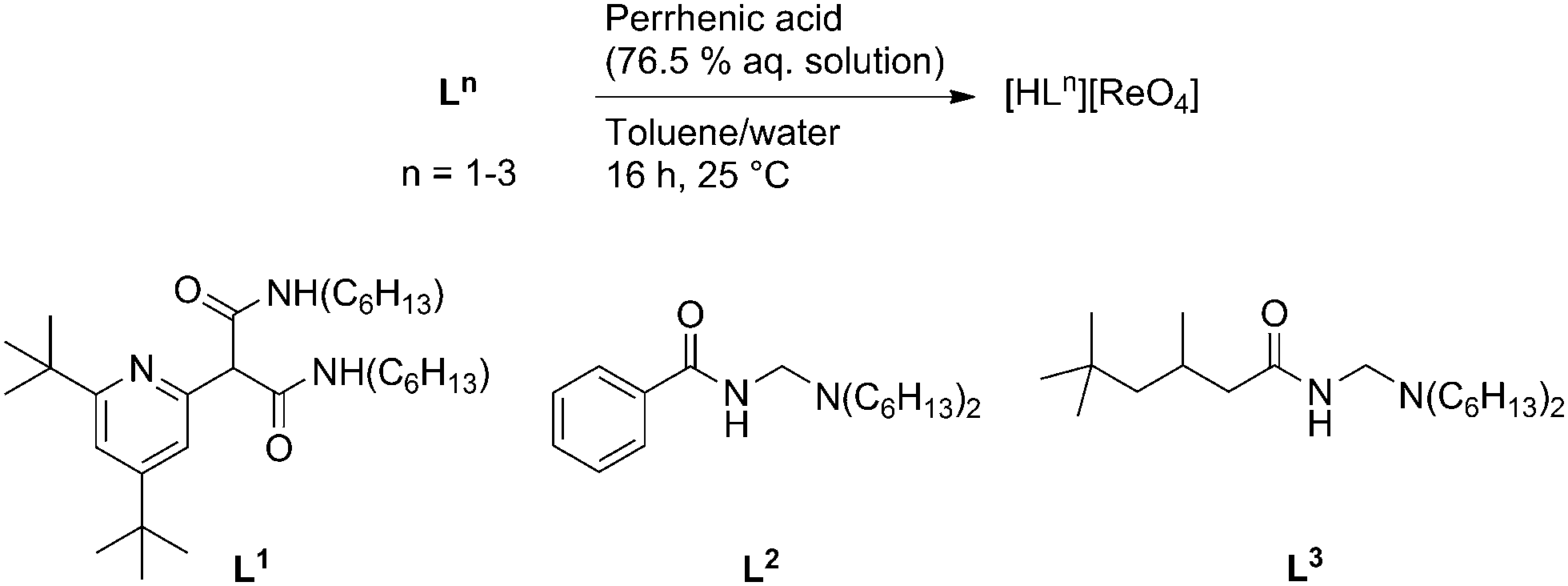 | ||
| Scheme 1 Synthesis of perrhenate SIPs [HLn][ReO4] (n = 1, 2, 3). | ||
Three toluene-soluble perrhenate SIPs [HLn][ReO4] (n = 1, 2, 3) were synthesized by liquid–liquid extraction of 76.5 wt% perrhenic acid in water with the corresponding receptors L1–L3 in toluene (Scheme 1).
The compounds were analysed by 1H- and 13C-NMR and IR spectroscopy and by elemental analysis (for details see ESI†). While the SIP [HL1][ReO4] is an off-white solid, [HL2][ReO4] and [HL3][ReO4] are colourless oils at room temperature. All of the SIPs are soluble in toluene and ethers, are insoluble in aliphatic hydrocarbons, water, and 50% H2O2, and decompose above 170 °C, according to thermogravimetric analysis (ESI†).
The solid-state structure of [HL1][ReO4] was determined by single-crystal X-ray diffraction of crystals grown from a 50/50 mixture of diethyl ether and n-hexane. The protonation of the receptor results in its organization due to intramolecular pyridinium-amide hydrogen bonding between the N1 donor and O1/O2 acceptor atoms (ESI†). These observations are analogous to our previous results and support the stabilisation and solubilisation of the proton through a six-membered ‘proton-chelate ring’.13 Expansion of the structure reveals a dimeric motif in which two protonated receptors interact with two [ReO4]− anions, along with a further two interactions with the tBu protons of adjacent molecules (Fig. 1). The rigid cationic receptor offers an array of seven N- and C-hydrogen bonds per [ReO4]− anion, with the strongest (and classical) hydrogen bonding seen between N2–H and N3–H to O6.
 | ||
| Fig. 1 The solid-state structure of [HL1][ReO4] showing the intermolecular N–H and C–H contacts to the perrhenate anion (displacement ellipsoids drawn at 50% probability). Bond distances (Å): Re1–O3 1.701(2), Re1–O4 1.715(2), Re1–O5 1.715(2), Re1–O6 1.738(2), N2⋯O6 2.919, N3⋯O6 3.013, C14⋯O6 3.377, C2⋯O4 3.357, C26⋯O4 3.585, C13⋯O3 3.489, C14⋯O3 3.242. | ||
Moreover, the central C14–H bond of the malonamide unit bridges O3 and O6, with further intermolecular C–H contacts to the O3 and O4 oxygen atoms of the perrhenate anion detected. These interactions lead to a distortion of the perrhenate anion from its optimal tetrahedral geometry and a distortion in the Re–oxo bond lengths, with Re1–O3 compressed and Re1–O6 elongated. These data indicate a change of the symmetry from Td to C2v for the perrhenate anion in the solid state and, as such, IR studies of the SIP were undertaken to support these observations (ESI,† Fig. S1). After deconvolution, the spectra show three signals for the asymmetric Re![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration (1000–800 cm−1), inconsistent with Td symmetry. In comparison to our previous studies7 the occurrence of three different signals for this vibration points to a C2v symmetric [ReO4]− anion, corroborating the above observations.
O stretching vibration (1000–800 cm−1), inconsistent with Td symmetry. In comparison to our previous studies7 the occurrence of three different signals for this vibration points to a C2v symmetric [ReO4]− anion, corroborating the above observations.
The solution structures of the perrhenate SIPs were probed using ESI-MS and NMR techniques. No ions are seen in the ESI-MS for samples dissolved in toluene so experiments were carried out using CH3CN as the diluent and at low inlet nozzle temperatures (50 °C). The ESI-MS of [HL1][ReO4] (Fig. 2) shows no ions that correlate with the X-ray crystal structure. However, the formation of aggregated assemblies is apparent, with the parent ion at 920 amu resulting from the pairing of [HL1]+ with L1. Furthermore, ions due to single or double perrhenate incorporation in assemblies of protonated and/or neutral receptors are seen at 1171, 1631, and 1883 amu. While these data do not correlate with the dinuclear structural motif identified in the solid state, it is clear that host–guest assemblies are formed in solution. For [HL2][ReO4] and [HL3][ReO4] in CH3CN (ESI,† Fig. S14 and S15), similar patterns are seen, but in these cases facile fragmentation of the receptor at the bridging methylene group is observed, resulting in more complex spectra.
 | ||
| Fig. 2 ESI-MS of [HL1][ReO4] in CH3CN. | ||
The formation of an organised receptor on protonation is further supported by the 1H-NMR spectrum of [HL1][ReO4] in CDCl3 which shows a new peak at 14.66 ppm for the pyridinium NH proton and all other protons shifted to higher frequency with the NCH2 (3.18 ppm) resonances of the hexylamine chains split into two; similar features were reported for the analogue [HL1]2[ZnCl4].13 As with the above ESI-MS data, the 1H NMR spectrum of [HL3][ReO4] indicates that some fragmentation of the receptor has occurred, with broad resonances at 7.9 and 6.5/5.9 ppm assignable to the separate ammonium and amide constituents of HL3 along with those due to the intact SIP. All three SIPs were analysed using DOSY NMR spectroscopy (ESI,† Fig. S5–S10). In C6D6, a single species is observed for [HL1][ReO4] with a calculated hydrodynamic radius of ca. 6.3 Å, larger than that of the free ligand and consistent with a degree of aggregation in solution. Similarly, for [HLn][ReO4] (n = 2, 3) in C6D6 single species with radii of ca. 6.6 and 6.7 Å, respectively are seen. In CH3CN, several species are observed for [HLn][ReO4] with radii varying between ca. 2.8 and 4.5 Å, indicating that more dynamic speciation occurs in this more polar solvent. It is therefore likely that the aggregated structure seen in the solid state is representative of the structures of the SIPs in non-polar solvents such as toluene.
The SIPs were used as catalysts for the two-phase epoxidation of cyclooctene with aqueous hydrogen peroxide (50%) without additional solvents. All of the SIPs are active catalysts, leading to nearly quantitative conversions of cyclooctene after 6–8 h reaction time at 70 °C (Fig. 3), with quantitative selectivity for epoxide, i.e. the possible by-product 1,2-cyclooctanediol is not formed. Significantly, in the absence of perrhenate conversion to the epoxide does not occur. This is the first example of the application of perrhenate as an epoxidation catalyst in an organic phase, resulting from its transfer from the aqueous phase into the hydrophobic medium. Significantly, under similar oxidative reaction conditions, other typical phase transfer agents such as crown ethers decompose, and quaternary ammonium salts are inactive. At the start of the reaction, [HL2][ReO4] and [HL3][ReO4] are more active than [HL1][ReO4] presumably because of their higher hydrophobicity. However, both [HL2] and [HL3] receptors are already partially fragmented during the SIP synthesis, and in presence of hydrogen peroxide they decompose entirely, which may be a reason for the decreasing activity over time. Hence, further catalytic investigations were carried out using [HL1][ReO4], which is the only SIP stable under oxidative (catalytic) conditions (ESI,† Fig. S11 and S12). While the other SIPs comprise mixtures of the fragmented cation (amide and ammonium), perrhenate, and intact [HLn][ReO4] (n = 2, 3) they are, however, still catalytically active, albeit to a lesser extent than [HL1][ReO4].
 | ||
Fig. 3 Kinetic plots of the epoxidation of cyclooctene using catalysts [HLn][ReO4] (n = 1–3) and aqueous H2O2 as oxidant. Reaction conditions: 70 °C, 5 mol% catalyst, molar ratio catalyst![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :substrate:oxidant 5:100:250. :substrate:oxidant 5:100:250. | ||
The reaction temperature has a significant effect on the catalyst activity (ESI,† Fig. S19). At 50 °C the yield of cyclooctene oxide is 33% after 6 h, in contrast to 100% conversion at 70 °C over the same time period; at 25 °C, no conversion is seen. Reducing the catalyst loading to 1 or 2.5 mol% also results in a lower conversion of cyclooctene with 34 and 80% conversions after 6 h, respectively, compared to 92% using 5 mol% loadings (ESI,† Fig. S20). Increasing the amount of oxidant has no effect on the catalytic activity of the SIPs (ESI,† Fig. S21). We have no indication for partial decomposition of H2O2 at these temperatures. When less than 1 equiv. H2O2 per cyclooctene is used, the conversion corresponds to the H2O2 consumed, yet the reaction is slower. Adding toluene to the reaction mixture increases the solubility of the catalyst in the organic phase, ensuring that the catalyst is dissolved even at decreasing cyclooctene concentrations. Monitoring the epoxidation of cyclooctene during an eight-hour reaction (ESI,† Fig. S22) confirms that the addition of toluene does not diminish activity and conversion compared to the neat reaction.
Based on our previous work and DFT calculations,7 the transfer of perrhenate from the aqueous to hydrophobic organic phase should activate H2O2 through H-bonding interactions, which in turn favours oxygen transfer to an olefin (Scheme 2). While we have not yet carried out DFT calculations on this current system, spectroscopic data suggest that a similar mechanism operates. When 17O-labelled [HL1][Re17O4] catalyst is used for the epoxidation of cyclooctene no 17O is seen in the 17O-NMR spectrum of the isolated cyclooctene oxide (ESI,† Fig. S13). This indicates that the oxygen transfer to the olefin does not originate from a Re–O species, but rather from an outer-sphere activation of H2O2, as shown in Scheme 2. Note that both the neutral receptor L1 and protonated [HL1][Br] do not exhibit catalytic activity under the applied reaction conditions (ESI,† Fig. S24), so the activity of the SIP can only be ascribed to the presence of perrhenate.
 | ||
| Scheme 2 Mechanism of the epoxidation of olefins catalysed by the perrhenate ion in ILs.7 | ||
Importantly, the biphasic system consisting of water/H2O2 and toluene/SIP/product facilitates the catalyst-product separation. The SIP catalyst and the product are extracted with toluene, and subsequent distillation of the solvent and product recovers the SIP. Leaching of the SIP catalyst into the aqueous phase does not occur; phase separation after 2.5 h reaction time (ca. 50% conversion of cyclooctene, see Fig. 3) and addition of fresh cyclooctene to the aqueous phase did not lead to further epoxide formation. As such, this procedure allows for catalyst reusability, rendering these supramolecular catalysts suitable for larger scale applications. Indeed, the SIP [HL1][ReO4] was recycled five times after 4 h reaction time and displayed no loss in activity within the error range of the data analyses (ESI,† Fig. S23).
To demonstrate the generality of these catalysts, we have studied the epoxidation of other alkenes, and, in nearly all cases investigated, good conversions are reached (Table 1). It is known that terminal alkenes, such as 1-octene and styrene are intrinsically more difficult to epoxidise than (cyclic) cis-alkenes and the ring opening of the products to diols (the only by-product) is, in these cases, facilitated due to steric reasons.8,9 Even though the overall performance obtained with the SIP catalysts is lower than that of known molecular epoxidation catalysts such as MTO (TOFmax = 39000 h−1 for cyclooctene as substrate at 0 °C),9,14–16 the SIPs exhibit the advantage of being more stable and recyclable; at 70 °C the catalytically active species formed from MTO and H2O2 decomposes rapidly.16
|
|
|||
|---|---|---|---|
| Entry | Substrate | Conv. [%] | Sel. [%] |
| Reaction conditions: 5 mol% catalyst, aq. H2O2 as oxidant, molar cat:substrate:oxidant ratio 5:100:250, 8 h, 70 °C. |
|||
| 1 | Cyclohexene | 89 | 99 |
| 2 | 1-Octene | 77 | 24 |
| 3 | Styrene | 70 | 44 |
| 4 | Allyl alcohol | 84 | 18 |
| 5 | Propene | 16 | 50 |

It is noteworthy that the epoxidation of allyl alcohol leads to a good conversion, yet low selectivity. This is likely due to the good miscibility of the epoxide product in water, which leads to over-oxidation and the formation of glycerol. Also, the SIPs catalyse the epoxidation of propene to propene oxide (PO). Although the conversion is low, it should be noted that there are only a few reports on propene epoxidation using molecular catalysts, all of which display similar conversions.5 In this case, although the catalytic performance of the SIP is lower than that of MTO, the system presented here is recyclable and so the SIPs outcompete MTO in the long term.
The transfer of the perrhenate anion into a hydrophobic medium enhances its supramolecular interactions, resulting in very robust and active catalysts for biphasic alkene epoxidation by hydrogen peroxide. This is the first report on the epoxidation of alkenes catalysed by perrhenate in organic solvents and exploits the concept of transferring compounds regarded as notoriously inactive in catalysis into an organic phase to significantly enhance catalytic performance. Furthermore, the back-transfer of the anion allows for straightforward separation and recovery of the catalyst, which clearly outperforms other immobilized molecular catalysts, so offering a simple, sustainable alternative to established molecular catalysts.
Notes and references
- M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1660 RSC; M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1734 RSC; K. Brak and E. N. Jacobsen, Angew. Chem., Int. Ed., 2013, 52, 534 CrossRef CAS PubMed; T. Ooi and K. Maruoka, Angew. Chem., Int. Ed., 2007, 46, 4222 CrossRef PubMed; J. W. Steed and J. L. Atwood, Supramolecular Chemistry., John Wiley & Sons, Chichester, UK, 2009 CrossRef; R. Breslow and L. E. Overman, J. Am. Chem. Soc., 1970, 92, 1075 CrossRef; S. Beckendorf, S. Asmus and O. García Mancheño, ChemCatChem, 2012, 4, 926 CrossRef.
- C. L. Liotta and H. P. Harris, J. Am. Chem. Soc., 1974, 96, 2250 CrossRef CAS; E. V. Dehmlow, Angew. Chem., Int. Ed., 1977, 16, 493 CrossRef; P. Viout, J. Mol. Catal., 1981, 10, 231 CrossRef; J.-F. Brière, S. Oudeyer, V. Dalla and V. Levacher, Chem. Soc. Rev., 2012, 41, 1696 RSC.
- D. J. Sam and H. E. Simmons, J. Am. Chem. Soc., 1972, 94, 4024 CrossRef CAS.
- G. De Faveri, G. Ilyashenko and M. Watkinson, Chem. Soc. Rev., 2011, 40, 1722 RSC; R. H. Ingle and N. K. K. Raj, J. Mol. Catal. A: Chem., 2008, 294, 8 CrossRef CAS PubMed; M. Bösing, A. Nöh, I. Loose and B. Krebs, J. Am. Chem. Soc., 1998, 120, 7252 CrossRef; K. Sato, M. Aoki and R. Noyori, Science, 1998, 281, 1646 CrossRef.
- M. C. A. van Vliet, I. W. C. E. Arends and R. A. Sheldon, J. Chem. Soc., Perkin Trans. 1, 2000, 377 RSC.
- D. Mandelli, M. C. A. van Vliet, U. Arnold, R. A. Sheldon and U. Schuchardt, J. Mol. Catal. A: Chem., 2001, 168, 165 CrossRef CAS; D. Veljanovski, A. Sakthivel, W. A. Herrmann and F. E. Kühn, Adv. Synth. Catal., 2006, 348, 1752 CrossRef.
- I. I. E. Markovits, W. A. Eger, S. Yue, M. Cokoja, C. J. Münchmeyer, B. Zhang, M.-D. Zhou, A. Genest, J. Mink, S.-L. Zang, N. Rösch and F. E. Kühn, Chem. – Eur. J., 2013, 19, 5972 CrossRef CAS PubMed.
- S. Huber, M. Cokoja and F. E. Kühn, J. Organomet. Chem., 2014, 751, 25 CrossRef CAS PubMed.
- S. A. Hauser, M. Cokoja and F. E. Kühn, Catal. Sci. Technol., 2013, 3, 552 CAS.
- D. Betz, A. Raith, M. Cokoja and F. E. Kühn, ChemSusChem, 2010, 3, 559 CrossRef CAS PubMed.
- E. A. Katayev, G. V. Kolesnikov and J. L. Sessler, Chem. Soc. Rev., 2009, 38, 1572 RSC; M. Saeki, Y. Sasaki, A. Nakai, A. Ohashi, D. Banerjee, A. C. Scheinost and H. Foerstendorf, Inorg. Chem., 2012, 51, 5814 CrossRef CAS PubMed.
- R. J. Ellis, J. Chartres, D. K. Henderson, R. Cabot, P. R. Richardson, F. J. White, M. Schröder, J. R. Turkington, P. A. Tasker and K. C. Sole, Chem. – Eur. J., 2012, 18, 7715 CrossRef CAS PubMed; J. R. Turkington, P. J. Bailey, J. B. Love, A. M. Wilson and P. A. Tasker, Chem. Commun., 2013, 49, 1891 RSC; A. M. Wilson, P. J. Bailey, P. A. Tasker, J. R. Turkington, R. A. Grant and J. B. Love, Chem. Soc. Rev., 2014, 43, 123 RSC.
- J. R. Turkington, V. Cocalia, K. Kendall, C. A. Morrison, P. Richardson, T. Sassi, P. A. Tasker, P. J. Bailey and K. C. Sole, Inorg. Chem., 2012, 51, 12805 CrossRef CAS PubMed.
- I. I. E. Markovits, M. H. Anthofer, H. Kolding, M. Cokoja, A. Pöthig, A. Raba, W. A. Herrmann, R. Fehrmann and F. E. Kühn, Catal. Sci. Technol., 2014, 4, 3845 CAS.
- H.-J. Lee, T.-P. Shi, D. H. Busch and B. Subramaniam, Chem. Eng. Sci., 2007, 62, 7282 CrossRef CAS PubMed.
- P. Altmann, M. Cokoja and F. E. Kühn, Eur. J. Inorg. Chem., 2012, 3235–3239 CrossRef CAS; F. E. Kühn, A. Scherbaum and W. A. Herrmann, J. Organomet. Chem., 2004, 689, 4149 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Full synthetic and characterisation details of the [HLn][ReO4] compounds as well as catalysis and recycling experiments. CCDC 1030881. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc10235e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |