
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Associative chemosensing by fluorescent macrocycle–dye complexes – a versatile enzyme assay platform beyond indicator displacement†
Frank
Biedermann
*,
Denisa
Hathazi
and
Werner M.
Nau
*
Department of Life Sciences and Chemistry, Jacobs University Bremen, Campus Ring 1, 28759 Bremen, Germany. E-mail: frankbiedermann@daad-alumni.de; w.nau@jacobs-university.de
First published on 15th January 2015
Abstract
A label-free in situ method to monitor reactions in real time by using fluorescent supramolecular chemosensors based on cucurbit[8]uril is presented. It allows sensing of enzymatic activity, inhibitor and activator screening, and analyte detection with unprecedented versatility and high sensitivity.
Methods for monitoring (bio)catalytic activity are indispensable for the elucidation of reaction mechanisms, the testing of inhibitors and activators, and for the activity screening of natural and tailor-made enzymes.1 Emission-based detection is almost always preferred, especially in pharmaceutical-industrial settings, because of its sensitivity, economic use, and safety. Only a few substrate–product pairs are intrinsically fluorescent, and even fewer are spectrally distinguishable from each other. Therefore, fluorescently labeled substrate analogues are often resorted to, at the expense of added costs and frequently non-transferable activity profiles.1a,b Supramolecular tandem enzyme assays, using a sensing ensemble composed of a macrocyclic host and an indicator dye, were previously introduced by us, allowing for label-free reaction monitoring.1b,2 These operate through an indicator displacement scheme,3 which requires a subtle balance of the affinities of the substrate, product, and fluorescent dye.1b,2 To address this shortcoming, we explored a complementary chemosensor design strategy, which was recently introduced for the detection and reaction monitoring of chiral aromatic analytes by induced circular dichroism.4 We now introduce self-assembled fluorescent chemosensors that associate with aromatic substrates and, thereby, allow the real-time monitoring of their enzymatic conversions. This establishes a general enzyme assay platform responsive to aryl moieties, as they are most abundant in drugs, amino acids, toxins, hormones, antibiotics, and colorants.5
The self-assembled fluorescent chemosensors can be obtained by the spontaneous inclusion of a suitable dicationic fluorescent dye (e.g., MDAP, MDPP, or PDI) into the large cavity of the macrocycle cucurbit[8]uril (CB8),6 see Scheme 1. The residual cavity space of such CB8-based chemosensors allows for the subsequent binding of an aryl-functionalized analyte which is typically accompanied by net fluorescence quenching.4,7 The magnitude of the intensity change is a function of the concentration, binding affinity (Ka), and quenching efficiency (QE) of the substrate–product couple, see also Table S1 (ESI†) for representative Ka and QE values. Importantly, the non-covalent and reversible chemosensor–analyte complexation is essentially instantaneous: it responds dynamically to analyte concentration changes.4 Consequently, the change in the concentrations of the substrate (S) and the product (P) in the course of an enzymatic reaction translates directly into alteration of the ratio of emissive (analyte-free, binary) and non-emissive (analyte-bound, ternary) chemosensor and, thus, a change in the measurable emission intensity (Iem). The only condition which needs to be fulfilled is differential affinities, that is, Ka(S) ≠ Ka(P), see Scheme 1c, which is almost always the case for chemical modifications taking place in the vicinity of the aryl anchor. Specifically, an increase in Iem is expected for Ka(S) > Ka(P), the substrate-selective enzyme assay, whereas for the product-selective variant, with Ka(S) < Ka(P), a decrease in Iem applies. While we successfully tested several fluorescent dyes (Scheme 1b), the dye can in principle be selected at will to match the demands of a user for a particular wavelength or sensitivity, or different dyes can be screened to achieve a quick optimization of the assay.
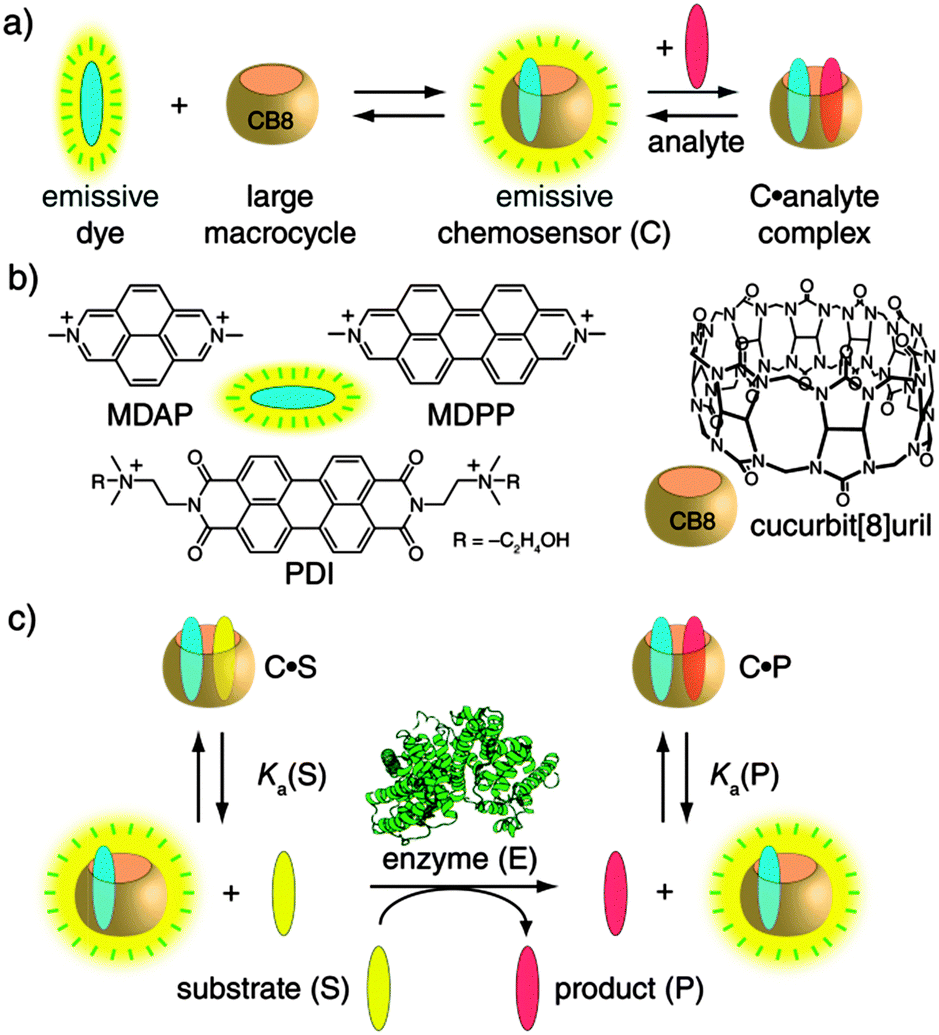 | ||
| Scheme 1 (a and b) Fluorescent chemosensors for aromatic analytes are self-assembled from the large macrocyclic host cucurbit[8]uril (CB8) and dicationic dyes. (c) Appearance of the product and the corresponding depletion of the substrate during the course of an enzymatic reaction are coupled to a change in the amount free and analyte-bound chemosensor. | ||
After we had confirmed that the chemosensors are chemically stable in the reaction mixtures, we demonstrated the versatility of this supramolecular approach to enzymatic reaction monitoring. For this purpose a wide range of enzymatic redox and hydrolysis reactions was tested, see Scheme 2. Representative results are surveyed below and all are contained in the ESI,† as are the quantitative aspects related to this supramolecular sensing concept. Our claim is that there is no other assay known which covers such a large range of enzymes, not to speak of the ease of use and the other advantages named above.
 | ||
| Scheme 2 Enzymatic (a) redox and (b) hydrolysis reactions that can be monitored with the associative chemosensors introduced herein. | ||
To benchmark our associative chemosensors, the kinetics of the horseradish peroxidase-(HRP)-catalysed oxidation of catechol with H2O2 was compared to the direct UV/vis recording of the resulting chromophoric 1,2-benzoquinone-catechol product-couple.8 Both methods yielded very similar kinetic traces (Fig. S1, ESI†). Furthermore, the variation of the enzyme concentration yielded the expected linear proportionality of the initial rates (Fig. S2, ESI†). Both observations confirm that the chemosensors do not interfere with the enzymatic activity and remain stable under the experimental conditions. The chemosensors show broad selectivity to many aromatic substrates, such that they are predestined for rapid substrate-screening studies. To demonstrate, the oxidation of naphthols and phenols (Fig. 1a and Fig. S3a, ESI†) was monitored by our emission-based method. Low μM substrate concentrations were sufficient, demonstrating high sensitivity. Additionally, different chemosensors could be used interchangeably (Fig. 1a). In line with intuition, the more electron-rich substrates were oxidized faster, e.g., 4-substituted phenols with R = –OMe > –Me > –H > –F (see Table S2, ESI† for numerical values and more examples.) The method can also be extended to the screening of reaction conditions, e.g., of the pH (Fig. 1b). In fact, the HRP-catalysed oxidation of Trp is known to proceed via a number of chemical intermediates,9 whose appearance depends on pH, which is in agreement with the observed complicated kinetic profiles (Fig. 1b). The use of different chemosensors with distinct responses to (some of) the intermediate(s) can in such cases provide additional kinetic information: For example, the HRP-catalyzed oxidation of aniline with H2O2, known to yield polyaniline in a step-wise process,10 was successfully monitored with all three receptors, that is, CB8·MDAP, CB8·MDPP, and CB8·PDI (Fig. S3b, ESI†). Comparison of the kinetic traces clearly exposed the multi-step nature of the reaction, for which at least two intermediates need to be invoked.
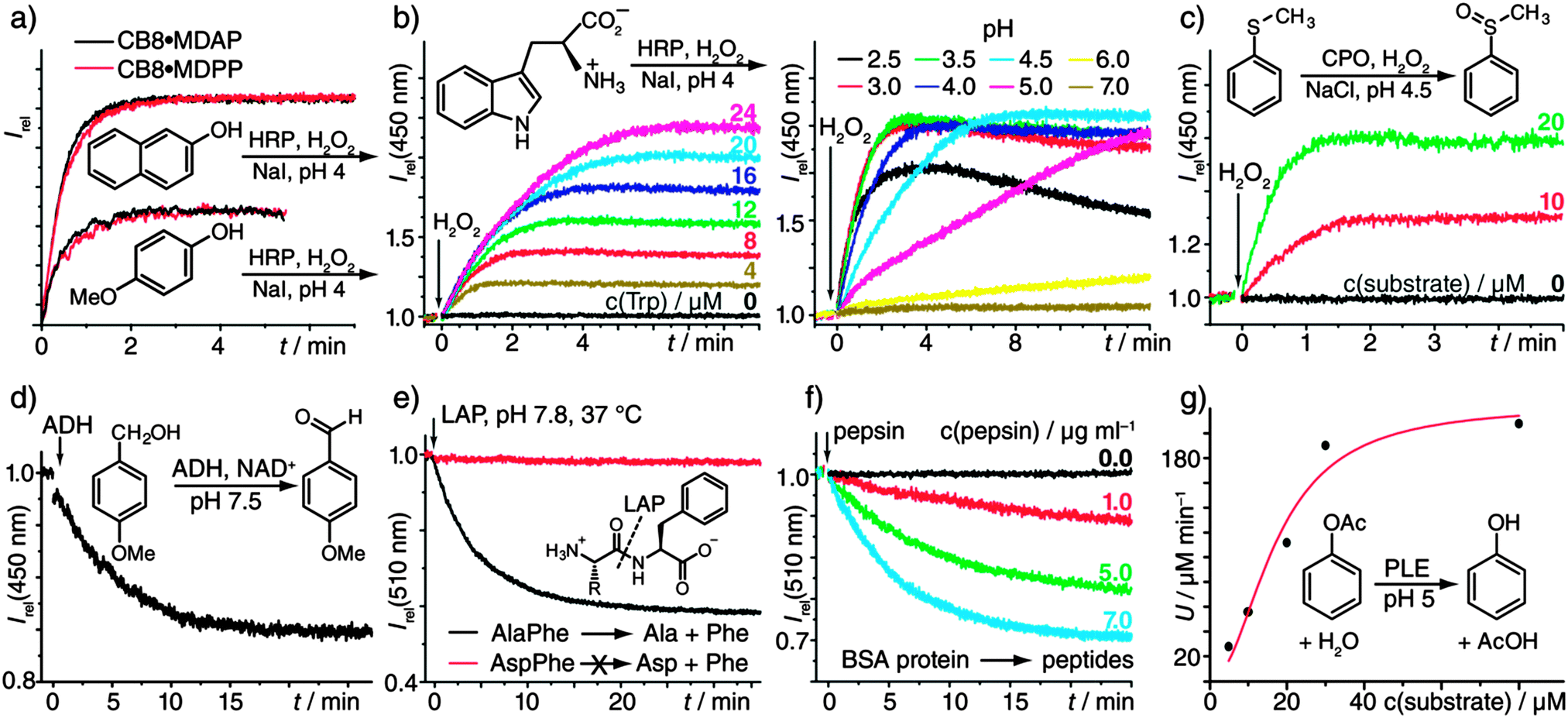 | ||
| Fig. 1 (a) HRP-catalysed (0.6 μg mL−1) oxidation of 2-naphthol and 4-methoxy phenol (each 46 μM) with H2O2 (100 μM) and NaI (120 μM), reported by two different chemosensors (each 5 μM). (b) HRP-catalysed (12 μg mL−1) oxidation of Trp with H2O2 (100 μM) and NaI (120 μM) in the presence of CB8·MDAP (5 μM). Left: varying Trp concentrations at pH 4. Right: pH variation with 20 μM Trp. (c) CPO-catalysed (25 μg mL−1) sulfoxidation with H2O2 (200 μM) and NaCl (100 μM) in the presence of CB8·MDAP (5 μM). (d) ADH-catalysed (3.8 μg mL−1) alcohol-oxidation (100 μM) with the cofactor NAD+ (1 mM) in the presence of CB8·MDAP (5 μM). (e) LAP-catalysed (50 μg mL−1) hydrolysis of dipeptides (20 μM) in the presence of CB8·MDPP (5 μM). (f) Pepsin-catalysed (0.0–7.0 μg mL−1) hydrolysis of bovine serum albumin (BSA) protein (80 μg mL−1) in the presence of CB8·MDPP (5 μM) at pH 2. (g) PLE-catalysed (80 μg mL−1) hydrolysis of phenyl acetate at pH 5 in the presence of CB8·MDAP (5 μM). The plot of the initial rates vs. the substrate concentration was fitted by a Michaelis–Menten function to yield a KM value of (25 ± 4.) μM. All reactions were carried out in 10 mM sodium phosphate buffer. See the Materials and methods section in the ESI† for further details. | ||
Chloroperoxidase (CPO) has a higher redox potential than HRP which allows for the oxidation of chloride ions to produce reactive intermediates, thus enabling the oxidation of organic molecules that are not subject to HRP-catalyzed reactions.11 For instance, the pharmaceutically important enzymatic production of a representative chiral sulfoxide12 by CPO could be followed in real time even at low μM substrate concentration with the chemosensor CB8·MDAP (Fig. 1c), which cannot be achieved by direct optical monitoring. In view of the success with HRP and CPO, it was not surprising that lactoperoxidase-catalysed reactions could also be monitored with the fluorescent receptors, see Fig. S4 (ESI†) for a representative example.
Copper-containing laccases utilize molecular dioxygen for the oxidation of various substrates and have therefore found widespread industrial use, e.g., for lignin degradation, waste-water cleaning, and in food production.13 Redox mediators such as 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) (ABTS) and N-hydroxyl-phthalimide (NHA) can significantly speed up the desired oxidation of a substrate.13c As is demonstrated in Fig. S5 (ESI†), it was possible to follow the representative laccase-catalysed oxidation of 4-chlorophenol and to screen for the activity of the mediators ABTS and NHA.‡
Alcohol dehydrogenases (ADHs) have industrial and pharmaceutical use for the oxidative conversion of alcohols to aldehydes and for the (stereoselective) reduction of ketones to alcohols.14 In contrast to the aforementioned enzymatic reactions, ADH requires a cofactor, NAD+/NADH,H+, that is commonly employed in large excess. Fortunately, neither NAD+ nor NADH,H+ interact strongly with the chemosensor CB8·MDAP and their absorption bands do not interfere with its excitation wavelength. Hence, the ADH-catalyzed oxidation of 4MeO-benzylalcohol to 4MeO-benzaldehyde could be readily monitored (Fig. 1d). A representative reduction reaction is shown in Fig. S6 (ESI†).
Moving from enzymatic redox to hydrolysis reactions, the exopeptidase L-leucine aminopeptidase (LAP) catalyses the hydrolysis of peptides and proteins starting from their N-terminus. In addition to its wide natural abundance, LAP found use in peptide sequencing.15Fig. 1e shows the kinetic profile for the LAP-catalyzed hydrolysis of AlaPhe into Ala and Phe, as was monitored in the presence of the chemosensor CB8·MDPP. Other examples including the hydrolysis of Trp-containing peptides (Fig. S7) and the comparison of the enzymatic activity when incubated with Mg2+ or Zn2+ (Fig. S8) are shown in the ESI.†
Carboxypeptidases are exopeptidases that cleave proteins and peptides C-terminally16 for which even substrate-labeled assays are scarce.17 The fluorescent chemosensor-based monitoring of carboxypeptidase A (CPA) activity on hippuryl-phenylalanine is shown in Fig. S9 (ESI†), providing a complementary monitoring method to our recently reported induced circular dichroism detection strategy.4
The aromatic amino acids of most proteins are located inside the protein hydrophobic core; they are thus inaccessible for direct binding to chemosensors.4 However, digestion of a protein with peptidase destroys its tertiary protein structure, which makes the aromatic recognition motifs sterically accessible. The digestion of bovine serum albumin (BSA) with pepsin at pH 2 serves as a model reaction, displaying the expected reduction in emission intensity in the presence of CB8·MDPP (Fig. 1f). The use of the chemosensor-based detection is likely a broadly applicable method to monitor the digestion of proteins since most proteins contain at least one Trp (or Phe) residue.
Esterases such as porcine liver esterase (PLE) have received interest for synthetic chemical purposes including chiral resolution, and also in agriculture, as well as the food and pharmaceutical industry.18 The applicability and compatibility of the receptor was demonstrated for the PLE-catalysed hydrolysis of the model substrates, phenyl acetate and butyl benzoate (Fig. 1g and Fig. S10, ESI†), including the determination of its Michaelis–Menten kinetics (Fig. 1g).
Phosphates catalyze the removal of phosphate groups from organic molecules, which alters their overall charge and, thus, their affinity for supramolecular hosts. Such charge-altering reactions are good candidates for dye-displacement assays.1b,2c Nevertheless, a phosphatase reaction can also be monitored with our chemosensing ensembles that complex preferentially non-charged over negatively charged analytes4,6,7b (product-selective assays). For instance, the dephosphorylation of phenyl and naphthyl phosphates by alkaline phosphatase (ALKP) is presented in Fig. S11 (ESI†) and shows the expected decrease in Iem as a function of substrate conversion.
Penicillinase belongs to the important enzyme class of β-lactamases, which are largely responsible for the resistance of certain bacteria to β-lactam antibiotics. It hydrolyses the β-lactam ring of benzylpenicillin, also known as penicillin G, followed by spontaneous decarboxylation. We could now monitor this enzymatic reaction by fluorescence in real time (Fig. S12, ESI†), offering a sensitive detection alternative to direct circular dichroism.19
While unsubstituted carbohydrates do not bind to the receptors, their aryl-substituted variants are complexed with mM to μM affinities.4 We have followed the hydrolysis of phenyl-β-D-galactopyranoside by β-galactosidase (β-Gal), a commonly studied reaction in cell biology. Also here, low μM concentrations of the substrate were sufficient for kinetic monitoring (Fig. S13, ESI†). Furthermore, pH-titration experiments revealed the highest enzymatic activity at around pH 5. The maximum is due to the involvement of a glutamic acid residue as a nucleophile in the rate-determining step, as has been previously deduced by using o-nitro-phenyl-β-D-galactopyranoside as a chromogenic labeled substrate.20
Associative chemosensing ensembles consisting of the large macrocycle cucurbit[8]uril and suitable fluorescent dyes provide a versatile platform to monitor in situ enzymatic reactions with μM sensitivity for a wide range of substrates carrying aromatic recognition motifs. We demonstrated the large scope of the method by employing 12 different enzymes catalyzing redox and hydrolysis reactions over a broad pH range, including peroxidases, dehydrogenases, laccases, peptidases, esterases, phosphatases, penicillinase, and β-galactosidase. All enzymatic conversions were followed in real time and enzyme-kinetic parameters are directly accessible. The broad selectivity of the receptors allows for activity screening of substrates and mediators; the extension to inhibitor screening is logical, albeit limited to inhibitors that do not (strongly) bind to the receptor. Important for sensing, the excitation and the emission can be chosen in the visible region of the spectrum and can be tuned through modification of the dye, keeping the macrocycle unchanged. The use of self-assembled CB8-dye receptors also enables the identification of reaction intermediates that would escape detection when observing the fluorescence/absorption of the substrate or product alone. We believe that the associative chemosensing strategy can find a wide range of applications in enzymatic reaction screening, in pharmaceutical research, white biotechnology, and also as an economic analytical tool in biochemical research labs.
This work was supported by the German Academic Exchange Service (F.B.) and the Deutsche Forschungsgemeinschaft (DFG Grant NA-686/5, WMN).
Notes and references
- (a) J.-L. Reymond, V. S. Fluxà and N. Maillard, Chem. Commun., 2009, 34 RSC; (b) R. N. Dsouza, A. Hennig and W. M. Nau, Chem. – Eur. J., 2012, 18, 3444 CrossRef CAS PubMed; (c) K. Drauz and H. Waldmann, Enzyme Catalysis in Organic Synthesis, Wiley-VCH, 2008 Search PubMed.
- (a) A. Hennig, H. Bakirci and W. M. Nau, Nat. Methods, 2007, 4, 629 CrossRef CAS PubMed; (b) G. Ghale, V. Ramalingam, A. R. Urbach and W. M. Nau, J. Am. Chem. Soc., 2011, 133, 7528 CrossRef CAS PubMed; (c) M. Florea and W. M. Nau, Org. Biomol. Chem., 2010, 8, 1033 RSC; (d) W. M. Nau, G. Ghale, A. Hennig, H. Bakirci and D. M. Bailey, J. Am. Chem. Soc., 2009, 131, 11558 CrossRef CAS PubMed; (e) D.-S. Guo, V. D. Uzunova, X. Su, Y. Liu and W. M. Nau, Chem. Sci., 2011, 2, 1722 RSC.
- (a) B. T. Nguyen and E. V. Anslyn, Coord. Chem. Rev., 2006, 250, 3118 CrossRef CAS PubMed; (b) S. L. Wiskur, H. Ait-Haddou, J. J. Lavigne and E. V. Anslyn, Acc. Chem. Res., 2001, 34, 963 CrossRef CAS PubMed.
- F. Biedermann and W. M. Nau, Angew. Chem., Int. Ed., 2014, 53, 5694 CrossRef CAS PubMed.
- G. R. Bickerton, G. V. Paolini, J. Besnard, S. Muresan and A. L. Hopkins, Nat. Chem., 2012, 4, 90 CrossRef CAS PubMed.
- (a) E. Masson, X. X. Ling, R. Joseph, L. Kyeremeh-Mensah and X. Y. Lu, RSC Adv., 2012, 2, 1213 RSC; (b) J. Lagona, P. Mukhopadhyay, S. Chakrabarti and L. Isaacs, Angew. Chem., Int. Ed., 2005, 44, 4844 CrossRef CAS PubMed.
- (a) F. Biedermann, E. Elmalem, I. Ghosh, W. M. Nau and O. A. Scherman, Angew. Chem., Int. Ed., 2012, 51, 7739 CrossRef CAS PubMed; (b) F. Biedermann, U. Rauwald, M. Cziferszky, K. A. Williams, L. D. Gann, B. Y. Guo, A. R. Urbach, C. W. Bielawski and O. A. Scherman, Chem. – Eur. J., 2010, 16, 13716 CrossRef CAS PubMed.
- A. Sadler, V. V. Subrahmanyam and D. Ross, Toxicol. Appl. Pharmacol., 1988, 93, 62 CrossRef CAS.
- N. M. Alexander, J. Biol. Chem., 1974, 249, 1946 CAS.
- Z. Jin, Y. Su and Y. Duan, Synth. Met., 2001, 122, 237 CrossRef CAS.
- H. M. de Hoog, M. Nallani, J. J. L. M. Cornelissen, A. E. Rowan, R. J. M. Nolte and I. W. C. E. Arends, Org. Biomol. Chem., 2009, 7, 4604 CAS.
- F. van de Velde, F. van Rantwijk and R. A. Sheldon, J. Mol. Catal. B: Enzym., 1999, 6, 453 CrossRef CAS.
- (a) S. Riva, Trends Biotechnol., 2006, 24, 219 CrossRef CAS PubMed; (b) S. Rodríguez Couto and J. L. Toca Herrera, Biotechnol. Adv., 2006, 24, 500 CrossRef PubMed; (c) O. V. Morozova, G. P. Shumakovich, S. V. Shleev and Y. I. Yaropolov, Appl. Biochem. Microbiol., 2007, 43, 523 CrossRef CAS.
- (a) J. D. Stewart, Curr. Opin. Chem. Biol., 2001, 5, 120 CrossRef CAS; (b) K. Nakamura and T. Matsuda, Enzyme Catalysis in Organic Synthesis, Wiley-VCH Verlag GmbH, 2008 Search PubMed; (c) C. A. Denard, J. F. Hartwig and H. Zhao, ACS Catal., 2013, 2856 CrossRef CAS.
- B. Wiederanders, J. Lasch, H. Kirschke, P. Bohley, S. Ansorge and H. Hanson, Eur. J. Biochem., 1973, 36, 504 CrossRef CAS PubMed.
- D. S. Auld and B. Holmquist, Biochemistry, 1974, 13, 4355 CrossRef CAS.
- A. Hennig, D. Roth, T. Enderle and W. M. Nau, ChemBioChem, 2006, 7, 733 CrossRef CAS PubMed.
- T. Panda and B. S. Gowrishankar, Appl. Microbiol. Biotechnol., 2005, 67, 160 CrossRef CAS PubMed.
- D. M. Long, Anal. Biochem., 1997, 247, 389 CrossRef CAS PubMed.
- (a) Y. Tanaka, A. Kagamiishi, A. Kiuchi and T. Horiuchi, J. Biochem., 1975, 77, 241 CAS; (b) J. Yuan, M. Martinez-Bilbao and R. E. Huber, Biochem. J., 1994, 299, 527 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials and methods and experimental details. See DOI: 10.1039/c4cc10227d |
| ‡ Both ABTS and NHA are only weakly complexed by the chemosensors. Besides, since the mediator is constantly reformed when the substrate remains, its concentration does not vary, at least at the beginning of the reaction, which is the most informative part of the kinetic trace. |
| This journal is © The Royal Society of Chemistry 2015 |