
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Advances in the molecular understanding of biological zinc transport†
Claudia A.
Blindauer
Department of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: c.blindauer@warwick.ac.uk; Fax: +44 (0)24 76 524112; Tel: +44 (0)24 76 528264
First published on 20th January 2015
Abstract
Between 5 and 10% of all proteins of a given organism are estimated to require zinc for function, and hence zinc is essential for almost any given metabolic process. It is therefore of great interest to understand major players and mechanisms that ensure the tight and correct control of zinc distribution and speciation in organisms and their individual cells. Significant progress has been made in recent years regarding 3-dimensional structures and modes of action of zinc sensor proteins, membrane-bound zinc transporters for cellular and sub-cellular uptake and efflux, as well as intracellular binding proteins. This feature article highlights advances in structures, zinc-binding sites and thermodynamics of proteins that are involved in zinc homeostasis and trafficking, including developments in understanding the metal selectivity of proteins.
1. Introduction: the growing importance of zinc in biological systems
Since the discovery of zinc as an essential element for fungi,1 animals2 including humans,3 plants and bacteria, and of the first enzyme to be shown to require zinc for activity in 1939,4 much has been learned about zinc in biological systems, ranging from effects on the whole organismal level, over the identification of important zinc-binding proteins, down to structural, thermodynamic, and kinetic details of zinc–protein interactions.5,6Zinc deficiency affects up to two billion people worldwide.3 Its multiple systemic effects include growth retardation, weight loss, infertility, mental and emotional disorders, impaired immune function, skin lesions and hair loss.3 The most salient recognition of the importance of zinc in human health was delivered by the 2008 Copenhagen Consensus conference, which ranked supplying zinc and vitamin A to over 100 million malnourished children as their highest priority solution to advance global welfare.7 Besides the drastic consequences of severe zinc deficiency and their alleviation by zinc supplementation,8 the more subtle impacts of zinc homeostasis on ageing,9,10 neurodegenerative diseases,11–13 cancer,14–16 the immune system,17 and energy metabolism18,19 are active study areas.
Apart from its crucial importance for human health, zinc plays also vital roles in the physiology of all other organisms. The impact on plants is illustrated by several-fold increases (up to 600%) in crop yields upon fertilisation of zinc-deficient soils in Anatolia with zinc.20 Zinc-deficient soils are widespread, and may contribute to zinc deficiency in humans, especially where they subsist on cereal-based diets which are rich in zinc-chelating phytate.21 Furthermore, the growth of eukaryotic phytoplankton, in particular coccolithophores and diatoms, has been suggested to be limited by zinc availability in certain regions of the oceans, with consequences for global carbon balances.22 The significance of zinc for bacteria is less well understood, but from the facts that most bacteria have dedicated systems for high-affinity zinc uptake23–25 and respond to zinc starvation,26–28 it may be concluded that zinc is also an essential nutrient for most if not all bacteria. This is also borne out by observations that zinc availability is actively reduced during a host's acute phase response to bacterial infection,29,30 and that zinc is required for full virulence for at least some pathogens.31–33 Influencing zinc homeostasis at the host–pathogen interface34 offers exciting new avenues for antimicrobial therapy, for example the inhibition of virulence factors such as anthrax lethal factor by zinc chelation.35 Conversely, it has also been recognised that free zinc is an unexpectedly potent cellular toxin,36 emphasising the importance of highly efficient homeostatic mechanisms.
For many of the above examples, one of the most exciting current fields in zinc biochemistry concerns understanding zinc “on the move” – in particular in the context of signalling.37,38 In terms of inter-cellular signalling, the “gluzinergic” neurons of the mammalian forebrain,13,39 and the “zinc sparks” emitted by fertilised oocytes40 are most notable. Along with the respective biological studies on the organismal and cellular level, progress in this area is being facilitated on the one hand by the development of dedicated fluorescent probes,37,41–45 including genetically encoded FRET sensors,46,47 and the associated imaging studies, and on the other by the discovery and characterisation of proteins involved in zinc transport and homeostasis.
1.1 Zinc homeostasis: general premises
Cells of all organisms accumulate total concentrations of zinc in the high micromolar range.48,49 It can be estimated that at least around 9% of all human proteins require zinc for correct activity,50 and up to 15% of all human proteins (8681 out of 56![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 376 structurally modelled target gene products) have been predicted to contain at least one “real” zinc-binding site.51 The respective proteins comprise zinc-dependent enzymes, thousands of zinc-finger proteins, proteins that are regulated by zinc, and proteins involved in zinc homeostasis. Several elegant studies52,53 have led to the conclusion that dissociation constants for zinc-binding proteins should be in the range of the prevailing “free” zinc concentrations in the compartments where these proteins fold or operate.48,49,54 Estimates for free Zn2+ range from picomolar55 to femtomolar48 in E. coli, and reliable estimates for eukaryotic cells are in the single-digit nanomolar to picomolar range.49,56 Although cells have considerable zinc-buffering capacity at their disposal,48,56 substantial deviations from these steady-state concentrations may occur on various time scales, be it as a consequence of changes in zinc levels in the environment of the cell, or in the course of physiological zinc signalling events.
376 structurally modelled target gene products) have been predicted to contain at least one “real” zinc-binding site.51 The respective proteins comprise zinc-dependent enzymes, thousands of zinc-finger proteins, proteins that are regulated by zinc, and proteins involved in zinc homeostasis. Several elegant studies52,53 have led to the conclusion that dissociation constants for zinc-binding proteins should be in the range of the prevailing “free” zinc concentrations in the compartments where these proteins fold or operate.48,49,54 Estimates for free Zn2+ range from picomolar55 to femtomolar48 in E. coli, and reliable estimates for eukaryotic cells are in the single-digit nanomolar to picomolar range.49,56 Although cells have considerable zinc-buffering capacity at their disposal,48,56 substantial deviations from these steady-state concentrations may occur on various time scales, be it as a consequence of changes in zinc levels in the environment of the cell, or in the course of physiological zinc signalling events.
Cells are dynamic systems; there is hence a permanent influx and efflux of compounds including metal ions such as zinc. The total zinc content of a cell is thus primarily controlled by the action of more or less specific membrane-bound transporters (Fig. 1).
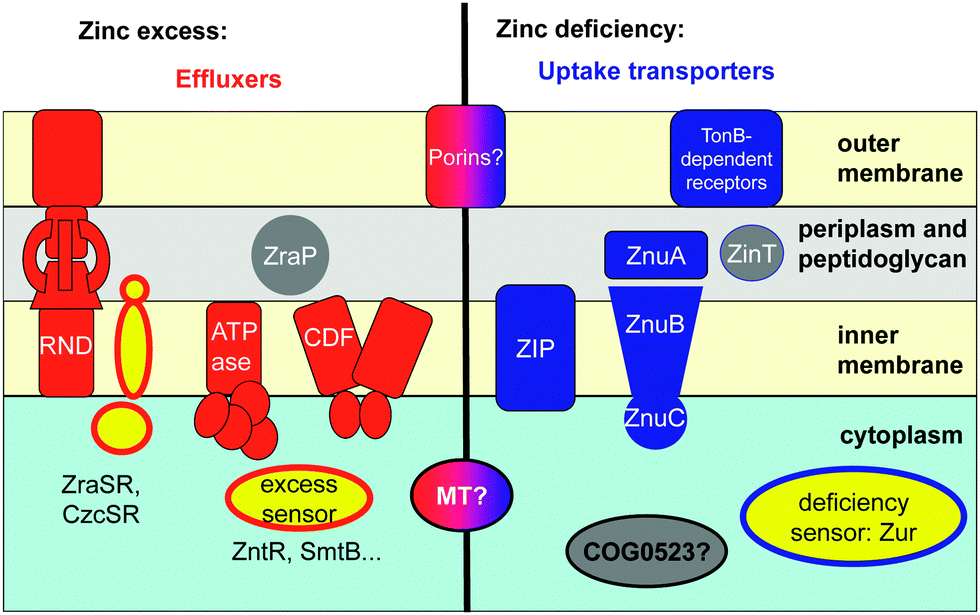 | ||
| Fig. 1 Major players in zinc homeostasis, exemplified for Gram-negative bacteria. Proteins involved in efflux are shown in red, those involved in uptake in blue. Chaperones are shown in grey, and sensor proteins in yellow. The two-component systems that regulate expression of RND systems or ZraP are also illustrated, with a membrane-spanning sensor histidine kinase and a cytosolic DNA-binding protein. ZIP and CDF transporters are also ubiquitously used for zinc uptake and efflux from the cytosol of eukaryotes. Zn-transporting ATPases are also known for plants, but not for mammals. ABC transporters, TonB-dependent receptors, and RND systems for zinc are only known for bacteria. The ExbB/ExbD proteins providing energy to the TonB receptor are not shown. Intracellular zinc-binding metallothioneins (MTs) may be of benefit in both excess and deficiency conditions, and outer-membrane porins may permit both uptake and efflux. | ||
Fluxes of zinc depend in the first instance on the abundance of the transporters in the appropriate location and on the concentration of zinc available for transport.57 The abundance of the transporters can be regulated on the transcriptional level; this is generally mediated by zinc-dependent transcription factors, also termed zinc sensor proteins, some of which are mentioned in Fig. 1 and Table S1 (ESI†). In eukaryotic systems, the activity of a transport protein may also be regulated post-transcriptionally58 and post-translationally, e.g. through metal-regulated protein trafficking.59 Zn2+ may also allosterically regulate the activity of the transporters (see Section 2.2). Furthermore, the importance of metal-modulated protein turnover is also increasingly recognised, especially for proteins involved in zinc sensing.60,61
For the regulation of free cytosolic Zn2+ concentrations, most eukaryotes and some bacteria synthesise metallothioneins, small cysteine-rich proteins with metal-binding properties ideally suited for intracellular zinc buffering (Section 2.3.1). A recent addition to the portfolio of intracellular zinc trafficking proteins is the COG0523 family (Section 2.3.2).
Besides the total cellular and free cytosolic Zn2+ concentrations, those of various cellular compartments also need to be maintained within desired levels, because they may contain Zn-dependent proteins. The granules for insulin storage and secretion by pancreatic β-cells are a prominent example, but a less well-known yet rather momentous fact is the finding that failure to maintain an adequate zinc level in the endoplasmatic reticulum triggers the unfolded protein response.62 Similarly, even zinc levels in the periplasm of Gram-negative bacteria are regulated by sensor proteins and (metallo-)chaperone proteins (see Section 2.3.3),28,63,64 perhaps to ensure that periplasmic zinc-requiring enzymes, e.g. alkaline phosphatase, can acquire their cofactor, whilst excluding Zn2+ from adventitious sites in other proteins.
While kinetic control is important in metal homeostasis, and some outcomes are achieved by the inclusion of irreversible steps,65 thermodynamics of metal binding are at the heart of metal homeostasis. Therefore, the accurate determination of affinity constants is exceptionally important. However, it has been observed previously66–68 that this endeavour is substantially less straightforward than might be expected. As a rule of thumb, dissociation constants for cytoplasmic Zn2+-binding proteins that are not at least nanomolar should be approached with scepticism, as such low affinities are not likely to be physiologically relevant under basal conditions. In contrast, micro- to nanomolar dissociation constants may be encountered in extracellular zinc-binding proteins as well as in membrane-bound transporters.
These rules pertain to both enzymes and homeostatic proteins, and pose an intriguing puzzle as to the interrelationship between thermodynamic and kinetic parameters of metal–ligand interactions, as enzymes and transporters need to achieve fundamentally different goals: firstly, a metal cofactor in an enzyme should not normally dissociate from the enzyme, whilst this is absolutely necessary in a metal-homeostatic protein, ideally on a fairly rapid time scale – in essence, fast kinetics of metal binding and release are a necessity for homeostatic proteins. An example that this is achievable with similar metal affinities has been given by comparing the enzyme carbonic anhydrase (half-life of Zn in its binding site on the order of years) with metallothioneins (see Section 2.3.1), in which half-lives are on the order of seconds.69 Few if any concrete data are available regarding on and off rates for Zn2+ binding by homeostatic proteins, but it has been argued that some dissociation reactions require the action of competing ligands to proceed at rates compatible with biological observations.61 The mobilisation of Zn2+ from a metallothionein by the glutathione/glutathione disulfide redox couple was one of the first illustrations of this principle.70 Secondly, Zn2+ in an enzyme needs to exert catalytic activity, but this is clearly to be avoided during zinc transport (as well as in structural sites). It is hence not surprising that the structures and properties of the binding sites of zinc sensors71 and zinc transporters65 are often significantly different from those in enzymes, although this is not always the case as will be seen in Section 2.
2. Structures and mechanisms of proteins involved in zinc homeostasis
The major components of cellular zinc homeostasis are summarised in Fig. 1. Broadly, zinc homeostatic proteins can be divided into sensors, membrane-bound transporters (Section 2.2), and intracellular binding proteins (Section 2.3). A thorough review on biological zinc sensing has recently appeared,72 structures and thermodynamics of several bacterial zinc sensor proteins have been discussed in detail in another excellent article,73 and a dedicated review on the eukaryotic zinc sensor MTF-1 is also available.74 Therefore, zinc sensor proteins will not be discussed in detail here, although some fundamental insights will be summarised in Section 2.1.2.1 Zinc metalloregulation: zinc-responsive sensor proteins and DNA sequences
The combined efforts of studying the in vivo function of metal-responsive proteins and detailed biophysical studies of purified proteins have led to major advances in the understanding of metalloregulation. Broadly, there are two types of zinc-responsive transcriptional regulators, those that counteract zinc deficiency, and those that counteract zinc excess/toxicity (Fig. 1). Sensors for zinc excess trigger the expression of proteins involved in efflux (e.g. ATPases, CDF proteins, RND proteins; Section 2.2; Table S1, ESI†) or sequestration (metallothioneins, ZraP, Section 2.3; Table S2, ESI†), whereas sensors for deficiency mediate enhanced expression of uptake transporters (ABC systems, porins, ZIP proteins; Section 2.2; Table S1, ESI†), putative chaperones (Section 2.3; Table S2, ESI†), and proteins that reduce cellular zinc requirements. For instance, many bacterial zinc regulons comprise genes involved in zinc uptake, but also genes for alternative ribosomal proteins devoid of zinc binding sites.75,76 This may suggest that the ribosome is a major zinc storage site in bacteria, and that this zinc can be mobilised in conditions of zinc starvation.The best-understood zinc sensors are those found in bacteria.77 Two general mechanisms are at work; sensors can either function as repressors or activators of gene transcription, in dependence on whether or not Zn2+ is bound. For example, the zinc excess sensor SmtB and related proteins are repressors in their Zn-free apo form, and de-repression occurs upon zinc binding. In contrast, the Zn-bound form of Zur, an uptake regulator responsive to zinc deficiency, is a repressor, inactivating gene transcription in the presence of sufficient Zn2+. The Zn-bound form of ZntR, an excess sensor, is an activator of gene transcription. In the most simple cases, sensor proteins bind to specific DNA sequences in the upstream region of genes that code for proteins involved in zinc homeostasis. Such recognition sequences on the DNA have different names in different phyla. In bacteria, they usually carry the names of their cognate sensor proteins. In some animals, “metal-response elements” (MREs) and zinc transcriptional response elements (ZTREs)78 are known, both of which mediate response to high zinc. 11-Base-pair ZREs (zinc-responsive elements) that mediate response to zinc deficiency are known for baker's yeast,79 and in plants, zinc-deficiency response elements (ZDREs) have been identified recently.80 In addition, putative MREs have also been predicted in plants, albeit without the associated protein(s) that recognise them.81 The sensor proteins from different phyla also differ significantly from each other. In mammals and insects, MREs with the core consensus sequence TGCRCNC (N = any nucleotide, R = A or G) are recognised by the zinc finger protein MTF-1;74 the 10-base-pair plant ZDREs are recognised by basic leucine zipper (bZIP) proteins,80 and in bacteria, a variety of sensor families recognise different inverted repeat sequences.82
Zinc sensing by transcriptional regulators almost always involves allostery:73,77 in the most simple cases, the binding of zinc elicits a conformational change, or stabilises a particular protein conformation, which increases or decreases the affinity to DNA. Many bacterial DNA-binding proteins exert their function as dimers, especially when interacting with inverted repeats or palindromic DNA sequences. In some cases, dimerisation is also influenced by Zn binding. The Zn2+ affinities of several zinc sensors have been measured by competition with metallochromic dyes83 and isothermal titration calorimetry,84 with most values around logK = 12–13 (i.e. picomolar KD).77In vivo reporter assays in E. coli yielded femtomolar values for activation of the sensors Zur and ZntR.48 Some of the related protein–DNA interactions have also been quantified, allowing the quantitative description of the various coupled equilibria (metal binding–DNA binding).73
In structural terms, one unifying feature of the zinc binding sites in sensor proteins is their high solvent exposure. This presumably facilitates fast binding kinetics, although these have not been measured for any zinc sensor. Regarding specificity, scrutiny of the structures for different metal sensor proteins reveals that metal sites are “optimised” for the cognate metal ion,67 both in terms of coordination geometries and of the HSAB principle (Pearson's principle of hard and soft acids and bases; see ref. 85 for an overview). Cu+ sensors harbour 2- and 3-coordinate sites, often comprising cysteines, Zn2+ sensors have 4-coordinate sites comprising mixtures of Cys, His and Asp, and Mn2+, Fe2+, Co2+, and Ni2+ sensors have 6-coordinate sites, with the Mn2+ sensors displaying an abundance of carboxylate residues. However, this will not, per se, prevent e.g. Cu+ binding to a Zn2+ sensor and vice versa. The first mechanism to prevent wrong sensing relates to the idea that intracellular metal concentrations and dissociation constants of the relevant metal–protein combinations are correlated.54 This may however not be sufficient – especially under conditions where the more competitive metal ion is in excess. In this case, allosteric changes can be metal-specific: other metals may bind to a particular sensor, but without eliciting the conformational change required to alter DNA-binding affinity. The coordination chemistry principle that is exploited here is preferred coordination geometry: for example, Zn2+ does bind to the Ni/Co excess sensor NmtR from Mycobacterium tuberculosis, but adopts tetrahedral rather than the required octahedral coordination geometry, and hence does not trigger the sensing mechanism.86
2.2 Proteins for uptake and efflux
Several proteins that mediate the transmembrane transport of zinc, along with transporters for other metal ions, have been discovered and characterised in the past decade.65 All transmembrane transport proteins are catalogued in the Transporter Classification Database (TCDB).87 This classification system works similarly to the EC system for enzyme classification, giving each protein a TCDB number. Channels and pores are class 1, electrochemical potential-driven transporters are class 2, and primary active transporters are class 3. The most important membrane transport protein classes for zinc and their TCDB numbers are compiled in Table S1 (ESI†); it should however be acknowledged that in some cases, information on transport mechanisms is still too limited for a conclusive classification.Because of its inherently stronger tendency for complex formation, and hence reduced mobility,88 transport mechanisms for Zn2+ (and those for other 3d row metal ions‡) differ significantly from those of the more mobile alkali and earth alkali metal ions.65 The higher affinities are also the likely reason for the fact that transmembrane transport of 3d row metal ions is considerably slower than that of the group Ia and IIa metal ions, with typically less than 10 ions per second.65 In principle, zinc transport can be active or passive, against or with Zn2+ gradients, although most systems that have been characterised in some detail tend to be active transporters. Fig. 1 shows cartoons for the most important types of efflux and uptake transporters. ATP-driven class 3 active transporters will be discussed first (Sections 2.2.1 and 2.2.2), followed by class 2 porters (Sections 2.2.3–2.2.5), with representatives from class 1 and others highlighted in Section 2.2.6.
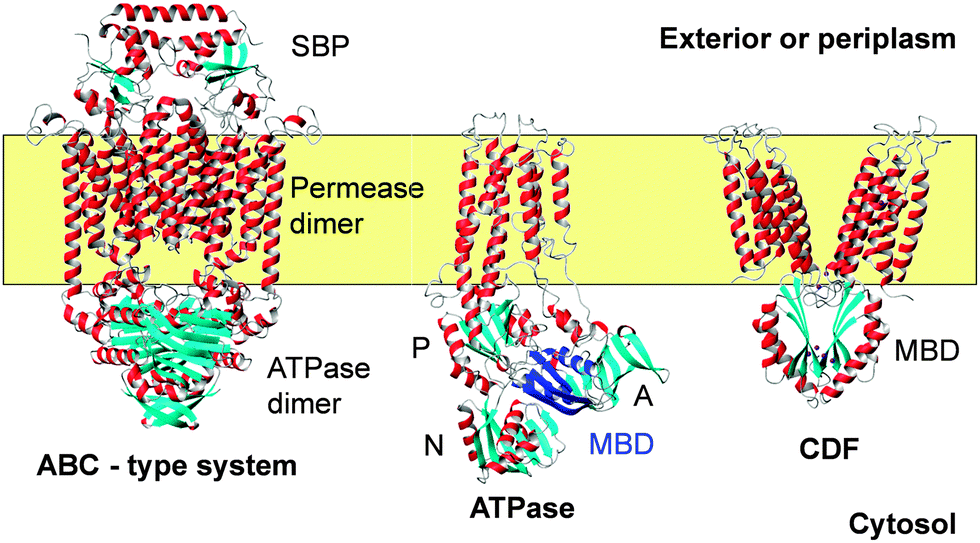 | ||
| Fig. 2 Backbone traces for examples for three membrane (yellow) transporters for metal ions including Zn2+. Helices are shown in red and grey, and β-strands are shown in cyan. The ABC transporter shown is BtuCD-F (pdb 4fi3)91 for cobalamin uptake. A detailed structure for a ZnuA substrate binding protein (SBP) is shown in Fig. 3. The ATPase is CopA from A. fulgidus. The structure is based on cryo-electron microscopy (pdb 3j09),108 and was chosen as it illustrates the position of the cytosolic MBD (highlighted in dark blue). Actuator (A), phosphorylation (P), and nucleotide-binding (N) domains are also shown. The metal-binding loop of the MBD is in close proximity to the actuator domain. The CDF protein is YiiP (FieF) from E. coli (pdb 3h90).128 The functional dimer is shown. The cytosolic domains have the same ferredoxin-like fold as MBDs from ATPases. A comparison of MBD folds and metal sites is provided in Fig. 5. | ||
The SBP forms a complex with the permease as shown in Fig. 2, which allows the delivery of the substrate into a channel formed by the permease.90 How ATP hydrolysis and Zn2+ transport are coupled for ZnuABC systems has not yet been determined, but it is likely that this will involve structural rearrangements at the permease dimer interface, with this interface providing a cavity with alternating access to outside and inside.92
SBPs all adopt a venus-fly trap structure, with the substrate-binding site between the two domains. Substrate binding may elicit a conformational change in the SBP, but this is not always the case. Furthermore, the sequences of many ZnuA proteins contain long His-rich stretches, which is perhaps the best hallmark to distinguish a “true” ZnuA from the related Mn-binding proteins, which are otherwise closely similar, including the location and identity of metal-binding His residues.24 The structure and role of these loops is a challenging puzzle in understanding the mode of action of ZnuA proteins. Several structures for ZnuA from E. coli are available (Fig. 3),93–95 with some striking variations in metal stoichiometry and coordination modes between these structures. The most recent study established that not just one, but two Zn2+ ions can be bound with significant affinity. Although the purified form contained only 1 mol. equiv. of Zn, it was possible to partially populate a second site by adding excess Zn2+, followed by gel filtration chromatography which in general removes any weakly bound metal ions. The presence of a second site with a dissociation constant below 20 nM was also corroborated by titration studies with Mag-Fura-2 as competing chelator.95 Only one of the protein-derived Zn ligands for this second site is visible in the structure, but some weak electron density suggests the presence of other ligands, most likely from the His-rich loop (residues 117–137), which is not resolved in any of the published structures. The latter indicates that these stretches are dynamic and/or disordered.
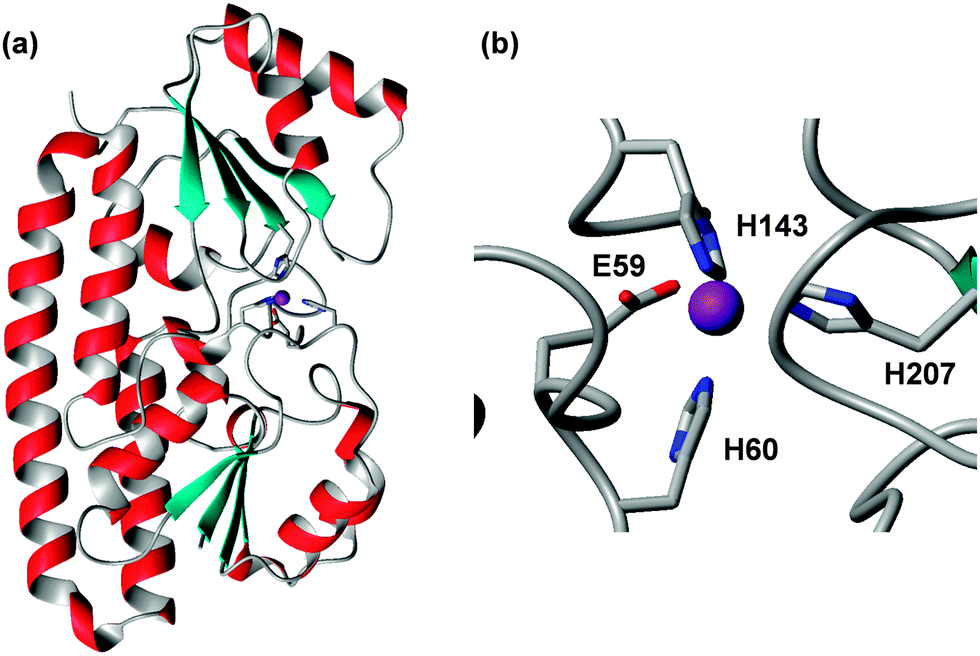 | ||
| Fig. 3 Overall structure (a) and zinc-binding site (b) in E. coli ZnuA (pdb 2osv).94 The Zn2+ ion is shown as a purple sphere, and is coordinated to three His and one Glu residue, the latter in a monodentate fashion. Variations to this motif are known (see text). The residues are numbered according to the UniProt sequence; the 2osv pdb file deviates from this by 18 residues. | ||
The primary zinc binding site in E. coli ZnuA is composed of three His residues and one Glu residue in two of the structures (2osv94 and 2ps095), or the same three His residues plus a water molecule, which is hydrogen-bonded to the Glu residue, in another (pdb 2ogw93) – in essence, the structures differ by inner- and outer-sphere coordination of the Glu residue. A further interesting variation is seen in the structure of ZnuA from Salmonella enterica (pdb 2xqv96); here, His60 is not coordinated to the zinc ion; instead, another His from the His-rich loop has taken its place. The latter structure was obtained by soaking apo-crystals with Zn2+, therefore it is not clear whether this mode would also be adopted in solution. ZnuA from the cyanobacterium Synechocystis adopts the same fold as the enterobacterial ZnuA's.97 However, Glu59 is replaced by a Pro residue, and the fourth coordination site is occupied by a water molecule, which is not hydrogen-bonded to another residue. A deletion mutant lacking the His-rich loop (residues 138–173) has been compared to the wild-type.98 Isothermal titration calorimetry (ITC) experiments established that the wild-type harboured two classes of zinc binding sites with affinities that differed by 2 orders of magnitude (KD(ITC) = 10 and 1000 nM; logKITC = 8 and 6). About up to 3 zinc ions could be bound with the weaker affinity, and the His-loop deletion mutant only retained the stronger binding site. Based on the fact that zinc loading of the high-affinity site was not affected by the loop deletion, a role of the loop in “chaperoning” zinc to the high-affinity site has been dismissed; instead, it was proposed that the loop may have a sensing/regulatory function that may slow down zinc uptake through the ABC system when periplasmic concentrations become 100 times higher than normal.98
Most of the work on the substrate-binding components of Zn-transporting ABC systems has been carried out for proteins from Gram-negative bacteria, but there are also candidate systems in Gram-positive bacteria, most prominently AdcA and AdcAII99 from Streptococci. Furthermore, a related periplasmic protein is present in the spirochaete Treponema pallidum and is the founding member of the TroA proteins.100 However, the metal specificity of proteins designated TroA is unclear, and in vitro TroAs are capable of binding a range of metal ions with significant affinity – as expected. It is very likely that some TroAs are, in vivo, Mn or Fe binding proteins, whilst others are truly orthologous to ZnuAs, but prediction of metal specificity is non-trivial.24 In the case of AdcA and AdcAII from Streptococcus pneumoniae, zinc specificity has been established in vivo.101
Recent work has demonstrated that the substrate-binding components may receive Zn2+ from other proteins. In some Gram-negative bacteria, the periplasmic zinc-binding protein ZinT (see Section 2.3.3) may deliver Zn2+ to ZnuA.102 In Streptococci, poly-His-triad (PhtD; see Section 2.2.6) proteins are located at the cell surface and have been proposed to deliver zinc to AdcAII.103 Significantly, although AdcA and AdcAII are functionally equivalent, they differ structurally: whilst AdcAII overall resembles typical ZnuAs, AdcA is a fusion protein of a ZnuA-like portion and a ZinT-like portion, thus it appears that the ZinT-like protein can either be independent or fused to its partner protein.
P1B-ATPases transport d-block metal ions (Table S1, ESI†), and are found in all kingdoms of life. No Zn-transporting ATPases are known for mammals, but several examples from plants and bacteria have been studied. Most Zn-transporting P1B-ATPases are involved in Zn2+ efflux from the cytosol, which in plants may also include transport into organelles.
Briefly, for P-type ATPases involved in metal efflux, the general Post–Albers reaction cycle involves four principal states (Fig. 4). In E1 and E1P states, an intra-membrane transport site is accessible from the cytosol, whereas in E2P and E2 states, this site is exposed to the exterior. The E1P state is reached by binding of Mg-ATP to the N domain and hydrolysis to ADP, with the remaining phosphate group transferred to an Asp residue in the P domain. It is thought that a concomitant conformational change prevents back-flow of the transported ion(s). A further, large conformational change leads to the E2P state, from which the metal substrate is released to the exterior. Hydrolysis of the covalently bound phosphate group gives the intermediate E2-Pi state; release of Pi yields the E2 state. A further conformational change closes the exit towards the exterior, and renders the inter-membrane metal binding site(s) accessible to metal substrate from the interior again (E1 state).90 The various conformational changes alter both the mutual orientation of the cytosolic domains, and that of the transmembrane helices, and are triggered by binding and releasing the species involved.
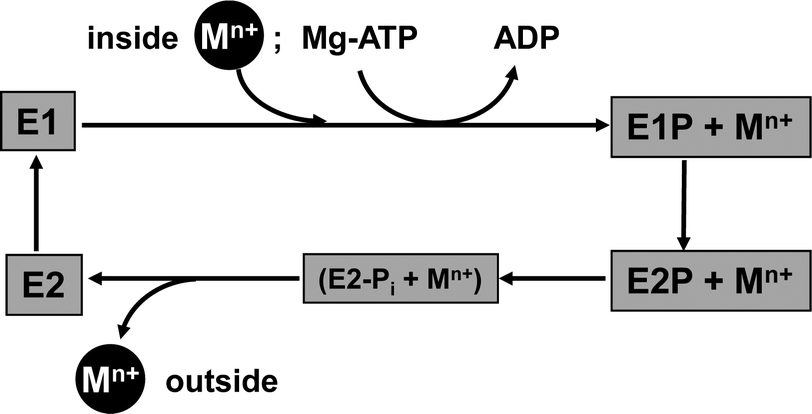 | ||
| Fig. 4 Major states in the catalytic cycle of metal-effluxing P-type ATPases. The “P” signifies the occurrence of a phosphorylated enzyme intermediate (E1P and E2P states). The E2-Pi state refers to a state in which the hydrolysed Pi group is still bound to the protein; this is usually modelled by AlF4−, and also informs on the E2 state. E2P states are commonly mimicked by BeF3− adducts. | ||
X-ray crystal structures of ZntA from Shigella sonnei in its Zn-free E2P and E2-Pi states have been determined recently (Fig. 5).105 Structures for the closely related Cu+-transporting ATPase, CopA from Legionella pneumophila,106,107 and a model based on cryo-electron microscopy of CopA from Archaeoglobus fulgidus (Fig. 2) are also available.108 Transmembrane helix 6 of many P1B-ATPases harbours a CPC motif where both Cys residues may provide a metal binding site. In bacterial Zn-ATPases, this site comprises at least one further conserved Asp residue from the C-terminal helix 8, as shown by metal binding studies in mutated ZntAs.109 The crystal structures of S. sonnei ZntA indeed show this residue (D714) close to the CPC motif (Fig. 5a). In the E2-Pi state only, which corresponds to a state after metal release, the carboxylate of D714 is hydrogen-bonded to the conserved K693; it is proposed that this switch may block/inhibit zinc binding to the intra-membrane site. Crucially, both Asp714 and Lys693 are absolutely essential for ATPase activity.110
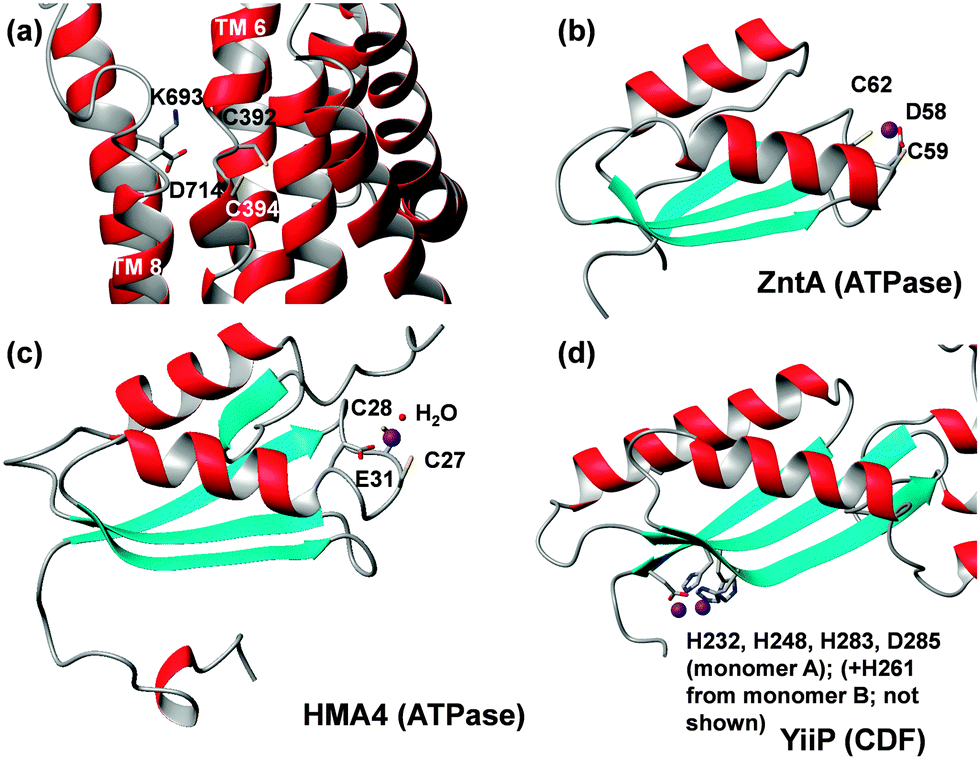 | ||
| Fig. 5 Metal binding residues and sites in ATPases and ferredoxin-like MBDs. (a) Transmembrane site of E. coli ZntA in the E2P state (pdb 4umv).105 Besides the two Cys residues, Asp714 is also suggested to be involved in metal binding. Alternatively, it can form a salt bridge with Lys693. ATPase MBDs contain a flexible metal-binding loop with DCxxC ((b), bacteria) or CCxxE ((c), plants) motifs between strand 1 and helix 1. In contrast, the metal-binding sites in the MBD of CDFs (d) are located at the opposite end of the domain, and coordinating residues are located in relatively rigid β-strands. Also see Fig. 6. | ||
Many of these ATPases also contain an N-terminal, cytosolic metal binding domain (MBD, sometimes also referred to as HMA, for heavy-metal associated domain). In none of the X-ray structures of the full-length proteins were these domains visible, likely due to disorder. The cryo-EM model of CopA has enabled location of this domain in the Cu-free E2 and E2P forms, where it interacts with N, P and A domains (Fig. 2). Contrastingly, the MBDs in both CopA and ZntA have also been docked in a different location,105,106 close to the membrane surface where an interaction with an amphipathic helix may take place. This helix is part of a proposed “platform” for metal entry into a funnel leading to the intra-membrane site.
Isolated MBDs of E. coli ZntA,111Synechocystis ZiaA,112 and A. thaliana HMA452 have been studied by solution NMR spectroscopy, but Zn-bound structures are only available for ZntA (pdb 1mwz111) and HMA4 (pdb 2kkh52) (Fig. 5b and c). All MBDs adopt a ferredoxin-like fold, and both ZntA and ZiaA MBDs bind Zn through a DCxxC motif, whilst the plant MBDs have a CCxxE motif. Thus, whilst the protein folds are very similar, the metal binding motifs are not, yet the overall composition (Cys, Cys, carboxylate) of the binding sites is still the same. It is thought that a fourth ligand is provided by water, and that the carboxylate binds in a monodentate fashion. Furthermore, the presence of three negatively charged residues may be sufficient to suppress the Lewis acidity of bound Zn2+.65
The MBD is not required for transport or ATPase activity, and mutating the two Cys in the DCxxC motif of ZntA to Ala did not negatively affect transport either, suggesting that the MBD did not block access to the intra-membrane site, and was also not required for Zn2+ delivery. However, mutation of the two Cys to Ser in E. coli ZntA reduced ATPase activity by ca. 50%,110 suggesting a regulatory function, with the apo-MBD inhibiting the enzyme in the absence of the substrate metal ion.
E. coli ZntA lacking the MBD has been studied by EXAFS, providing structural information on the transmembrane (TM) site.113 The best fit was obtained with 2 S ligands at 2.30 Å and 2 N/O ligands at 2.0 Å, hence this site is likely to be four-coordinate. Affinities have also been determined for both MBD and TM site, and were found to be similar,109 consistent with the idea that both sites are formed by two Cys and one carboxylate ligand.
Both HMA452 and ZiaA112 MBDs bind Cu+ more tightly than Zn2+. In either organism, there are additional ATPases with higher affinity towards Cu+,52,112 and these systems have been used to illustrate that “specificity” can be achieved through “relative affinities”:114 as long as there is sufficient Cu-binding capacity provided by dedicated proteins, other proteins meant to bind or deal with less competitive metal ions are left free to do so. Furthermore, the cytosolic copper chaperone ScAtx1 cannot interact with ZiaA, but direct protein–protein interactions, as well as Cu+ exchange, occur with two copper-transporting ATPases, PacS and CtaA.112 Similar dedicated interactions between ATPase and copper chaperone are common for Cu+ pathways, but unknown for zinc. It is thus possible that at least some MBDs provide an additional selectivity filter – both through their tailored metal sites, as well as through their protein surfaces.
Finally, Zn-transporting ATPases from plants (e.g. HMA2 and HMA4 from A. thaliana) also harbour Cys- and His-rich C-terminal stretches that are located in the cytosol.115 The Cys residues are arranged in characteristic patterns including CC and CCx3C motifs, and some of the His residues occur consecutively with no intervening other residues – resembling engineered His-tags. Like the His-rich stretches in ZnuAs, these sections are likely to be structurally disordered, and their role is unclear, although it is known that Zn binding elicits a conformational change that may influence interactions with the other cytosolic domains and affect their activity.116 Indeed, mutant A. thaliana HMA4 lacking its Cys/His-rich C-terminal tail was more efficient at pumping Zn2+ and Cd2+, consistent with an inhibitory function of this portion when not fully occupied.117 The same study also reported that up to 10 Zn2+ ions could be bound to this section.
000 patients has shown that loss-of-function mutation of ZnT8 in fact reduced the risk for diabetes.123 Non-functional ZnT4 is responsible for the “lethal milk” syndrome,124 due to a failure to load secretory vesicles in mammary epithelial glands with zinc, whilst mutant ZnT2 can lead to transient neonatal zinc deficiency.125 ZnT3 in the brain loads Zn2+ into pre-synaptic vesicles of zincergic neurons, and its deficiency is associated with cognitive dysfunction.126
The structure of a zinc- and iron-transporting CDF protein, YiiP (FieF) from E. coli, has been determined (Fig. 2, 3 and 6).127 YiiP is a homo-dimer with 6 transmembrane helices per monomer. Each monomer also comprises a cytosolic metal-binding domain (MBD) with a ferredoxin-like fold, similar to that found in many P1B-ATPases – but with completely different metal-binding sites (Fig. 5).115 In the dimer, the two cytosolic MBD are linked by two bridging zinc ions (Fig. 6), the binding residues of which are conserved in other Zn/Fe-CDFs.115 It has been proposed that the two zinc ions are necessary to keep the two cytosolic domains together, which otherwise would be driven apart by charge repulsion.128 The isolated cytosolic domain of CzrB from Thermus thermophilus also formed dimers, with similar Zn-binding sites.129 Size-exclusion HPLC analysis indicated that for the full-length protein, zinc did not change the oligomerisation state, but FRET analysis demonstrated that a conformational change, probably involving “en-bloc” movement of entire domains, occurred upon zinc binding. This is also consistent with a recent study using cryo-electron microscopy.130 The 13 Å structure of Zn-free YiiP revealed a conformation different to that found in the Zn-bound structure, consistent with pivoting and/or scissoring of the transmembrane domains. An outward-facing conformation in the presence of Zn, and an inward-facing conformation in its absence was proposed. It is possible that these domain movements are at least partially mediated by the cytosolic MBDs. An ITC study suggested that a CDF protein from the bacterium Maricaulis maris lacking the MBD was still capable of binding Zn2+ and Cd2+ with micromolar affinity.131 Truncated versions of Cupriavidus metallidurans§ CzcD and E. coli ZitB lacking the MBD also were still capable of metal transport in vivo, but less so – hence the C-terminal MBDs were required for full functionality.132 All observations are consistent with the notion that the cytosolic domains may act as regulatory sensors for excess cytosolic zinc, and allosterically promote Zn2+ efflux by the transport domains.
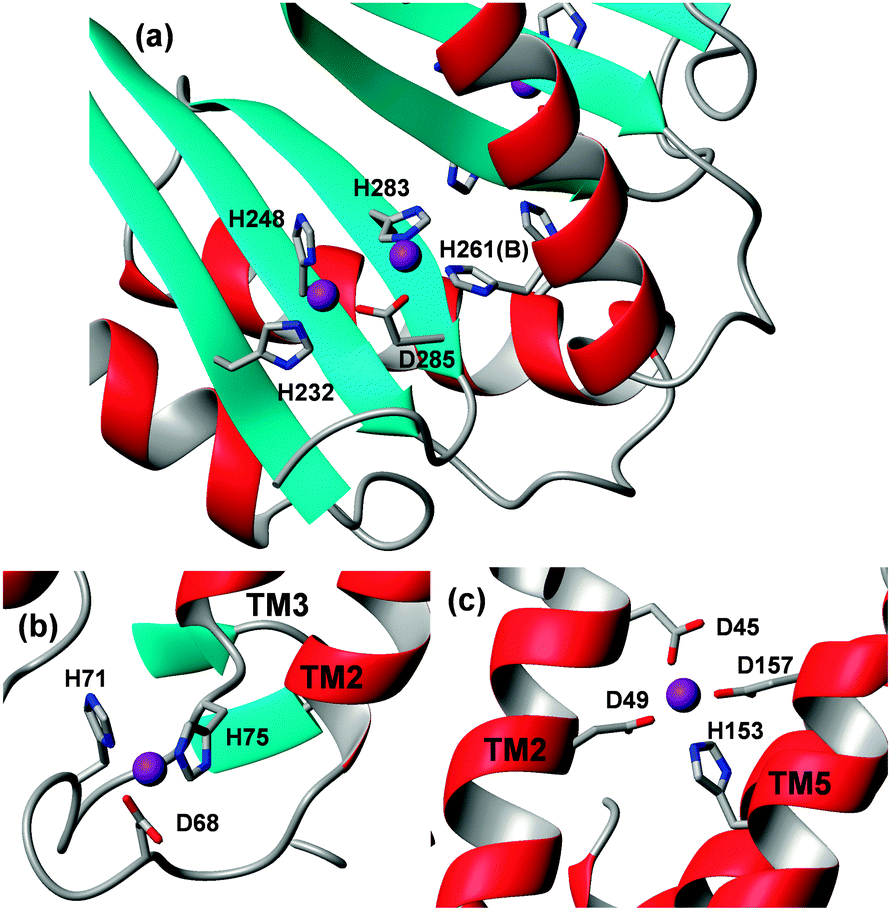 | ||
| Fig. 6 Details of Zn-binding sites in E. coli YiiP. (a) The binuclear, inter-protomer binding site in the cytosolic MBD. Four metal-binding residues are provided by all three β-strands, giving a fairly rigid scaffold for Zn2+ coordination. The fifth ligand (H261) comes from helix 2 of the second monomer. The tetrahedral coordination for both Zn2+ ions is completed by two water molecules (not shown). (b) A further cytosolic site is located in a loop between TM2 and TM3; the fourth ligand is a water molecule (not shown). (c) The four-coordinate transmembrane transport site is formed by two residues from helix TM2 and two from TM5. | ||
In the YiiP structure, two further zinc ions were bound per monomer; one in the cytosolic loop connecting TM helices 2 and 3, formed of two His, one Asp, and one water ligand, and one intra-membrane site. The latter is the actual transport site; helix 2 provides two Asp ligands, and helix 5 an Asp and a His residue (Fig. 6). The second cytosolic site may modulate the packing of helices, and hence influence the transport site.
The binding of Zn2+, Cd2+ and Hg2+ to YiiP were studied by ITC.133 At least two sets of binding sites for Zn2+ were evident, with KD(ITC) = 3 μM and 159 μM, and there were also indications for a further binding site with higher affinity that was not adequately captured by the ITC experiments. It is possible that this high-affinity site corresponds to at least one of the inter-protomer sites located in the MBD, and that one of the weaker sites corresponds to the transport site. An ITC study of a CDF protein from the hyperthermophile Aquifex aeolicus indicated that both Zn2+ and Cd2+ binding was endothermic,134 suggesting that structural changes were required to accommodate the ions. Again, this would fit with the metal binding sites at the protomer interface, and be consistent with a regulatory role.
Contrary to their exclusive use for zinc in mammals, members of the CDF family in other phyla (plants, invertebrates, fungi, bacteria) also transport Fe2+ and Mn2+. Understanding the selectivity of CDF proteins is an active research area. Sequence analysis has suggested that these can be organised into three groups,135 Zn-CDF, Fe/Zn-CDF, and Mn-CDF according to the principal transported metal ion(s). Hallmarks for metal specificity identified in the latter study included likely metal-coordinating residues in TM helices 2 and 5, as well as presence or absence of a His-rich cytosolic segment – similar to those stretches found in ZnuA's and Zn-transporting P-type ATPases (see Sections 2.2.1 and 2.2.2). E. coli YiiP belongs to the Fe/Zn-CDF group, and does not contain such a segment. A recent study on human ZnT5 and ZnT8 demonstrated that neither are capable of transporting Cd2+, but that a single His-to-Asp mutation in TM2 resulted in proteins with similar Zn2+ transport activity as the wild-type, but that could also promote the efflux of Cd2+.136 Metal specificity in CDF proteins from plants (termed metal tolerance proteins, MTPs) has also been investigated, mainly by complementation assays using wild-type and mutant proteins in Zn-sensitive yeast. One study identified several non-coordinating residues in the His-rich loop as well as in TM helix 3 of A. thaliana MTP1 as determinants for Zn selectivity over Co2+.137 Another study on A. thaliana MTP1 identified N-terminal Cys residues as essential for conferring Zn tolerance to a sensitive yeast strain.138 Mutation of several non-coordinating residues in TM helices 2 and 5 enhanced Zn transport, as did deletion of 12 N-terminal residues. Deletion of 28 residues from the N-terminus made no difference, but deletion of 55 N-terminal residues comprising two conserved Cys residues led to a non-functional protein. A range of other mutations, including deletion of the His-rich loop, led to loss of Zn selectivity. We have shown that the C-terminal cytosolic metal-binding domain (MBD) for plant MTPs is sufficient for achieving a similar phylogenetic clustering as that obtained when using the full length proteins, and sequence comparisons focusing on the likely MBD metal-binding residues suggested that at least part of the selectivity may be mediated by the MBDs.115 Great caution always has to be applied when inferring substrate selectivity from protein sequence data, and our proposition has not been experimentally tested yet.
Their wide distribution and importance in human diseases notwithstanding,120,140,143 very little biophysical data and no X-ray structures for any ZIP protein are available. Despite the tremendous progress in the study of other membrane proteins, including those for zinc transport, in recent years, ZIP proteins have proven to be extremely difficult to express and purify in a functional form. Consequently, the mechanisms of transport and determinants of metal specificity are not yet well understood, although a recent breakthrough, described in more detail below, has begun to remedy this situation.144 Most ZIP proteins are predicted to have 8 transmembrane helices, and many, irrespective from which phylum, comprise histidine-rich cytosolic loops. Extracellular loops tend to be short, with the exception of some animal ZIPs (e.g. human ZIP5, 6 and 10) which have large (ca. 200–300 amino acid residues) ectodomains which are evolutionarily related to prion proteins.145 Due to the difficulties with studying full length ZIP proteins, some in vitro metal binding studies have been carried out on fragments. The peptide PHGHGHGHGP from the large intracellular histidine-rich loop of IRT1 from A. thaliana has been studied by ITC.146 IRT1 is regulated by a requirement for iron, and can transport Fe2+, but also Mn2+, Zn2+, Co2+ and Cd2+.147 Thermodynamic parameters measured for the 2+ ions from Mn through to Zn, plus Cd2+ and Fe3+, followed the same trends as small-molecule chelators such as trien, but the loss of entropy was considerably larger. The pH- and buffer-independent stability constant for Zn2+ (logK = 6.21–6.69) was surprisingly low. A. thaliana IRT1 also contains an extracellular metal-binding loop. The Ac-(95)MHVLPDSFEMLSSICLEENPWHK(117)-NH2 peptide derived from this loop, and an N-terminal fragment of human ZIP13 have been studied by NMR, mass spectrometry (MS) and potentiometry.148 The latter technique yielded stability constants of logK = 14.75 for the IRT1 loop, which contains two His, one Cys, and several carboxylate groups, and 17.85 for the ZIP13 fragment that contained four Cys residues. These numbers are stoichiometric, not conditional constants, i.e. the conditional constants valid at neutral pH will be lower. MS analysis demonstrated that only 1:1 complexes were observed, and 1H and 13C NMR spectroscopy showed that both metal-free and metal-bound peptides were disordered. The selective broadening of a number of resonances supported the coordination of Zn2+ by His, Cys and Glu in the IRT1 peptide. It is not yet known whether these protein regions contribute to selectivity; this will require studies of intact mutant proteins in a membrane environment.
Biochemical transport assays have shown that transport of Zn2+ by human ZIP1 and ZIP2 is stimulated by HCO3−,140 as is that of human ZIP8.149 Most recently, the bacterial ZIPB from Bordetella bronchiseptica has been purified and reconstituted in proteoliposomes, providing the so far most detailed biophysical study of a ZIP protein.144 Purified ZIPB was dimeric. Zinc fluxes were monitored by stopped-flow fluorometry utilising various metallochromic dyes, which demonstrated that zinc transport by this protein obeyed a first order rate law. This suggests passive electrodiffusion. Zn2+ was the only divalent cation that elicited an electrogenic effect. Electric membrane potentials generated by K+ were able to drive Zn2+ transport in both directions, i.e. in a voltage-dependent manner. In addition, transport was shown to be pH dependent, but not driven by the proton-motive force.144 No enhancement of transport was observed in the presence of HCO3−, and other anions inhibited transport. Taken together, these results demonstrated that ZIPB functions as a Zn2+ (and Cd2+) selective channel, and suggested that transport is driven by in vivo Zn2+ gradients. Since the free, hence electroactive, concentrations of Zn2+ inside the cytosol are extremely low (pico- to femtomolar), gradient-driven electrodiffusion through a selective channel can provide an efficient uptake mechanism – providing that the free Zn2+ concentration in the extracellular (or organellar) medium is higher than the cytosolic concentration. This may often be the case in multicellular eukaryotes, but depending on their environment, is perhaps less common for unicellular organisms. It is also noteworthy that even though ZIPB works as a channel, Zn2+ transport was very slow – even slower than that measured for the secondary active transporter YiiP (Section 2.2.3). Furthermore, if more ZIP proteins turn out to be channels rather than porters, a re-classification will be required.
Human ZIP4, the protein linked to the skin disease Acrodermatitis enteropathica, also has an N-terminal extracellular domain which is rich in His and Cys residues.150 hZIP4 was heterologously expressed in Xenopus oocytes, and zinc transport was monitored using radioactive 65Zn2+. Analysis of the kinetics of uptake yielded two KM values of 76 nM and 1.4 μM. The only metal ions that could compete with Zn2+ were Cu2+ and Ni2+, but in contrast to other ZIP proteins, not Cd2+ when present at micromolar concentrations. E. coli ZupT was shown to also promote the uptake of Fe2+, Co2+, and Mn2+.151 Plant ZIP proteins have also been reported to be relatively non-selective, promoting the uptake of Zn2+, Cd2+, and Fe2+.152 It is becoming clear that the metal selectivities of ZIP proteins may vary quite considerably, and it will be interesting to see how and where this selectivity is determined.
RND systems are composed of three proteins: in the case of metal-transporting RND pumps, the “A” component is trimeric,|| spans the inner membrane and reaches into the periplasm; the “C” component is also trimeric, spans the outer membrane and also reaches into the periplasm; and six periplasmic “B” adaptor proteins are necessary to link these two trimers (Fig. 1 and 7).158,159
The inner membrane CusA protein can use the proton-motive force for metal translocation, even in the absence of the “B” and “C” components,160 although there is debate as to whether RND-mediated transport from the cytosol (“transenvelope”) is of major importance, or whether the main compartment from which metals are transported is the periplasm.158 Several findings support the latter proposition, not least the fact that the expression of RND systems is often regulated by a two-component sensor system.157 The inner-membrane-spanning sensor histidine kinases of these systems sense the composition of the periplasm, not the cytosol (as illustrated in Fig. 1).
Several structures of RND systems are available, including ZneA156 and ZneB155 from Cupriavidus metallidurans. The structures of CusC161 and the CusBA complex162 from E. coli also have been determined recently, permitting insights on the entire tripartite complex. Trimeric CusC forms an α/β-barrel (Fig. 7). The outer-membrane spanning portion consists of a porin-like 12-stranded β-barrel, whilst the 100 Å long periplasmic tube is formed from 12 α-helices. The “A” component (CusA or ZneA) comprises three domains. 3 × 12 α-helices span the inner membrane; the periplasmic pore or porter domain consist of four sub-domains, and the “outer-membrane factor docking” domain has a similar diameter to that of the “C” component, likely enabling direct interactions between the two. This domain is also thought to be the exit funnel for substrates. The pore domain is accessible from the periplasm. For the related RND protein AcrB, three conformational states have been observed in crystal structures: “open access”, “bound”, and “extrusion”, with each “A” type protomer adopting one of these conformations.163 It is thought that this enables “functional rotation” and is related to energy coupling of zinc transport. In the ZneA structure determined at low pH (5.2), two of the three protomers were partially occupied by Zn at a site comprising E136, D602, E610, D645, and D658, termed the proximal site, located in the centre of the pore domains. In the structure at higher pH (7.5), all three proximal sites were occupied, and one second, distal site (D172, E599 from one protomer and E72 from another), located near the exit funnel, was also detectable (Fig. 7). Both sites were thought to mediate transfer to ZneC.156 No channel for zinc transport was detected in the ZneA inner-membrane domain, contrary to a methionine-lined channel in CusA.160 Therefore, the ZneA structure is compatible with export from the periplasm only.
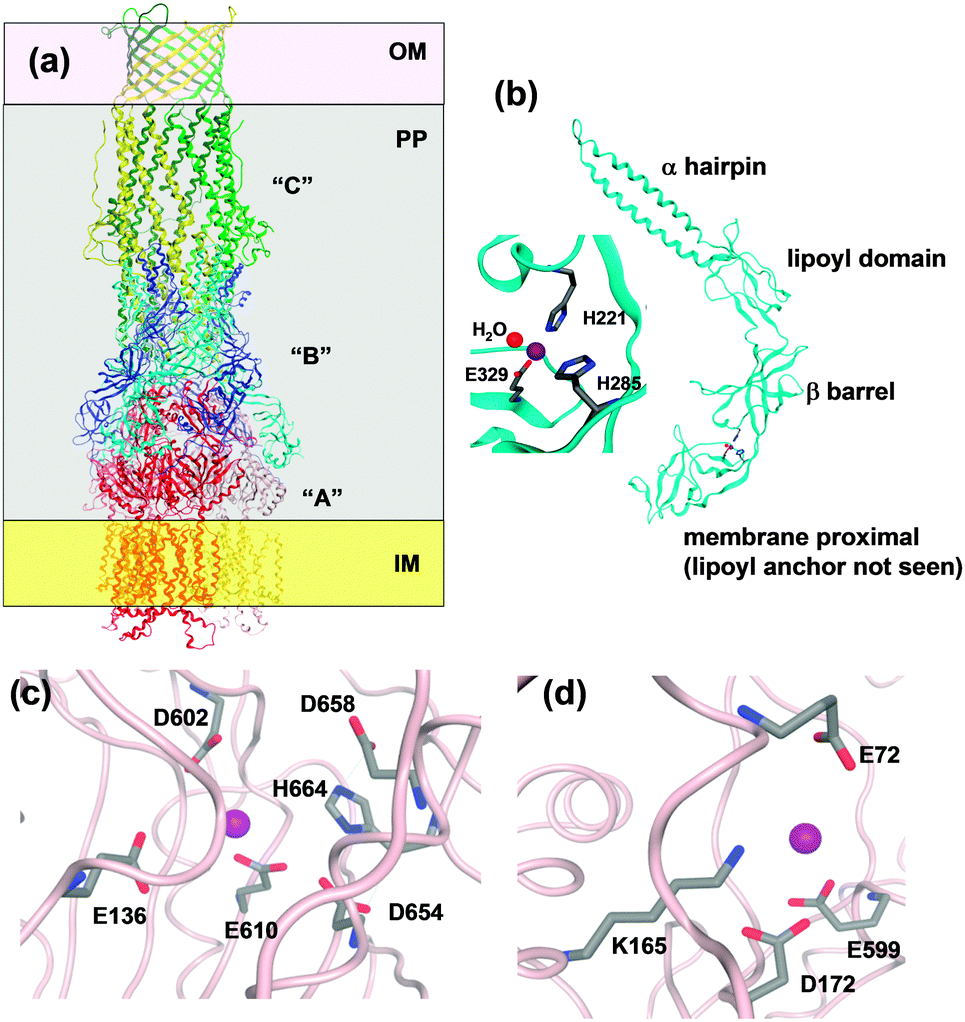 | ||
| Fig. 7 Overall structure of an RND efflux system and resolved zinc binding sites in ZneA and ZneB. (a) Sketch using CusBA and C (pdb 3ne5162 and 3pik161). The three CusC monomers are drawn in yellow, light and dark green, the six CusB monomers in blue and cyan, and the three CusA monomers in red, dark pink, and light pink. OM = outer membrane, IM = inner membrane, and PP = periplasm. (b) Overall structure and zinc binding site in ZneB (pdb 3lnn).155 (c) Residues within a 4.5 Å radius of the “proximal” Zn site in one of the ZneA protomers (pdb 4k0j). In addition, E610 and D658 which have also been implicated in Zn binding156 are shown. (d) Residues within a 4.5 Å radius of the “distal” Zn site in one of the ZneA protomers. | ||
Finally, the periplasmic adaptor protein “B” (e.g. CusB and ZneB) consists of four domains, namely an α-hairpin (or 3-helix bundle in the case of CusB) domain which interacts with the helices of the periplasmic portion of the “C” protein, a lipoyl domain consisting of two 4-stranded β-sheets, a small 6-stranded β-barrel, and the membrane-proximal domain which is anchored to the inner membrane through an N-terminal lipoyl tail (Fig. 7). The ZneB structure contains Zn2+ bound to a His2Glu site between the membrane-proximal and β-barrel domains (Fig. 7), and Zn2+ binding induces conformational changes, observed in both crystal and solution states.155 The location of the zinc site enables a hinge-like movement, and thus the adaptor protein could alter the distance between the “A” and the “C” periplasmic funnels. For the CusBA complex, a direct transfer of the metal ion from the “B” to the “A” component has been dismissed, as in the assembled complex, the metal sites in the two proteins are too far apart.162 Instead, it can be envisaged that Zn2+ binding to sites in ZneB and ZneA stabilise conformations that allow the collection of Zn2+ in the ZneA pore, followed by extrusion of excess Zn2+ through ZneC from the periplasm. This suggests that both ZneA and ZneB have an active role in Zn2+ flux control, with ZneA providing energy through proton antiport coupled to conformational changes, and ZneB also operating as part of a “switch” mechanism.158 The apparent dissociation constant of Zn2+–ZneB has been reported as 3 μM; it is possible that this corresponds to a set point at which the transport efficiency of the RND system changes.
ZntB in γ-proteobacteria. A third zinc efflux protein found in enterobacteria and other pathogenic γ-proteobacteria, such as Vibrio cholerae and Yersinia pestis, is ZntB,164 which belongs to the CorA family of metal transporters (TC 1.A.35). ZntB is an inner-membrane protein like the ATPase ZntA. Although it is classified as a channel, ZntB can export Zn2+ against its concentration gradient. It has been suggested that this process may be driven by H+ antiport, but no data to support this hypothesis have been published yet. If true, then ZntB will need to be reclassified as a class 2 porter.
CorA family proteins form homo-pentamers. The first 266 amino acids of Salmonella typhimurium ZntB form a large cytosolic domain, with the remaining 61 residues forming two transmembrane helices.165 The structure of the large cytosolic soluble domains of Vibrio parahaemolyticus (pdb 3ck6)166 and S. typhimurium ZntB (pdb 3nwi167) (Fig. 8) have been determined. The two pdb entries show different structures of the “funnel” formed between the five monomers: the cylindrical channel with a diameter of 12 Å of the S. typhimurium structure is thought to represent an open form, whilst the conical shape of the pore in the V. parahaemolyticus structure is thought to show a closed conformation. The differences in channel structure come about by using alternative interaction surfaces between the monomers. Each monomer in the S. typhimurium structure, which was obtained by crystallisation in the presence of 1 mM ZnCl2, contains two zinc ions, bound by one Cys and one His residue (Fig. 8). Site 3 also has a Glu residue nearby, but its sidechain atoms were not resolved. ITC measurements of both cytosolic domain and full-length ZntB indicated apparent Zn2+ dissociation constants in the micromolar range, similar to numbers found for other Zn2+ transporters.
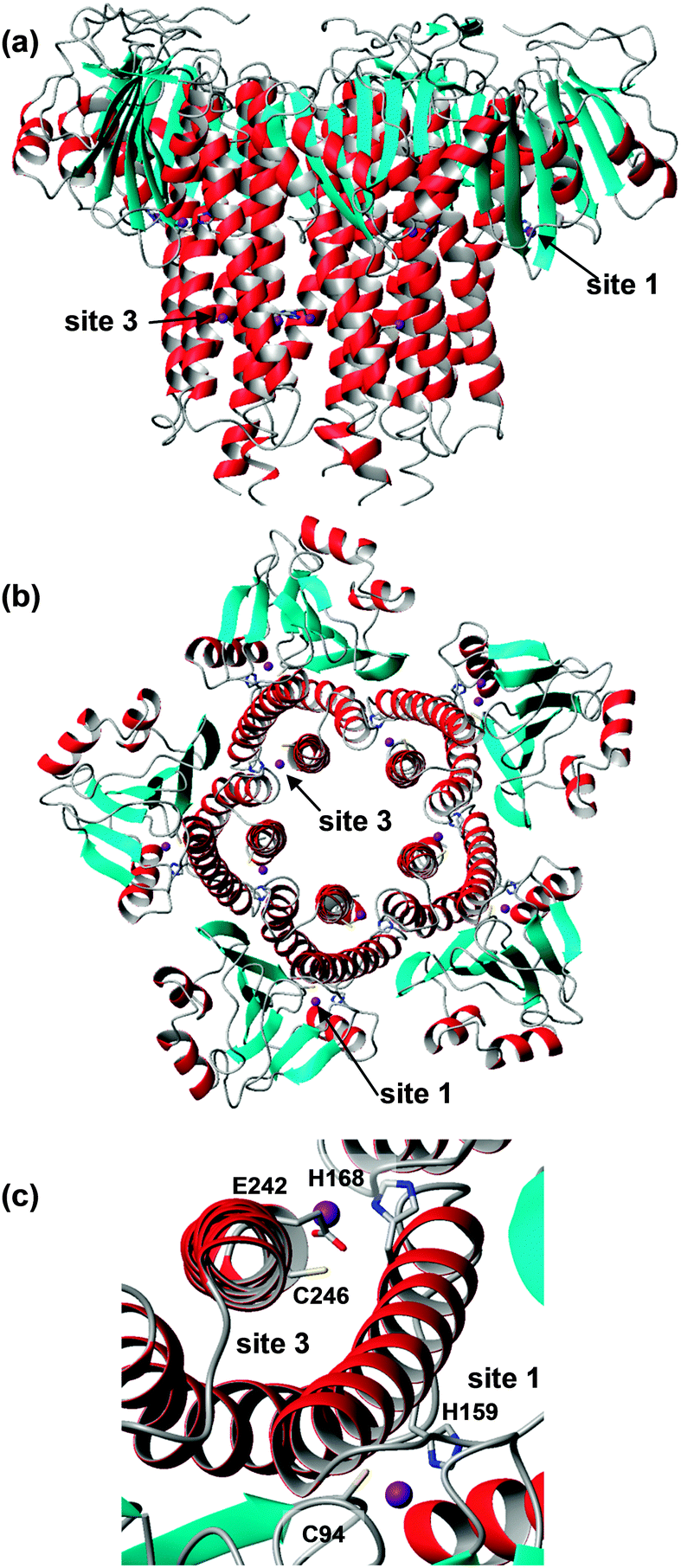 | ||
| Fig. 8 Overall structure and zinc-binding sites of soluble domain of Salmonella typhimurium ZntB (pdb 3nwi). (a) Pentameric assembly; the transmembrane domain would be located below the lower end of the structure. (b) Pentameric assembly, seen from the membrane side and showing locations of sites 1 and 3. (c) Detail of sites 1 and 3. The sidechain of E242 is not resolved in the X-ray structure, but has been reconstructed using Swiss pdb viewer. | ||
The zinc sites found in S. typhimurium ZntB are not conserved in ZntB from V. parahaemolyticus and no metal sites were resolved in its structure. Instead, many chloride ions were detected. The latter observations, together with electrostatic potential calculations, had previously led to the suggestion that metal transport in ZntB is mostly mediated by electrostatic forces – and hence perhaps would not require dedicated zinc binding sites. The likely 3-coordinate site 3 in S. typhimurium ZntB, together with its micromolar dissociation constant, suggests that ZntB from different bacteria may have different mechanisms.
Polyhistidine triad proteins. Polyhistidine triad (Pht) proteins are a family of surface proteins recently discovered in pathogenic Gram-positive Streptococci.168 Pht proteins interact with components of the immune system; they can bind complement factor H, impairing the deposition of complement C3 on the bacterial cell surface, and are thus required for pneumococcal virulence. They are currently being considered for their potential use in novel vaccines. Their expression is zinc-regulated, via the AdcR sensor protein.169 It has been suggested that zinc-dependent expression confers the ability to adapt protein expression to different host environments.
The name of these proteins stems from the presence of 5–6 instances of a HxxHxH motif. A crystal structure for a 54-amino acid segment of PhtA (pdb 2cs7)170 and a solution NMR structure for a 137-amino acid segment of PhtD (pdb 3zfj; Fig. 9),103 homologous members of this family, are available. Both comprise one Zn2+ ion, coordinated by three His and one Glu residue.
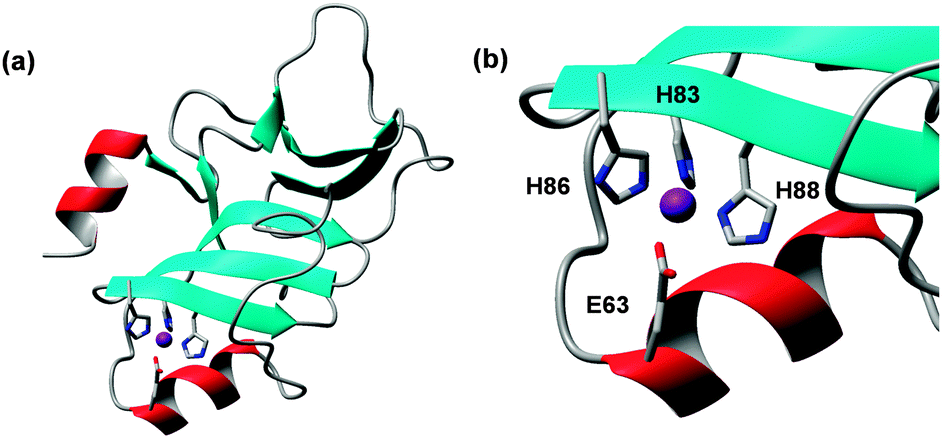 | ||
| Fig. 9 Structure of PhtD from Streptococcus pneumoniae (pdb 3zfj). (a) Overall fold of one Pht domain. (b) Details of zinc binding site. | ||
The phtD gene occurs in an operon with adcII, one of the extracellular components of the AdcABC transporter (see Section 2.2.1). The proteins AdcAII and PhtD interact in vivo and in vitro, and directional transfer of Zn2+ from PhtD to AdcAII has been demonstrated.103 This, together with the regulation by AdcR, would be concordant with a role in zinc uptake at low zinc levels, rather than protection against toxicity, although the finding that excess zinc leads to the upregulation of Pht proteins seems to contradict this hypothesis.169
Outer-membrane channels in Gram-negative bacteria. Whilst the substrate-binding proteins (AdcA/ZitS) in Gram-positive bacteria are essentially exposed to the cell exterior, this is not the case for the periplasmic ZnuA proteins. The presence of periplasmic zinc sensors, briefly mentioned in Section 2.2.5, points towards an at least partial regulation of metal levels within the periplasm, but this implies a certain level of control over transport across the outer membrane. Very little is known about zinc uptake across this barrier; mostly, it seems to have been assumed that Zn2+ enters the periplasm by diffusion along a concentration gradient through non-specific pores. This may indeed be the case in zinc-replete conditions, but this notion becomes problematic when exterior free Zn levels become extremely low. Two of the most zinc-deprived environments are the open ocean (where free Zn2+ can be as low as single-digit picomolar), and mammalian host tissues and plasma invaded by pathogenic bacteria.
In support of a more active role of the outer membrane, several bacterial outer-membrane proteins have been shown to be zinc-regulated, including TonB-dependent receptors from the cyanobacterium Anabaena PCC 7120,26 the soil γ-proteobacterium Pseudomonas protegens,171 the opportunistic pathogen Acinetobacter baumannii,172 and the pathogenic Neisseria meningitidis.173 TonB-dependent receptors usually transport organic substrates, including siderophores for iron uptake. The upregulation of TonB-dependent receptors in response to zinc deprivation has led to suggestions that they may function in the transport of “zincophores” – biological zinc chelators analogous to siderophores. In addition, the expression of several porins of Pseudomonas protegens,171 that of the OprD porin in Pseudomonas aeruginosa,174 and at least one cyanobacterial porin from Synechococcus WH8102175 was shown to be zinc-dependent. In the latter case, zinc-binding ability was demonstrated by capturing the native protein on an immobilised zinc affinity chromatography column. Further work, including electrophysiological and structural studies, is required for a better understanding of the roles of bacterial outer-membrane proteins in zinc uptake.
2.3 Intracellular zinc trafficking
The past 15 years have seen an enormous increase in the identification and study of proteins necessary for the assembly of more or less complex metalloproteins such as Cu,Zn-superoxide dismutase, cytochrome c oxidase, iron–sulfur cluster proteins, nitrogenase and hydrogenase. The biosynthetic pathways for the latter proteins require an assembly line of proteins, some of which have the sole function of metal binding and transfer and/or final insertion into the metalloprotein. Whilst many players in these assembly lines have been identified for Mo, Fe, Co, Ni, and Cu ions, zinc chaperones had remained conspicuously absent. This is not surprising, given the need to populate hundreds to thousands of different proteins: a dedicated chaperone per protein (as is the case for, e.g., copper) is clearly not a viable strategy. Instead, it is now thought that intracellular zinc is buffered and “muffled”, with the muffling at least partially mediated by metallothioneins (Section 2.3.1).56 In addition, a new family branch of putative metallochaperones of the COG0523 cluster, from a variety of organisms, has been implicated in zinc homeostasis.176 Finally, periplasmic zinc “chaperones” have been discovered, indicating a previously unexpected level of control over zinc concentrations in this bacterial compartment.The characteristic features of an MT are low molecular weight (usually less than 10 kDa), a high cysteine content (15–30%), scarcity or absence of aromatic residues, and spectroscopic evidence of metal–thiolate cluster formation. Fig. 10 shows some examples for zinc-binding MTs; it is clear that most of these feature very little secondary structure.
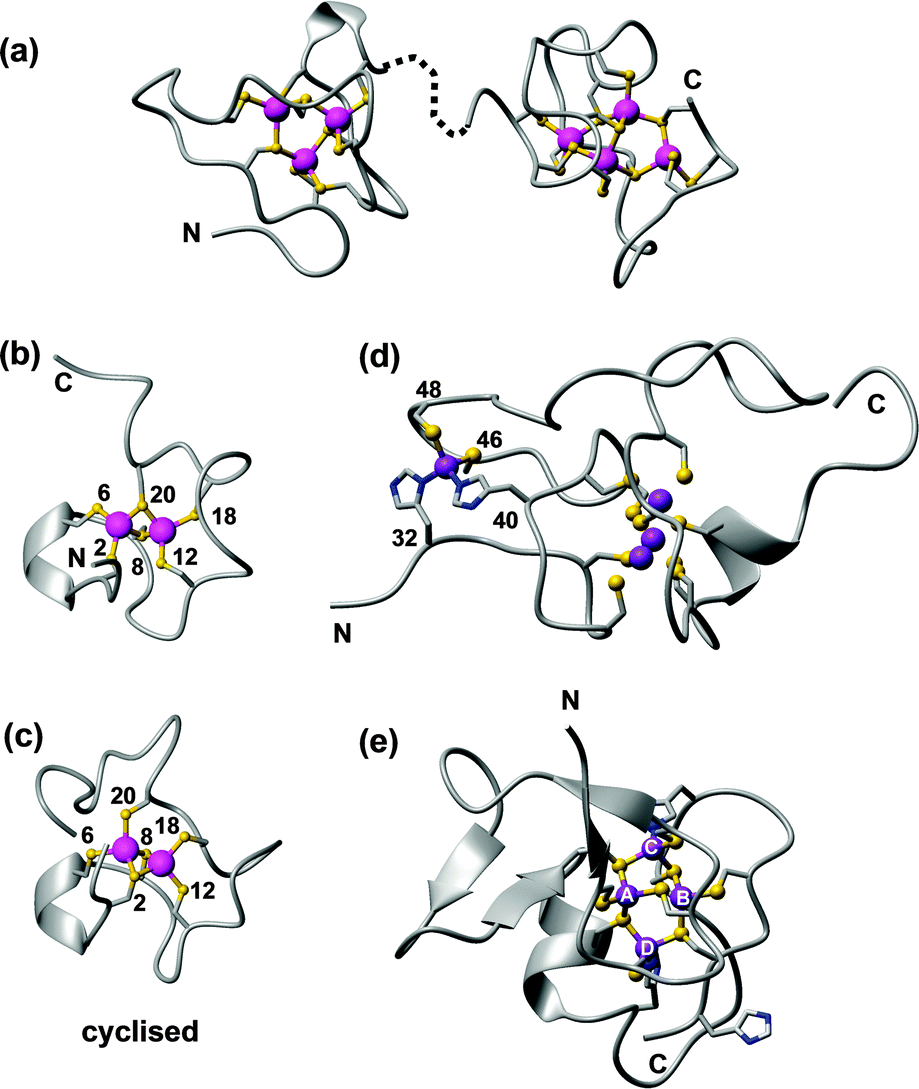 | ||
| Fig. 10 Examples of zinc-binding metallothionein structures. (a) The two solution structures of the Cd4-α (1mrt) and Cd3-β (2mrt) domain of rat MT2, mapped on the X-ray structure (4mt2).193 (b) One possible model for the γ domain of wheat EC in its Cd2 form (pdb 2l61).211 (c) Cyclised γ domain of wheat EC (Cd2 form; pdb 2mfp).212 (d) Domain 2 of wheat EC (Zn4 form, pdb 2kak),208 with residue numbers for the mononuclear His2Cys2 site. (e) SmtA from Synechococcus sp. PCC 7942 showing the Zn4Cys9His2 cluster and the zinc finger fold (pdb 1jjd). The zinc finger site A is inert towards metal exchange, site C is the most reactive. | ||
Indeed, MTs have only ordered structure when metals are bound – in essence, with respect to protein structure, the metal clusters fulfil the same stabilising role as a hydrophobic core would in other proteins. The presence of metal–thiolate clusters, relatively high solvent accessibility for at least some metal ions in the clusters, coupled with metal-dependent protein folding, leads to a peculiar combination of high thermodynamic stability with high kinetic lability – ideal prerequisites for intracellular zinc trafficking proteins. It is noted that the measurement of affinity constants of a protein with, e.g., 20 thiolate groups and seven metal binding sites is far from trivial, which might explain continuing disagreement about respective values,189,190 even though there is overall agreement on the conclusion that free thiols from partially metallated MTs are present in vivo, and participate in zinc homeostasis.191,192
The way to the first 3D structures for an MT was long and took at least one wrong turn: the first X-ray structure published was in fact incorrect, and it needed the advent of protein NMR spectroscopy to set the record straight.193 Besides requiring the development of the most important homonuclear 1H,1H correlation experiments still in use today, heteronuclear 1H,113Cd NMR experiments were essential for defining the correct metal–cysteine connectivities,194 which in turn are indispensable for defining the complete structure of an MT. Most biological and biophysical work has been carried out on mammalian MTs, but the past decade has witnessed prolific research activity to understand MTs from other phyla.188,195–197 These studies have highlighted the enormous diversity in structures, properties and functions of these intriguing proteins. Up until 2001, two types of clusters were known for Zn-binding MTs: an M3Cys9 and an M4Cys11 cluster (Fig. 10a). These two general arrangements can be achieved by diverse primary sequences, and utilising different M-Cys connectivity patterns. It has remained impossible to predict these patterns from primary sequence alone. Despite all methodological progress, the dedicated 1H,111/113Cd NMR experiments introduced in the 80ies are still required for their determination.
The structure determination of the first bacterial MT, SmtA from Synechococcus sp. PCC 7942, brought a new level of structural variation.198 SmtA contains only nine Cys residues, but binds four Zn2+ or Cd2+ ions. Two further ligands are provided by the imidazole side chains of two His residues, giving an M4Cys9His2 cluster (Fig. 10e), the structure of which closely resembles that of the M4Cys11 clusters in mammalian MTs,199 but with replacement of two terminal Cys residues by His. The two histidines have different roles in the cluster. His40 is located at the division line between the N-terminal “zinc-finger-like” portion of SmtA, which contains significant secondary structure, and the C-terminal “MT-like” portion, which lacks secondary structure like other MTs. His40 is essential to order this C-terminal section, as shown by studying a His40Cys mutant, in which structural order in the C-terminal section was markedly decreased.200 His49 is part of the most solvent-exposed and reactive metal site C, and reduces the redox lability of this site.201 Histidines are present in many other MTs202 but do not always participate in metal binding. For example, His55 in SmtA is not coordinated to a metal ion. The knowledge of important structural features has enabled the discovery of further bacterial MTs from cyanobacteria, pseudomonads and other α- and γ-proteobacteria, some of which have been characterised in vitro.203 The four Cys residues defining the zinc finger site A are strictly conserved, whilst a loop providing some of the residues for the most solvent-exposed site C is variable. Since this site governs the reactivity of the MT,201 it seems likely that different bacterial MTs have evolved to operate in different conditions.
Whether the zinc finger in SmtA engages in any biomolecular interactions has remained unknown, but it has a crucial effect on the dynamic behaviour of the protein. Using high-resolution mass spectrometry and the 67Zn isotope, the Zn2+ self-exchange reaction was studied, revealing that site A was inert towards metal exchange.204 This is due to the inaccessibility of this site, and indeed, the reaction of Zn4SmtA with an excess of Cd2+ did not lead to the expected full exchange, but yielded a Cd3ZnSmtA species. This product is entirely kinetically controlled; Cd4SmtA can be readily generated from apo-SmtA by reconstitution with Cd2+, and adopts a well-ordered structure similar to that of Zn4SmtA.205
Such isostructural replacement of Zn2+ by Cd2+, whilst common in mammalian and a few other structurally characterised MTs, is not a necessary occurrence; a notable exception is the plant type 4 MT EC from wheat.206–210 Wheat EC is a two-domain MT; the N-terminal 20-residue domain contains an M2Cys6 cluster,211 and folds equally well with either Zn2+ or Cd2+ (Fig. 10b). A designed cyclised version of this domain also adopts similar structures with Zn2+ and Cd2+, and allowed the unambiguous determination of 113Cd–Cys connectivities, with Cys2 and Cys8 as bridging residues (Fig. 10c).212 The C-terminal domain is with 47 residues considerably larger, and only adopts ordered structure in the presence of four Zn2+ ions, whilst the Cd4 form is disordered.208,209 The reason for this differential behaviour is the presence of a mononuclear Cys2His2 site in this domain (Fig. 10d); occupation of this site is important for the folding of the entire domain. We have proposed that according to the HSAB principle, the four Cd2+ ions that can bind to this domain do not utilise the two His residues, but instead form alternative clusters with the remaining eleven Cys residues, which are sufficient for a Cd4Cys11 cluster as found in other MTs.209
Specificity for a less competitive metal ion can normally not be engendered by absolute thermodynamic stability. In the case of thiolate ligands, the order of stability is Zn2+ < Cd2+ < Cu+; this is evidenced by differences in pH stability and metal-replacement titrations.178 Nevertheless, the example of wheat EC has shown that differential behaviour towards similar metal ions may be achieved by different protein folding and dynamics, mediated by the most favourable metal–ligand combinations. A more dynamic structure may have implications for in vivo protein stability; indeed, proteolysis as a mechanism for metal release has been recently demonstrated for other plant MTs,213 and in general, less well-folded proteins are more prone to proteolytic degradation. It remains to be seen whether protein-folding-mediated metal discrimination has any consequences in vivo.
The concept of “relative affinities”114 in action has been illustrated by the pair of MTs from C. elegans.214–216 The genome of this soil nematode harbours exactly two genes for MTs. Mtl-1 is constitutively expressed in the pharynx of the worm, whilst mtl-2 is strongly induced by external Cd2+, which is quite abundant in many soils. Exposure of an equimolar mixture of Zn7MTL-1 and Zn6MTL-2 to sub-stoichiometric Cd2+ led to preferential incorporation of Cd2+ into MTL-2.216 This was in full accordance with affinity data measured for Zn- and Cd-loaded MTL-1 and MTL-2.215 Whilst both MTs display almost equal affinity towards Zn (KDZn = 10−12 M), their affinities towards Cd2+ differ by almost two orders of magnitude (KDCd = 7.9 × 10−14 M (MTL-1) and 10−15 M (MTL-2)). This is only partially due to the presence of three Zn-coordinating His residues in MTL-1, but in the absence of structural data, the full molecular basis for reduced Cd2+ affinity of MTL-1 remains to be elucidated.
Major recent advances in understanding the reactions of MTs, including those described above, were possible by employing native Electrospray Ionisation Mass Spectrometry (ESI-MS),217 the only technique that can simultaneously detect all metallo-species present in a mixture. This has been employed for characterising metallo-species resulting from recombinant expression, and led to several discoveries, including the formation of mixed and undermetallated metallo-species if the growth media contain an excess of the “wrong” metal ion,218,219 and that under such circumstances, often sulfide ions are recruited in the E. coli cytosol to complete the metal–thiolate clusters.220 Similar cluster expansion can also be achieved by chemical means.221
Apart from work on composition, metal affinities, metal “preferences”, and structures, the most important biophysical studies of MTs concern their reactivity in reactions with likely physiological relevance: metal uptake (metallation),222 metal release/metal transfer201,217 and metal exchange.223 Thioneins (metal-free, or apo-MTs) can acquire metal ions from other proteins, for example the zinc finger transcription factor TF-IIIa,224,225 Cd-loaded carbonic anhydrase226 and the Zn-bound insulin hexamer.227 In turn, metallated MTs can also transfer their cargo to other molecules, including chelators such as EDTA201 and apo-proteins.228 In the case of zinc-dependent enzymes, Zn-MTs can activate these, and zinc transfer from Zn-MT to several enzymes has been shown to occur in cell extracts from mouse heart.229 Metal release may be elicited by oxidation of the cysteine thiols,230,231 and whether or not a protein may acquire Zn2+ in the presence of MT depends, amongst other factors, on the redox state of the cell, and the thionein/metallothionein ratio.232
A prevailing question regarding the metal-binding properties of MTs concerns cooperativity within individual clusters. Various earlier experiments had indicated that metal release, e.g. to EDTA, occurs in a cooperative fashion. Providing that the species observed are in equilibrium and hence thermodynamic products, this would indicate that partially formed clusters are, relative to other available alternatives (for example mixtures of fully formed clusters and “empty” proteins), less stable. If that is the case, then species formed at sub-stoichiometric levels of Zn2+ added to apo-proteins should also reflect this. Curiously, most ESI-MS studies have demonstrated a lack of cooperativity in metal uptake reactions. An early study of rabbit MT-3 indicated non-cooperative binding, with no preference for either 3- or 4-metal species observed, which would be expected if the Zn3Cys9 or Zn4Cys11 cluster was formed cooperatively.233 Over-metallated species with 8 and 9 Zn ions bound were also observed; this could be due the fact that at least the β domain of MT-3 is optimised for binding Cu+ rather than a divalent ion.234 Subsequently, rabbit MT-2 was shown to also not form either cluster preferentially, but the fully metallated Zn7 form was dominant.233 Finally, titrations of human apo-MT-1a with various metal ions including Zn2+ using a stopped-flow approach showed that this MT also binds these metal ions non-cooperatively, as yet again the observed metallo-species did not indicate a preference for complete clusters.235,236
Since MTs can be populated by cadmium in vivo,177,178 it is of interest to study the replacement of Zn2+ by Cd2+. Spectrophotometric analysis of the kinetics of Cd2+/Zn2+ exchange in rabbit MT-2 as well as the isolated α domain revealed that metal exchange occurs via an associative mechanism, with the incoming Cd2+ initially binding to the fully metallated MT, followed by exchange with a bound Zn2+.237 This is consistent with the observation of a Cd5 species for the α domain of MT-2.223 Analysis of the products of exchange by 111Cd NMR spectroscopy indicated preferential occupation of particular binding sites with Cd2+.237 One of the sites in C. elegans MTL-1 did not react at all, leading to the formation of a stable Cd6Zn1MTL-1 species,216 which is also the major form isolated when MTL-1 is expressed in the presence of excess Cd2+.215 In contrast to the kinetic origin of the Cd3Zn species for SmtA, the preferential formation of Cd6Zn1MTL-1 has thermodynamic roots; the site in question is likely a His3Cys site, with very low affinity towards the soft Cd2+.215
Inter-protein metal exchange reactions of particular current interest concern the reactions of zinc-loaded mammalian MT-3 with copper-loaded proteins and peptides present in the brain, all with relationships to neurodegenerative diseases.238 The amyloid-beta peptide forms toxic oligomers in Alzheimer's disease. Thought to be due to redox cycling between Cu+ and Cu2+, the associated generation of reactive oxygen species, and modulation of aggregation, amyloid-beta cytotoxicity is exacerbated by copper, but overall toxicity can be alleviated by extracellular Zn7MT-3.239 The mechanism for this protective effect involves “swapping” zinc with copper, leading to the formation of Zn-amyloid-beta, and mixed, oxidised Zn,Cu-MT-3, in which copper is redox-silent. Metal swapping is rapid, even though it probably occurs not by direct interaction between the proteins, but through free Cu2+.240 Zn2+ transfer from Zn7MT-3 to amyloid-beta can also be promoted by hydrogen peroxide.241 Either way, Zn2+ release from MT-3 leads to the formation of fibrillar aggregates of amyloid-beta, with different morphology to those formed in the absence of Zn2+. The physiological significance of these zinc-induced changes in aggregation and morphology is not yet clear, and the role of MT-3 in the pathogenesis of Alzheimer's disease has remained controversial.242 The redox activity of copper-loaded alpha-synuclein243 and prion protein244 is also silenced by Zn7MT-3.
In summary, many MTs have clear roles in intracellular zinc homeostasis and are crucially involved in maintaining adequate intracellular Zn2+ levels. They also provide a link between these levels and the cellular redox balance. No other metalloprotein family has been studied so extensively regarding possible metal uptake, release, and transfer reactions. However, although this is not within the scope of this review, it should not be forgotten that MTs are not restricted to these roles.
The yciC gene from Bacillus subtilis was the first representative of a Zur-regulated COG0523 member,245 and yciC expression is important to support bacterial growth under zinc starvation conditions. How the YciC protein achieves this is not known, but one hypothesis is that the protein could help to optimise zinc usage by rapid re-distribution between old and new proteins – a strategy that, for iron, has been dubbed as metal “hot-bunking”.246
Two COG0523 proteins from E. coli, YeiR247 and YjiA,248 have been studied recently in vitro. Like B. subtilis YciC, YeiR also protects the bacterium from the effects of zinc starvation, whilst YjiA has not been allocated a particular metal specificity, but is thought to have a role in the response to carbon starvation. Both proteins bind various metal ions, including Zn2+, Ni2+ and Co2+. Metal ions modulate the GTPase activity of COG0523 proteins; Zn2+ enhanced the GTPase activity of YeiR, but inhibited that of YjiA. Quantitative metal-binding studies of these proteins can be quite complex, as metal binding induces dimerisation. Up to three Zn2+ ions per monomer were found to bind to YeiR, as measured by inductively-coupled plasma atomic emission spectroscopy (ICP-AES), whilst a two-metal species dominated the ESI-MS spectrum. Dissociation constants of ≪100 nM, 43 nM and 408 nM were estimated (for the highest affinity site) or determined by competition with the metallochromic dye Mag-Fura-2. The location and identity of the zinc-binding sites on YeiR has not been comprehensively determined. Upon mutation of the three Cys residues in a CXCC motif conserved in COG0523 members (but not other G3E GTPases), the zinc content only decreased to 2.6 molar equivalents, suggesting that at least two unperturbed sites remained.
YjiA was shown to bind Ni2+, Co2+, and Zn2+ with micromolar affinities, but with different stoichiometries: one Co, two Ni, and up to four Zn ions. GDP decreased the stoichiometry in each case, by about one molar equivalent. YjiA did not crystallise in the presence of metal ions; therefore, metal-binding sites were determined by soaking apo-crystals in 3 mM ZnSO4. Four sites were observed in the structure (pdb 3ixm;248Fig. 11a and b) – one site bridging two monomers, two surface sites, and one internal site involving one of the Cys residues of the CXCC motif and two Glu residues. Mutation of the residues of this internal site decreased the Co and Ni stoichiometry in solution, but confoundingly not that of Zn. Mutation of the CXCC motif in YjiA altered metal sensitivity of the GTPase activity: whilst any response to Ni was lost, Zn still inhibited the mutant proteins – in parallel to the findings regarding metal:protein stoichiometry. Further studies are needed to elucidate the biological and the molecular function of these intriguing proteins.
 | ||
| Fig. 11 Proteins with possible “zinc chaperone” function. (a) Overall fold of YjiA from E. coli (pdb 4ixm) (b) Detail of the internal zinc binding site. (c) Decameric ZraP from Salmonella typhimurium (pdb 3lay); no zinc-bound structure is available. (d) Overall structure of ZinT from E. coli (pdb 1txl), with the His3–H2O site shown. | ||
Exposure of E. coli to excessive zinc leads to high expression levels of the periplasmic protein ZraP.28,64 Its expression is regulated by the ZraSR two-component system (Fig. 1).249 ZraP has been shown to bind zinc,250 and a metalloproteomic study demonstrated that in zinc excess conditions, the large majority of cellular zinc in E. coli was bound to ZraP.251 The catalytic molecular chaperone activity of ZraP is Zn-dependent, and Zn also affects its oligomerisation state, with higher oligomers (10–20) observed in the presence of zinc. A structure for ZraP from Salmonella typhimurium is available, but without bound Zn (pdb 3lay; Fig. 11c).252 Despite its expression being upregulated in response to zinc excess and its demonstrated sequestration properties, ZraP is not required for zinc resistance.249 In contrast, ZraP, Spy and CpxP, all molecular chaperones, are all required for resistance against polymyxin B, a host-generated antibacterial cationic peptide.
Extreme zinc starvation experiments in E. coli have demonstrated a role for the periplasmic protein ZinT (formerly known as YodA).28,253 The expression of ZinT is Zur-regulated. ZinT is thought to facilitate zinc acquisition in extreme depletion conditions, and may be able to supply Zn2+ to the ZnuABC system.254 Structures for ZinT proteins are available (Fig. 11d).102,255,256 The protein adopts a lipocalin-like fold, and the zinc binding site bears resemblance to those found in some ZnuA proteins: three His residues plus one water molecule. Zinc binding has very little effect on the structure. In addition, ZinTs contain a His-rich N-terminal sequence (HGHHAHG), which was not resolved in any structure. Competition experiments of E. coli ZinT and Mag-Fura-2 indicated a KD below 20 nM,28 and a KD of 2.2 nM was determined for Salmonella enterica ZinT.102 The interaction between Salmonella ZinT and ZnuA has been studied. Mixing a Zn–ZinT complex with apo-ZnuA led to the formation of a ternary complex observable by analytical ultracentrifugation. The complex could not be crystallised, but a model was generated based on small-angle X-ray scattering data. The model suggested that the ZnuA His-rich loop may be capable of helping to extract Zn2+ from the ZinT binding site.102
ZinT proteins are not present in all Gram-negative bacteria, but homologues are present at the cell surface of several Gram-positive bacteria, including Bacillus subtilis and Streptococci.75 In the latter, ZinT is one domain of the AdcA protein, the second domain being homologous to ZnuA. Together with the evidence obtained for ZinT–ZnuA interactions in Gram-negative bacteria, it seems likely that ZinT is one of the surface or periplasmic proteins (or domains) that supply either extracellular or periplasmic Zn2+ to AdcA (Streptococci) or ZnuA (other Gram-positive and Gram-negative bacteria).
3. Conclusions and outlook
Research regarding structures and thermodynamics of proteins involved in the homeostasis of essential metal ions including zinc has been flourishing over the past decade. The largest part of biophysical work has been carried out on prokaryotic systems, mainly because eukaryotic systems often tend to be technically more challenging and more complex. The comparable ease of combining in vitro and in vivo approaches, and of genetic and genomic manipulations, for prokaryotic systems has particularly favoured rapid progress in our understanding of metal homeostasis. Some major players in zinc homeostasis, including ZIP and CDF transporters, are conserved from bacteria to man, and here, studies of bacterial representatives also greatly promote understanding of the eukaryotic proteins. In contrast, systems such as the ZnuABC zinc uptake transporters and the poly-histidine triad proteins are restricted to prokaryotes, and considering the importance of metal ions at the host–pathogen interface, this may open new opportunities in antibacterial therapy.A survey of all structurally characterised metal binding sites considered in this review reveals that most are formed by either three or four amino acid side-chains, with coordination numbers of four and tetrahedral geometry prevailing – quite similar to structural and catalytic sites. Surprisingly, there are even examples for sites with the same ligand composition as catalytic sites, for example in Synechocystis ZnuA and Cupriavidus metallidurans ZneB, sites are formed by thee His or two His and one carboxylate, respectively, with a fourth site free for an external ligand – motifs normally found in hydrolytic enzymes such as carbonic anhydrase and many metalloproteases. It remains unclear why these sites are not known to exhibit at least some degree of catalytic activity. In addition, apart from an obvious impact of the degree of surface-exposure of a particular site, our understanding of if and why most transport sites are considerably more labile than catalytic zinc sites is still patchy. At present, there is still a scarcity of kinetic data for zinc transporters, and zinc binding kinetics in general.257 It has however been noted that for example in the case of sensor proteins “the prevailing view on metal-mediated molecular regulation in terms of ‘on and off control’ might be oversimplified”.258
ATPases, CDF transporters and ZneCBA systems all seem to comprise regulatory zinc binding sites, the former two in the cytosolic domains, the latter in the periplasm, i.e. the compartments from which transport occurs. All three systems are active transporters, i.e. require energy. It would make sense that these pumps are inactivated as long as no zinc needs to be transported. Furthermore, many transmembrane zinc transporters from essentially all families harbour histidine-rich sequences which have been largely refractory to structural studies, and their mechanistic significance has remained mostly obscure.259 Large numbers of His-rich sequences and proteins have been identified in prokaryotes.260 Although many His-rich proteins may play roles unrelated to zinc homeostasis, similarities between these and MTs have been noted. Like for MTs, zinc binding to His-rich sequences has been observed to lead to conformational restriction.146 It is conceivable that in the case of zinc transporters, this may impact on protein–protein or domain–domain interactions, and also play a role in metal-dependent regulation of transporter activity.
Excitingly, the very first example of a structurally characterised de novo designed transmembrane transporter is a zinc transporter involving a homo-tetrameric helical bundle forming two His2Glu4 di-zinc sites. Although structurally unrelated to any of the zinc transporters described above, “Rocker” is able to use a proton gradient to transport Zn2+ against its concentration gradient.261
Finally, it is clear that studies on isolated proteins are only one albeit substantial part of a much larger picture, and need to be combined with studies on cellular and organismal levels. Renewed efforts to quantitatively describe biological systems, and the emerging role of zinc as a signalling agent will require data on time-dependence of molecular events as well as an enhanced quantitative understanding of zinc probes in cellular environments. Recent studies of zinc chelators and several probes have shown that contrary to common assumptions, the probes TSQ (6-methoxy-8-p-toluenesulfonamido-quinoline),262 Zinquin,263 sensors of the ZnAF family264 as well as the chelator TPEN265 are all capable to bind or sense protein-bound zinc besides the desired interaction with free or labile cellular zinc. This may lead to over-estimation of zinc concentrations and fluxes.
In summary, despite the tremendous acceleration of discovery, functional studies, and structure elucidation, ample scope remains for future studies on molecular mechanisms in zinc trafficking and homeostasis.
Acknowledgements
The author thanks the BBSRC (grant no. BB/J006467/1) and the Royal Society (Olga Kennard Fellowship to CAB 2004-9).Notes and references
- J. L. Raulin, Ann. Sci. Nat., Bot. Biol. Veg., 1869, 11, 293–299 Search PubMed.
- W. R. Todd, C. A. Elvehjem and E. B. Hart, Am. J. Physiol., 1934, 107, 146–156 CAS.
- A. S. Prasad, Adv. Nutr., 2013, 4, 176–190 CrossRef CAS PubMed.
- I. Keilin and T. Mann, Nature, 1939, 144, 442–443 CrossRef.
- W. Maret, Adv. Nutr., 2013, 4, 82–91 CrossRef CAS PubMed.
- W. Maret, J. Inorg. Biochem., 2012, 111, 110–116 CrossRef CAS PubMed.
- http://www.copenhagenconsensus.com/copenhagen-consensus-2008/outcomes .
- A. S. Prasad, Curr. Opin. Clin. Nutr. Metab. Care, 2009, 12, 646–652 CrossRef CAS PubMed.
- E. Mocchegiani, L. Costarelli, A. Basso, R. Giacconi, F. Piacenza and M. Malavolta, Curr. Pharm. Des., 2013, 19, 1753–1764 CAS.
- C. P. Wong and E. Ho, Mol. Nutr. Food Res., 2012, 56, 77–87 CAS.
- H. Kozlowski, M. Luczkowski, M. Remelli and D. Valensin, Coord. Chem. Rev., 2012, 256, 2129–2141 CrossRef CAS PubMed.
- S. S. Leal, H. M. Botelho and C. M. Gomes, Coord. Chem. Rev., 2012, 256, 2253–2270 CrossRef CAS PubMed.
- B. K. Y. Bitanihirwe and M. G. Cunningham, Synapse, 2009, 63, 1029–1049 CrossRef CAS PubMed.
- A. S. Prasad, F. W. Beck, D. C. Snell and O. Kucuk, Nutr. Cancer, 2009, 61, 879–887 CrossRef CAS PubMed.
- M. Murakami and T. Hirano, Cancer Sci., 2008, 99, 1515–1522 CrossRef CAS PubMed.
- R. B. Franklin and L. C. Costello, J. Cell. Biochem., 2009, 106, 750–757 CrossRef CAS PubMed.
- H. Haase and L. Rink, Annu. Rev. Nutr., 2009, 29, 133–152 CrossRef CAS PubMed.
- F. Chimienti, Nutr. Res. Rev., 2013, 26, 1–11 CrossRef CAS PubMed.
- J. P. Barnett, C. A. Blindauer, O. Kassaar, S. Khazaipoul, E. M. Martin, P. J. Sadler and A. J. Stewart, Biochim. Biophys. Acta, Gen. Subj., 2013, 1830, 5456–5464 CrossRef CAS PubMed.
- I. Cakmak, J. Trace Elem. Med. Biol., 2009, 23, 281–289 CAS.
- H. H. Sandstead, Adv. Nutr., 2013, 4, 76–81 CrossRef CAS PubMed.
- F. M. M. Morel, J. R. Reinfelder, S. B. Roberts, C. P. Chamberlain, J. G. Lee and D. Yee, Nature, 1994, 369, 740–742 CrossRef CAS.
- K. Hantke, Curr. Opin. Microbiol., 2005, 8, 196–202 CrossRef CAS PubMed.
- C. A. Blindauer, Chem. Biodiversity, 2008, 5, 1990–2013 CAS.
- J. P. Barnett, A. Millard, A. Z. Ksibe, D. J. Scanlan, R. Schmid and C. A. Blindauer, Front. Microbiol., 2012, 3, 142 Search PubMed.
- M. Napolitano, M. A. Rubio, J. Santamaria-Gomez, E. Olmedo-Verd, N. J. Robinson and I. Luque, J. Bacteriol., 2012, 194, 2426–2436 CrossRef CAS PubMed.
- D. H. Nies, J. Bacteriol., 2012, 194, 2407–2412 CrossRef CAS PubMed.
- A. I. Graham, S. Hunt, S. L. Stokes, N. Bramall, J. Bunch, A. G. Cox, C. W. McLeod and R. K. Poole, J. Biol. Chem., 2009, 284, 18377–18389 CrossRef CAS PubMed.
- E. K. LeGrand and J. Alcock, Q. Rev. Biol., 2012, 87, 3–18 CrossRef.
- B. D. Corbin, E. H. Seeley, A. Raab, J. Feldmann, M. R. Miller, V. J. Torres, K. L. Anderson, B. M. Dattilo, P. M. Dunman, R. Gerads, R. M. Caprioli, W. Nacken, W. J. Chazin and E. P. Skaar, Science, 2008, 319, 962–965 CrossRef CAS PubMed.
- S. Shafeeq, O. P. Kuipers and T. G. Kloosterman, Mol. Microbiol., 2013, 88, 1047–1057 CrossRef CAS PubMed.
- D. Osman and J. S. Cavet, in Adv. Microb. Physiol., ed. R. K. Poole, Academic Press Ltd-Elsevier Science Ltd, London, 2011, vol. 58, pp. 175–232 Search PubMed.
- G. Porcheron, A. Garenaux, J. Proulx, M. Sabri and C. M. Dozois, Front. Cell. Infect. Microbiol., 2013, 3, 90 Search PubMed.
- J. J. Braymer and D. P. Giedroc, Curr. Opin. Chem. Biol., 2014, 19C, 59–66 CrossRef PubMed.
- F. Li, I. Chvyrkova, S. Terzyan, N. Wakeham, R. Turner, A. K. Ghosh, X. J. C. Zhang and J. Tang, Appl. Microbiol. Biotechnol., 2012, 94, 1041–1049 CrossRef CAS PubMed.
- R. A. Bozym, F. Chimienti, L. J. Giblin, G. W. Gross, I. Korichneva, Y. A. Li, S. Libert, W. Maret, M. Parviz, C. J. Frederickson and R. B. Thompson, Exp. Biol. Med., 2010, 235, 741–750 CrossRef CAS PubMed.
- T. Fukada, S. Yamasaki, K. Nishida, M. Murakami and T. Hirano, JBIC, J. Biol. Inorg. Chem., 2011, 16, 1123–1134 CrossRef CAS PubMed.
- W. Maret, BioMetals, 2011, 24, 411–418 CrossRef CAS PubMed.
- C. J. Frederickson, M. Hershfinkel and L. J. Giblin, in Synaptic Plasticity and Transsynaptic Signaling, ed. P. K. Stanton, C. Bramham and H. E. Scharfman, Springer, New York, 2005, pp. 123–137 Search PubMed.
- A. M. Kim, M. L. Bernhardt, B. Y. Kong, R. W. Ahn, S. Vogt, T. K. Woodruff and T. V. O'Halloran, ACS Chem. Biol., 2011, 6, 716–723 CrossRef CAS PubMed.
- M. M. Henary, Y. G. Wu and C. J. Fahrni, Chem. – Eur. J., 2004, 10, 3015–3025 CrossRef CAS PubMed.
- M. Taki, J. L. Wolford and T. V. O'Halloran, J. Am. Chem. Soc., 2004, 126, 712–713 CrossRef CAS PubMed.
- H. Woo, Y. You, T. Kim, G. J. Jhon and W. Nam, J. Mater. Chem., 2012, 22, 17100–17112 RSC.
- W. Chyan, D. Y. Zhang, S. J. Lippard and R. J. Radford, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 143–148 CrossRef CAS PubMed.
- R. J. Radford and S. J. Lippard, Curr. Opin. Chem. Biol., 2013, 17, 129–136 CrossRef CAS PubMed.
- P. Chabosseau, E. Tuncay, G. Meur, E. A. Bellomo, A. Hessels, S. Hughes, P. R. V. Johnson, M. Bugliani, P. Marchetti, B. Turan, A. R. Lyon, M. Merkx and G. A. Rutter, ACS Chem. Biol., 2014, 9, 2111–2120 CrossRef CAS PubMed.
- B. J. McCranor, R. A. Bozym, M. I. Vitolo, C. A. Fierke, L. Bambrick, B. M. Polster, G. Fiskum and R. B. Thompson, J. Bioenerg. Biomembr., 2012, 44, 253–263 CrossRef CAS PubMed.
- C. E. Outten and T. V. O'Halloran, Science, 2001, 292, 2488–2492 CrossRef CAS PubMed.
- A. Krezel and W. Maret, JBIC, J. Biol. Inorg. Chem., 2006, 11, 1049–1062 CrossRef CAS PubMed.
- C. Andreini, I. Bertini and A. Rosato, Acc. Chem. Res., 2009, 42, 1471–1479 CrossRef CAS PubMed.
- M. Brylinski and J. Skolnick, Proteins, 2011, 79, 735–751 CrossRef CAS PubMed.
- M. Zimmermann, O. Clarke, J. M. Gulbis, D. W. Keizer, R. S. Jarvis, C. S. Cobbett, M. G. Hinds, Z. G. Xiao and A. G. Wedd, Biochemistry, 2009, 48, 11640–11654 CrossRef CAS PubMed.
- S. Tottey, K. J. Waldron, S. J. Firbank, B. Reale, C. Bessant, K. Sato, T. R. Cheek, J. Gray, M. J. Banfield, C. Dennison and N. J. Robinson, Nature, 2008, 455, 1138–1142 CrossRef CAS PubMed.
- A. W. Foster and N. J. Robinson, BMC Biol., 2011, 9, 3 CrossRef PubMed.
- D. Wang, O. Hosteen and C. A. Fierke, J. Inorg. Biochem., 2012, 111, 173–181 CrossRef CAS PubMed.
- R. A. Colvin, W. R. Holmes, C. P. Fontaine and W. Maret, Metallomics, 2010, 2, 306–317 RSC.
- D. H. Nies, Science, 2007, 317, 1695–1696 CrossRef CAS PubMed.
- B. P. Weaver, J. D. Beattie, T. Kambe and G. K. Andrews, Biol. Chem., 2007, 388, 1301–1312 CrossRef CAS PubMed.
- J. Dufner-Beattie, Y. M. Kuo, J. Gitschier and G. K. Andrews, J. Biol. Chem., 2004, 279, 49082–49090 CrossRef CAS PubMed.
- M. Pruteanu, S. B. Neher and T. A. Baker, J. Bacteriol., 2007, 189, 3017–3025 CrossRef CAS PubMed.
- P. T. Chivers, J. Bacteriol., 2007, 189, 2953–2954 CrossRef CAS PubMed.
- C. D. Ellis, F. Wang, C. W. MacDiarmid, S. Clark, T. Lyons and D. J. Eide, J. Cell Biol., 2004, 166, 325–335 CrossRef CAS PubMed.
- D. Wang and C. A. Fierke, Metallomics, 2013, 5, 372–383 RSC.
- C. Appia-Ayme, A. Hall, E. Patrick, S. Rajadurai, T. A. Clarke and G. Rowley, Biochem. J., 2012, 442, 85–93 CrossRef CAS PubMed.
- J. M. Argüello, D. Raimunda and M. Gonzalez-Guerrero, J. Biol. Chem., 2012, 287, 13510–13517 CrossRef PubMed.
- Z. Xiao and A. G. Wedd, Nat. Prod. Rep., 2010, 5, 768–789 RSC.
- K. J. Waldron, J. C. Rutherford, D. Ford and N. J. Robinson, Nature, 2009, 460, 823–830 CrossRef CAS PubMed.
- I. Zawisza, M. Rozga and W. Bal, Coord. Chem. Rev., 2012, 256, 2297–2307 CrossRef CAS PubMed.
- W. Maret, J. Trace Elem. Med. Biol., 2005, 19, 7–12 Search PubMed.
- W. Maret and B. L. Vallee, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 3478–3482 CrossRef CAS.
- L. A. Finney and T. V. O'Halloran, Science, 2003, 300, 931–936 CrossRef CAS PubMed.
- S. Choi and A. J. Bird, Metallomics, 2014, 6, 1198–1215 RSC.
- A. J. Guerra and D. P. Giedroc, Arch. Biochem. Biophys., 2012, 519, 210–222 CrossRef CAS PubMed.
- V. Günther, U. Lindert and W. Schaffner, Biochim. Biophys. Acta, 2012, 1823, 1416–1425 CrossRef PubMed.
- E. M. Panina, A. A. Mironov and M. S. Gelfand, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 9912–9917 CrossRef CAS PubMed.
- H. Nanamiya and F. Kawamura, Biosci., Biotechnol., Biochem., 2010, 74, 451–461 CrossRef CAS PubMed.
- H. Reyes-Caballero, G. C. Campanello and D. P. Giedroc, Biophys. Chem., 2011, 156, 103–114 CrossRef CAS PubMed.
- L. J. Coneyworth, K. A. Jackson, J. Tyson, H. J. Bosomworth, E. van der Hagen, G. M. Hann, O. A. Ogo, D. C. Swann, J. C. Mathers, R. A. Valentine and D. Ford, J. Biol. Chem., 2012, 287, 36567–36581 CrossRef CAS PubMed.
- H. Zhao, E. Butler, J. Rodgers, T. Spizzo, S. Duesterhoeft and D. Eide, J. Biol. Chem., 1998, 273, 28713–28720 CrossRef CAS PubMed.
- A. G. Assunção, D. P. Persson, S. Husted, J. K. Schjørring, R. D. Alexander and M. G. Aarts, Metallomics, 2013, 9, 1110–1116 RSC.
- I. M. Evans, L. N. Gatehouse, J. A. Gatehouse, N. J. Robinson and R. R. D. Croy, FEBS Lett., 1990, 262, 29–32 CrossRef CAS.
- D. Osman and J. S. Cavet, Nat. Prod. Rep., 2010, 27, 668–680 RSC.
- M. L. VanZile, X. H. Chen and D. P. Giedroc, Biochemistry, 2002, 41, 9765–9775 CrossRef CAS PubMed.
- N. E. Grossoehme and D. P. Giedroc, J. Am. Chem. Soc., 2009, 131, 17860–17870 CrossRef CAS PubMed.
- C. Blindauer, Biochemistry, 2012, 34, 4–13 CAS.
- J. S. Cavet, W. M. Meng, M. A. Pennella, R. J. Applehoff, D. P. Giedroc and N. J. Robinson, J. Biol. Chem., 2002, 277, 38441–38448 CrossRef CAS PubMed.
- The Transporter Classification Database, M. H. Saier, V. S. Reddy, D. G. Tamang and A. Vastermark, Nucleic Acids Res., 2014, 42, D251–D258 CrossRef CAS PubMed , http://www.tcdb.org/.
- J. J. R. Fraústo da Silva and R. J. P. Williams, The biological chemistry of the elements: the inorganic chemistry of life, Oxford University Press, 2nd edn, 2001 Search PubMed.
- B. F. Weston, A. Brenot and M. G. Caparon, Infect. Immun., 2009, 77, 2840–2848 CrossRef CAS PubMed.
- J. S. Klein and O. Lewinson, Metallomics, 2011, 3, 1098–1108 RSC.
- V. M. Korkhov, S. A. Mireku and K. P. Locher, Nature, 2012, 490, 367–372 CrossRef CAS PubMed.
- P. M. Jones, M. L. O'Mara and A. M. George, Trends Biochem. Sci., 2009, 34, 520–531 CrossRef CAS PubMed.
- B. R. Chandra, M. Yogavel and A. Sharma, J. Mol. Biol., 2007, 367, 970–982 CrossRef CAS PubMed.
- H. Li and G. Jogl, J. Mol. Biol., 2007, 368, 1358–1366 CrossRef CAS PubMed.
- L. A. Yatsunyk, J. A. Easton, L. R. Kim, S. A. Sugarbaker, B. Bennett, R. M. Breece, I. I. Vorontsov, D. L. Tierney, M. W. Crowder and A. C. Rosenzweig, JBIC, J. Biol. Inorg. Chem., 2008, 13, 271–288 CrossRef CAS PubMed.
- A. Ilari, F. Alaleona, P. Petrarca, A. Battistoni and E. Chiancone, J. Mol. Biol., 2011, 409, 630–641 CrossRef CAS PubMed.
- S. Banerjee, B. X. Wei, M. Bhattacharyya-Pakrasi, H. B. Pakrasi and T. J. Smith, J. Mol. Biol., 2003, 333, 1061–1069 CrossRef CAS PubMed.
- B. X. Wei, A. M. Randich, M. Bhattacharyya-Pakrasi, H. B. Pakrasi and T. J. Smith, Biochemistry, 2007, 46, 8734–8743 CrossRef CAS PubMed.
- E. Loisel, L. Jacquamet, L. Serre, C. Bauvois, J. L. Ferrer, T. Vernet, A. M. Di Guilmi and C. Durmort, J. Mol. Biol., 2008, 381, 594–606 CrossRef CAS PubMed.
- Y. H. Lee, R. K. Deka, M. V. Norgard, J. D. Radolf and C. A. Hasemann, Nat. Struct. Biol., 1999, 6, 628–633 CrossRef CAS PubMed.
- L. Bayle, S. Chimalapati, G. Schoehn, J. Brown, T. Vernet and C. Durmort, Mol. Microbiol., 2011, 82, 904–916 CrossRef CAS PubMed.
- A. Ilari, F. Alaleona, G. Tria, P. Petrarca, A. Battistoni, C. Zamparelli, D. Verzili, M. Falconi and E. Chiancone, Biochim. Biophys. Acta, Gen. Subj., 2014, 1840, 535–544 CrossRef CAS PubMed.
- B. Bersch, C. Bougault, L. Roux, A. Favier, T. Vernet and C. Durmort, PLoS One, 2013, 8 Search PubMed.
- J. M. Argüello, E. Eren and M. Gonzalez-Guerrero, BioMetals, 2007, 20, 233–248 CrossRef PubMed.
- K. T. Wang, O. Sitsel, G. Meloni, H. E. Autzen, M. Andersson, T. Klymchuk, A. M. Nielsen, D. C. Rees, P. Nissen and P. Gourdon, Nature, 2014, 514, 518–522 CrossRef CAS PubMed.
- P. Gourdon, X. Y. Liu, T. Skjorringe, J. P. Morth, L. B. Moller, B. P. Pedersen and P. Nissen, Nature, 2011, 475, 59–65 CrossRef CAS PubMed.
- M. Andersson, D. Mattle, O. Sitsel, T. Klymchuk, A. M. Nielsen, L. B. Moller, S. H. White, P. Nissen and P. Gourdon, Nat. Struct. Mol. Biol., 2014, 21, 43–48 CAS.
- G. S. Allen, C. C. Wu, T. Cardozo and D. L. Stokes, Structure, 2011, 19, 1219–1232 CrossRef CAS PubMed.
- S. J. Dutta, J. Liu, Z. Hou and B. Mitra, Biochemistry, 2006, 45, 5923–5931 CrossRef CAS PubMed.
- J. Okkeri and T. Haltia, Biochim. Biophys. Acta, Bioenerg., 2006, 1757, 1485–1495 CrossRef CAS PubMed.
- L. Banci, L. Bertini, S. Ciofi-Baffoni, L. A. Finney, C. E. Outten and T. V. O'Halloran, J. Mol. Biol., 2002, 323, 883–897 CrossRef CAS.
- L. Banci, I. Bertini, S. Ciofi-Baffoni, L. Poggi, M. Vanarotti, S. Tottey, K. J. Waldron and N. J. Robinson, JBIC, J. Biol. Inorg. Chem., 2010, 15, 87–98 CrossRef CAS PubMed.
- D. Raimunda, P. Subramanian, T. Stemmler and J. M. Argüello, Biochim. Biophys. Acta, Biomembr., 2012, 1818, 1374–1377 CrossRef CAS PubMed.
- K. J. Waldron and N. J. Robinson, Nat. Rev. Microbiol., 2009, 1, 25–35 CrossRef PubMed.
- C. A. Blindauer and R. Schmid, Metallomics, 2010, 2, 510–529 RSC.
- E. Eren, D. C. Kennedy, M. J. Maroney and J. M. Argüello, J. Biol. Chem., 2006, 281, 33881–33891 CrossRef CAS PubMed.
- L. Baekgaard, M. D. Mikkelsen, D. M. Sorensen, J. N. Hegelund, D. P. Persson, R. F. Mills, Z. Yang, S. Husted, J. P. Andersen, M. J. Buch-Pedersen, J. K. Schjoerring, L. E. Williams and M. G. Palmgren, J. Biol. Chem., 2010, 285, 31243–31252 CrossRef CAS PubMed.
- Y. Chao and D. Fu, J. Biol. Chem., 2004, 279, 12043–12050 CrossRef CAS PubMed.
- E. Ohana, E. Hoch, C. Keasar, T. Kambe, O. Yifrach, M. Hershfinkel and I. Sekler, J. Biol. Chem., 2009, 284, 17677–17686 CrossRef CAS PubMed.
- T. Kambe, Biosci., Biotechnol., Biochem., 2011, 75, 1036–1043 CrossRef CAS PubMed.
- L. P. Huang and S. Tepaamorndech, Mol. Aspects Med., 2013, 34, 548–560 CrossRef CAS PubMed.
- F. Chimienti, Nutr. Res. Rev., 2013, 26, 1–11 CrossRef CAS PubMed.
- J. Flannick, G. Thorleifsson, N. L. Beer, S. B. R. Jacobs, N. Grarup, N. P. Burtt, A. Mahajan, C. Fuchsberger, G. Atzmon, R. Benediktsson, J. Blangero, D. W. Bowden, I. Brandslund, J. Brosnan, F. Burslem, J. Chambers, Y. S. Cho, C. Christensen, D. A. Douglas, R. Duggirala, Z. Dymek, Y. Farjoun, T. Fennell, P. Fontanillas, T. Forsen, S. Gabriel, B. Glaser, D. F. Gudbjartsson, C. Hanis, T. Hansen, A. B. Hreidarsson, K. Hveem, E. Ingelsson, B. Isomaa, S. Johansson, T. Jorgensen, M. E. Jorgensen, S. Kathiresan, A. Kong, J. Kooner, J. Kravic, M. Laakso, J.-Y. Lee, L. Lind, C. M. Lindgren, A. Linneberg, G. Masson, T. Meitinger, K. L. Mohlke, A. Molven, A. P. Morris, S. Potluri, R. Rauramaa, R. Ribel-Madsen, A.-M. Richard, T. Rolph, V. Salomaa, A. V. Segre, H. Skarstrand, V. Steinthorsdottir, H. M. Stringham, P. Sulem, E. S. Tai, Y. Y. Teo, T. Teslovich, U. Thorsteinsdottir, J. K. Trimmer, T. Tuomi, J. Tuomilehto, F. Vaziri-Sani, B. F. Voight, J. G. Wilson, M. Boehnke, M. I. McCarthy, P. R. Njolstad, O. Pedersen, L. Groop, D. R. Cox, K. Stefansson, D. Altshuler and corporate authors: Go-T2D Consortium; T2D-GENES Consortium, Nat. Genet., 2014, 46, 357–363 CrossRef CAS PubMed.
- L. P. Huang and J. Gitschier, Nat. Genet., 1997, 17, 292–297 CrossRef CAS PubMed.
- I. Lasry, Y. A. Seo, H. Ityel, N. Shalva, B. Pode-Shakked, F. Glaser, B. Berman, I. Berezovsky, A. Goncearenco, A. Klar, J. Levy, Y. Anikster, S. L. Kelleher and Y. G. Assaraf, J. Biol. Chem., 2012, 287, 29348–29361 CrossRef CAS PubMed.
- S. Ayton, P. Lei and A. I. Bush, Free Radical Biol. Med., 2013, 62, 76–89 CrossRef CAS PubMed.
- M. Lu and D. Fu, Science, 2007, 317, 1746–1748 CrossRef CAS PubMed.
- M. Lu, J. Chai and D. Fu, Nat. Struct. Mol. Biol., 2009, 16, 1063–1067 CAS.
- V. Cherezov, N. Hofer, D. M. E. Szebenyi, O. Kolaj, J. G. Wall, R. Gillilan, V. Srinivasan, C. P. Jaroniec and M. Caffrey, Structure, 2008, 16, 1378–1388 CrossRef CAS PubMed.
- N. Coudray, S. Valvo, M. H. Hu, R. Lasala, C. Kim, M. Vink, M. Zhou, D. Provasi, M. Filizola, J. Tao, J. Fang, P. A. Penczek, I. Ubarretxena-Belandia and D. L. Stokes, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2140–2145 CrossRef CAS PubMed.
- D. Russell and T. Soulimane, FEBS Lett., 2012, 586, 4332–4338 CrossRef CAS PubMed.
- A. Anton, A. Weltrowski, C. J. Haney, S. Franke, G. Grass, C. Rensing and D. H. Nies, J. Bacteriol., 2004, 186, 7499–7507 CrossRef CAS PubMed.
- Y. Chao and D. Fu, J. Biol. Chem., 2004, 279, 17173–17180 CrossRef CAS PubMed.
- D. Goswami, J. Kaur, S. Surade, E. Grell and H. Michel, Biol. Chem., 2012, 393, 617–629 CrossRef CAS PubMed.
- B. Montanini, D. Blaudez, S. Jeandroz, D. Sanders and M. Chalot, BMC Genomics, 2007, 8, 16 CrossRef PubMed.
- E. Hoch, W. Lin, J. Chai, M. Hershfinkel, D. Fu and I. Sekler, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7202–7207 CrossRef CAS PubMed.
- D. Podar, J. Scherer, Z. Noordally, P. Herzyk, D. Nies and D. Sanders, J. Biol. Chem., 2012, 287, 3185–3196 CrossRef CAS PubMed.
- M. Kawachi, Y. Kobae, S. Kogawa, T. Mimura, U. Krämer and M. Maeshima, FEBS J., 2012, 279, 2339–2356 CrossRef CAS PubMed.
- M. L. Guerinot, Biochim. Biophys. Acta, 2000, 1465, 190–198 CrossRef CAS.
- J. Jeong and D. J. Eide, Mol. Aspects Med., 2013, 34, 612–619 CrossRef CAS PubMed.
- L. Hudek, L. A. Pearson, A. Michalczyk, B. A. Neilan and M. L. Ackland, Appl. Microbiol. Biotechnol., 2013, 97, 8649–8662 CrossRef CAS PubMed.
- M. Cerasi, J. Z. Liu, S. Ammendola, A. J. Poe, P. Petrarca, M. Pesciaroli, P. Pasquali, M. Raffatellu and A. Battistoni, Metallomics, 2014, 6, 845–853 RSC.
- D. L. Knoell and M. J. Liu, Int. J. Vitam. Nutr. Res., 2010, 80, 271–277 CrossRef CAS PubMed.
- W. Lin, J. Chai, J. Love and D. Fu, J. Biol. Chem., 2010, 285, 39013–39020 CrossRef CAS PubMed.
- S. Ehsani, H. R. Huo, A. Salehzadeh, C. L. Pocanschi, J. C. Watts, H. Wille, D. Westaway, E. Rogaeva, P. H. St. George-Hyslop and G. Schmitt-Ulms, Prog. Neurobiol., 2011, 93, 405–420 CrossRef CAS PubMed.
- N. E. Grossoehme, S. Akilesh, M. L. Guerinot and D. E. Wilcox, Inorg. Chem., 2006, 45, 8500–8508 CrossRef CAS PubMed.
- G. Vert, N. Grotz, F. Dedaldechamp, F. Gaymard, M. L. Guerinot, J. F. Briat and C. Curie, Plant Cell, 2002, 14, 1223–1233 CrossRef CAS.
- S. Potocki, D. Valensin, F. Camponeschi and H. Kozlowski, J. Inorg. Biochem., 2013, 127, 246–252 CrossRef CAS PubMed.
- Z. Liu, H. Li, M. Soleimani, K. Girijashanker, J. M. Reed, L. He, T. P. Dalton and D. W. Nebert, Biochem. Biophys. Res. Commun., 2008, 365, 814–820 CrossRef CAS PubMed.
- S. Antala and R. E. Dempski, Biochemistry, 2012, 51, 963–973 CrossRef CAS PubMed.
- G. Grass, S. Franke, N. Taudte, D. H. Nies, L. M. Kucharski, M. E. Maguire and C. Rensing, J. Bacteriol., 2005, 187, 1604–1611 CrossRef CAS PubMed.
- U. Krämer, I. N. Talke and M. Hanikenne, FEBS Lett., 2007, 581, 2263–2272 CrossRef PubMed.
- F. Long, C. C. Su, H. T. Lei, J. R. Bolla, S. V. Do and E. W. Yu, Philos. Trans. R. Soc., B, 2012, 367, 1047–1058 CrossRef CAS PubMed.
- E. Y. Valencia, V. S. Braz, C. Guzzo and M. V. Marques, BMC Microbiol., 2013, 13 Search PubMed.
- F. De Angelis, J. K. Lee, J. D. O'Connell, L. J. W. Miercke, K. H. Verschueren, V. Srinivasan, C. Bauvois, C. Govaerts, R. A. Robbins, J. M. Ruysschaert, R. M. Stroud and G. Vandenbussche, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 11038–11043 CrossRef CAS PubMed.
- J. E. Pak, E. N. Ekende, E. G. Kifle, J. D. O'Connell, F. De Angelis, M. B. Tessema, K. M. Derfoufi, Y. Robles-Colmenares, R. A. Robbins, E. Goormaghtigh, G. Vandenbussche and R. M. Stroud, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 18484–18489 CrossRef CAS PubMed.
- G. Dieppois, V. Ducret, O. Caille and K. Perron, PLoS One, 2012, 7, 12 Search PubMed.
- E. H. Kim, D. H. Nies, M. M. McEvoy and C. Rensing, J. Bacteriol., 2011, 193, 2381–2387 CrossRef CAS PubMed.
- P. Hinchliffe, M. F. Symmons, C. Hughes and V. Koronakis, Annu. Rev. Microbiol., 2013, 67, 221–242 CrossRef CAS PubMed.
- F. Long, C. C. Su, M. T. Zimmermann, S. E. Boyken, K. R. Rajashankar, R. L. Jernigan and E. W. Yu, Nature, 2010, 467, 484–488 CrossRef CAS PubMed.
- H. T. Lei, J. R. Bolla, N. R. Bishop, C. C. Su and E. W. Yu, J. Mol. Biol., 2014, 426, 403–411 CrossRef CAS PubMed.
- C. C. Su, F. Long, M. T. Zimmermann, K. R. Rajashankar, R. L. Jernigan and E. W. Yu, Nature, 2011, 470, 558–562 CrossRef CAS PubMed.
- S. Murakami, R. Nakashima, E. Yamashita and A. Yamaguchi, Nature, 2002, 419, 587–593 CrossRef CAS PubMed.
- A. J. Worlock and R. L. Smith, J. Bacteriol., 2002, 184, 4369–4373 CrossRef CAS.
- A. M. Caldwell and R. L. Smith, J. Bacteriol., 2003, 185, 374–376 CrossRef CAS.
- K. Tan, A. Sather, J. L. Robertson, S. Moy, B. Roux and A. Joachimiak, Protein Sci., 2009, 18, 2043–2052 CrossRef CAS PubMed.
- Q. Wan, M. F. Ahmad, J. Fairman, B. Gorzelle, M. de la Fuente, C. Dealwis and M. E. Maguire, Structure, 2011, 19, 700–710 CrossRef CAS PubMed.
- C. D. Plumptre, A. D. Ogunniyi and J. C. Paton, Trends Microbiol., 2012, 20, 485–493 CrossRef CAS PubMed.
- A. D. Ogunniyi, M. Grabowicz, L. K. Mahdi, J. Cook, D. L. Gordon, T. A. Sadlon and J. C. Paton, FASEB J., 2009, 23, 731–738 CrossRef CAS PubMed.
- A. Riboldi-Tunnicliffe, N. W. Isaacs and T. J. Mitchell, FEBS Lett., 2005, 579, 5353–5360 CrossRef CAS PubMed.
- C. K. Lim, K. A. Hassan, A. Penesyan, J. E. Loper and I. T. Paulsen, Environ. Microbiol., 2013, 15, 702–715 CrossRef CAS PubMed.
- M. I. Hood, B. L. Mortensen, J. L. Moore, Y. F. Zhang, T. E. Kehl-Fie, N. Sugitani, W. J. Chazin, R. M. Caprioli and E. P. Skaar, PLoS Pathog., 2012, 8, e1003068 CAS.
- M. Stork, M. P. Bos, I. Jongerius, N. de Kok, I. Schilders, V. E. Weynants, J. T. Poolman and J. Tommassen, PLoS Pathog., 2010, 6, e1000969 Search PubMed.
- M. C. Conejo, I. Garcia, L. Martinez-Martinez, L. Picabea and A. Pascual, Antimicrob. Agents Chemother., 2003, 47, 2313–2315 CrossRef CAS.
- J. P. Barnett, D. J. Scanlan and C. A. Blindauer, Metallomics, 2014, 6, 1254–1268 RSC.
- C. E. Haas, D. A. Rodionov, J. Kropat, D. Malasarn, S. S. Merchant and V. de Crecy-Lagard, BMC Genomics, 2009, 10 Search PubMed.
- M. Margoshes and B. L. Vallee, J. Am. Chem. Soc., 1957, 79, 4813–4814 CrossRef CAS.
- J. H. R. Kägi and B. L. Vallee, J. Biol. Chem., 1961, 236, 2435–2442 Search PubMed.
- G. Isani and E. Carpene, Biomolecules, 2014, 4, 435–457 CrossRef CAS PubMed.
- E. Freisinger, Dalton Trans., 2008, 6663–6675 RSC.
- E. Freisinger, JBIC, J. Biol. Inorg. Chem., 2011, 16, 1035–1045 CrossRef CAS PubMed.
- O. I. Leszczyszyn, H. T. Imam and C. A. Blindauer, Metallomics, 2013, 5, 1146–1169 RSC.
- X. Q. Wan and E. Freisinger, Metallomics, 2009, 1, 489–500 RSC.
- O. Schicht and E. Freisinger, Inorg. Chim. Acta, 2009, 362, 714–724 CrossRef CAS PubMed.
- B. Dolderer, H.-J. Hartmann and U. Weser, in Met. Ions Life Sci., ed. A. Sigel, H. Sigel and R. K. O. Sigel, RSC Publishing, Cambridge, UK, 2009, vol. 5, pp. 83–105 Search PubMed.
- C. A. Blindauer, in Met. Ions Life Sci., ed. A. Sigel, H. Sigel and R. K. O. Sigel, RSC Publishing, Cambridge, UK, 2009, vol. 5, pp. 51–81 Search PubMed.
- C. A. Blindauer, JBIC, J. Biol. Inorg. Chem., 2011, 16, 1011–1024 CrossRef CAS PubMed.
- C. A. Blindauer and O. I. Leszczyszyn, Nat. Prod. Rep., 2010, 27, 720–741 RSC.
- A. Krezel and W. Maret, J. Am. Chem. Soc., 2007, 129, 10911–10921 CrossRef CAS PubMed.
- M. A. Namdarghanbari, J. Meeusen, G. Bachowski, N. Giebel, J. Johnson and D. H. Petering, J. Inorg. Biochem., 2010, 104, 224–231 CrossRef CAS PubMed.
- A. Krezel and W. Maret, Biochem. J., 2007, 402, 551–558 CrossRef CAS PubMed.
- D. H. Petering, J. Y. Zhu, S. Krezoski, J. Meeusen, C. Kiekenbush, S. Krull, T. Specher and M. Dughish, Exp. Biol. Med., 2006, 231, 1528–1534 CAS.
- W. Braun, M. Vašák, A. H. Robbins, C. D. Stout, G. Wagner, J. H. R. Kägi and K. Wüthrich, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 10124–10128 CrossRef CAS.
- M. H. Frey, G. Wagner, M. Vašák, O. W. Sorensen, D. Neuhaus, E. Wörgötter, J. H. R. Kägi, R. R. Ernst and K. Wüthrich, J. Am. Chem. Soc., 1985, 107, 6847–6851 CrossRef CAS.
- M. Capdevila, R. Bofill, O. Palacios and S. Atrian, Coord. Chem. Rev., 2012, 256, 46–62 CrossRef CAS PubMed.
- Metallothioneins: Chemical and Biological Challenges, JBIC, J. Biol. Inorg. Chem., 2011, 16, 975–1134 CrossRef PubMed.
- Metallothioneins and Related Chelators, ed. A. Sigel, H. Sigel and R. K. O. Sigel, RSC Publishing, Cambridge, vol. 5 of Metal Ions Life Sci., 2009 Search PubMed.
- C. A. Blindauer, M. D. Harrison, J. A. Parkinson, A. K. Robinson, J. S. Cavet, N. J. Robinson and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 9593–9598 CrossRef CAS PubMed.
- C. A. Blindauer and P. J. Sadler, Acc. Chem. Res., 2005, 38, 62–69 CrossRef CAS PubMed.
- C. A. Blindauer, M. T. Razi, D. J. Campopiano and P. J. Sadler, JBIC, J. Biol. Inorg. Chem., 2007, 12, 393–405 CrossRef CAS PubMed.
- O. I. Leszczyszyn, C. D. Evans, S. E. Keiper, G. Z. L. Warren and C. A. Blindauer, Inorg. Chim. Acta, 2007, 360, 3–13 CrossRef CAS PubMed.
- C. A. Blindauer, J. Inorg. Biochem., 2008, 102, 507–521 CrossRef CAS PubMed.
- C. A. Blindauer, M. D. Harrison, A. K. Robinson, J. A. Parkinson, P. W. Bowness, P. J. Sadler and N. J. Robinson, Mol. Microbiol., 2002, 45, 1421–1432 CrossRef CAS.
- C. A. Blindauer, N. C. Polfer, S. E. Keiper, M. D. Harrison, N. J. Robinson, P. R. R. Langridge-Smith and P. J. Sadler, J. Am. Chem. Soc., 2003, 125, 3226–3227 CrossRef CAS PubMed.
- C. A. Blindauer, M. D. Harrison, J. A. Parkinson, N. J. Robinson and P. J. Sadler, in 10th International Symposium on Metal Ions in Biology and Medicine, ed. P. Collery, I. Maymard, T. Theophanides, L. Khassanova and T. Collery, John Libby Eurotext, 2008, vol. 10, pp. 167–173.
- E. A. Peroza and E. Freisinger, JBIC, J. Biol. Inorg. Chem., 2007, 12, 377–391 CrossRef CAS PubMed.
- O. I. Leszczyszyn, R. Schmid and C. A. Blindauer, Proteins: Struct., Funct., Bioinf., 2007, 68, 922–935 CrossRef CAS PubMed.
- E. A. Peroza, R. Schmucki, P. Güntert, E. Freisinger and O. Zerbe, J. Mol. Biol., 2009, 387, 207–218 CrossRef CAS PubMed.
- O. I. Leszczyszyn, C. R. J. White and C. A. Blindauer, Mol. BioSyst., 2010, 6, 1592–1603 RSC.
- E. A. Peroza, A. Al Kaabi, W. Meyer-Klaucke, G. Wellenreuther and E. Freisinger, J. Inorg. Biochem., 2009, 103, 342–353 CrossRef CAS PubMed.
- J. Loebus, E. A. Peroza, N. Bluthgen, T. Fox, W. Meyer-Klaucke, O. Zerbe and E. Freisinger, JBIC, J. Biol. Inorg. Chem., 2011, 16, 683–694 CrossRef CAS PubMed.
- K. Tarasava, S. Johannsen and E. Freisinger, Molecules, 2013, 18, 14414–14429 CrossRef PubMed.
- E. A. Peroza, A. D. Cabral, X. Q. Wan and E. Freisinger, Metallomics, 2013, 5, 1204–1214 RSC.
- R. Bofill, R. Orihuela, M. Romagosa, J. Domenech, S. Atrian and M. Capdevila, FEBS J., 2009, 276, 7040–7069 CrossRef CAS PubMed.
- S. Zeitoun-Ghandour, J. M. Charnock, M. E. Hodson, O. I. Leszczyszyn, C. A. Blindauer and S. R. Stürzenbaum, FEBS J., 2010, 277, 2531–2542 CrossRef CAS PubMed.
- O. I. Leszczyszyn, S. Zeitoun-Ghandour, S. R. Stürzenbaum and C. A. Blindauer, Chem. Commun., 2011, 47, 448–450 RSC.
- O. I. Leszczyszyn and C. A. Blindauer, Phys. Chem. Chem. Phys., 2010, 12, 13408–13418 RSC.
- O. Palacios, S. Atrian and M. Capdevila, JBIC, J. Biol. Inorg. Chem., 2011, 16, 991–1009 CrossRef CAS PubMed.
- R. Bofill, M. Capdevila and S. Atrian, Metallomics, 2009, 1, 229–234 RSC.
- M. Capdevila, J. Domenech, A. Pagani, L. Tio, L. Villarreal and S. Atrian, Angew. Chem., Int. Ed., 2005, 44, 4618–4622 CrossRef CAS PubMed.
- T. Huber and E. Freisinger, Dalton Trans., 2013, 42, 8878–8889 RSC.
- K. E. R. Duncan and M. J. Stillman, J. Inorg. Biochem., 2006, 100, 2101–2107 CrossRef PubMed.
- K. E. Rigby Duncan, C. W. Kirby and M. J. Stillman, FEBS J., 2008, 275, 2227–2239 CrossRef PubMed.
- J. Zeng, B. L. Vallee and J. H. R. Kägi, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 9984–9988 CrossRef CAS.
- M. Huang, C. F. Shaw and D. H. Petering, J. Inorg. Biochem., 2004, 98, 639–648 CrossRef CAS PubMed.
- J. Ejnik, A. Munoz, T. Gan, C. F. Shaw and D. H. Petering, JBIC, J. Biol. Inorg. Chem., 1999, 4, 784–790 CrossRef CAS.
- J. Zaia, D. Fabris, D. Wei, R. L. Karpel and C. Fenselau, Protein Sci., 1998, 7, 2398–2404 CrossRef CAS PubMed.
- C. Jacob, W. Maret and B. L. Vallee, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 3489–3494 CrossRef CAS.
- W. Feng, J. Cai, W. M. Pierce, R. B. Franklin, W. Maret, F. W. Benz and Y. J. Kang, Biochem. Biophys. Res. Commun., 2005, 332, 853–858 CrossRef CAS PubMed.
- W. Maret, J. Nutr., 2000, 130, 1455S–1458S CAS.
- S. Zeitoun-Ghandour, O. I. Leszczyszyn, C. A. Blindauer, F. M. Geier, J. G. Bundy and S. R. Stürzenbaum, Mol. BioSyst., 2011, 7, 2397–2406 RSC.
- A. Krezel and W. G. Maret, JBIC, J. Biol. Inorg. Chem., 2008, 13, 401–409 CrossRef CAS PubMed.
- P. Palumaa, I. Tammiste, K. Kruusel, L. Kangur, H. Jornvall and R. Sillard, Biochim. Biophys. Acta, Proteins Proteomics, 2005, 1747, 205–211 CrossRef CAS PubMed.
- E. Artells, O. Palacios, M. Capdevila and S. Atrian, FEBS J., 2014, 281, 1659–1678 CrossRef CAS PubMed.
- K. L. Summers, D. E. K. Sutherland and M. J. Stillman, Biochemistry, 2013, 52, 2461–2471 CrossRef CAS PubMed.
- D. E. K. Sutherland, K. L. Summers and M. J. Stillman, Biochemistry, 2012, 51, 6690–6700 CrossRef CAS PubMed.
- J. Ejnik, C. F. Shaw and D. H. Petering, Inorg. Chem., 2010, 49, 6525–6534 CrossRef CAS PubMed.
- G. Meloni, P. Faller and M. Vašák, J. Biol. Chem., 2007, 282, 16068–16078 CrossRef CAS PubMed.
- G. Meloni, V. Sonois, T. Delaine, L. Guilloreau, A. Gillet, J. Teissie, P. Faller and M. Vašák, Nat. Chem. Biol., 2008, 4, 366–372 CrossRef CAS PubMed.
- J. T. Pedersen, C. Hureau, L. Hemmingsen, N. H. H. Heegaard, J. Ostergaard, M. Vašák and P. Faller, Biochemistry, 2012, 51, 1697–1706 CrossRef CAS PubMed.
- J. Durand, G. Meloni, C. Talmard, M. Vašák and P. Faller, Metallomics, 2010, 2, 741–744 RSC.
- Y. Manso, J. Carrasco, G. Comes, G. Meloni, P. A. Adlard, A. I. Bush, M. Vašák and J. Hidalgo, Cell. Mol. Life Sci., 2012, 69, 3683–3700 CrossRef CAS PubMed.
- G. Meloni and M. Vašák, Free Radical Biol. Med., 2011, 50, 1471–1479 CrossRef CAS PubMed.
- G. Meloni, A. Crameri, G. Fritz, P. Davies, D. R. Brown, P. M. H. Kroneck and M. Vašák, ChemBioChem, 2012, 13, 1261–1265 CrossRef CAS PubMed.
- S. E. Gabriel, F. Miyagi, A. Gaballa and J. D. Helmann, J. Bacteriol., 2008, 190, 3482–3488 CrossRef CAS PubMed.
- M. A. Saito, E. M. Bertrand, S. Dutkiewicz, V. V. Bulygin, D. M. Moran, F. M. Monteiro, M. J. Follows, F. W. Valois and J. B. Waterbury, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2184–2189 CrossRef CAS PubMed.
- C. E. Blaby-Haas, J. A. Flood, V. de Crecy-Lagard and D. B. Zamble, Metallomics, 2012, 4, 488–497 RSC.
- A. M. Sydor, M. Jost, K. S. Ryan, K. E. Turo, C. D. Douglas, C. L. Drennan and D. B. Zamble, Biochemistry, 2013, 52, 1788–1801 CrossRef CAS.
- C. Appia-Ayme, A. Hall, E. Patrick, S. Rajadurai, T. A. Clarke and G. Rowley, Biochem. J., 2012, 442, 85–93 CrossRef CAS PubMed.
- M. Noll, K. Petrukhin and S. Lutsenko, J. Biol. Chem., 1998, 273, 21393–21401 CrossRef CAS PubMed.
- A. M. Sevcenco, M. W. H. Pinkse, H. T. Wolterbeek, P. Verhaert, W. R. Hagen and P. L. Hagedoorn, Metallomics, 2011, 3, 1324–1330 RSC.
- E. V. Filippova, G. Minasov, L. Shuvalova, J. Winsor, I. Dubrovska, L. Papazisi and W. F. Anderson, pdb 3lay, released 19/01/2010.
- M. P. Hensley, T. S. Gunasekera, J. A. Easton, T. K. Sigdel, S. A. Sugarbaker, L. Klingbeil, R. M. Breece, D. L. Tierney and M. W. Crowder, J. Inorg. Biochem., 2012, 111, 164–172 CrossRef CAS PubMed.
- P. Petrarca, S. Ammendola, P. Pasquali and A. Battistoni, J. Bacteriol., 2010, 192, 1553–1564 CrossRef CAS PubMed.
- G. David, K. Blondeau, M. Schiltz, S. Penel and A. Lewit-Bentley, J. Biol. Chem., 2003, 278, 43728–43735 CrossRef CAS PubMed.
- S. Eswaramoorthy, S. Swaminathan and S. K. Burley, New York SGX Research Center for Structural Genomics, pdb 1txl, released 20/07/2004.
- C. A. Blindauer, J. Inorg. Biochem., 2013, 121, 145–155 CrossRef CAS PubMed.
- U. Heinz, L. Hemmingsen, M. Kiefer and H. W. Adolph, Chem. – Eur. J., 2009, 15, 7350–7358 CrossRef CAS PubMed.
- H. Kozlowski, S. Potocki, M. Remelli, M. Rowinska-Zyrek and D. Valensin, Coord. Chem. Rev., 2013, 257, 2625–2638 CrossRef CAS PubMed.
- T. F. Cheng, W. Xia, P. W. Wang, F. J. Huang, J. W. Wang and H. Z. Sun, Metallomics, 2013, 5, 1423–1429 RSC.
- N. H. Joh, T. Wang, M. P. Bhate, R. Acharya, Y. Wu, M. Grabe, M. Hong, G. Grigoryan and W. F. DeGrado, Science, 2014, 346, 1520–1524 CrossRef CAS PubMed.
- J. W. Meeusen, H. Tomasiewicz, A. Nowakowski and D. H. Petering, Inorg. Chem., 2011, 50, 7563–7573 CrossRef CAS PubMed.
- A. B. Nowakowski and D. H. Petering, Inorg. Chem., 2011, 50, 10124–10133 CrossRef CAS PubMed.
- A. Staszewska, E. Kurowska and W. Bal, Metallomics, 2013, 5, 1483–1490 RSC.
- J. W. Meeusen, A. Nowakowski and D. H. Petering, Inorg. Chem., 2012, 51, 3625–3632 CrossRef CAS PubMed.
- A. D. McNaught and A. Wilkinson, IUPAC, Compendium of Chemical Terminology, Blackwell Scientific Publications, Oxford, 2nd edn (the “Gold Book”), 1997 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Tables S1 and S2. See DOI: 10.1039/c4cc10174j |
| ‡ Frequently misquoted in current literature, zinc is strictly speaking not a transition metal, as the only ionic form occurring in nature, Zn2+, does still have a completely filled 3d shell.266 |
| § Previously known as Ralstonia metallidurans, and Alcaligenes eutrophus. |
| ¶ Note that the “ZIP” abbreviation in these two families is coincidental. |
| || In its narrower sense, the “RND” label refers to the inner-membrane-associated component of the tripartite pumps only. |
| This journal is © The Royal Society of Chemistry 2015 |