
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ring-opening copolymerization (ROCOP): synthesis and properties of polyesters and polycarbonates
Shyeni
Paul†
,
Yunqing
Zhu†
,
Charles
Romain
,
Rachel
Brooks
,
Prabhjot K.
Saini
and
Charlotte K.
Williams
*
Dept. Chemistry, Imperial College London, London SW7 2AZ, UK. E-mail: c.k.williams@imperial.ac.uk
First published on 17th February 2015
Abstract
Controlled routes to prepare polyesters and polycarbonates are of interest due to the widespread application of these materials and the opportunities provided to prepare new copolymers. Furthermore, ring-opening copolymerization may enable new poly(ester–carbonate) materials to be prepared which are inaccessible using alternative polymerizations. This review highlights recent advances in the ring-opening copolymerization catalysis, using epoxides coupled with anhydrides or CO2, to produce polyesters and polycarbonates. In particular, the structures and performances of various homogeneous catalysts are presented for the epoxide–anhydride copolymerization. The properties of the resultant polyesters and polycarbonates are presented and future opportunities highlighted for developments of both the materials and catalysts.
 Back (left-right): Yunqing Zhu and Charles Romain Front (left to right): Rachel Brooks, Shyeni Paul, Charlotte K. Williams and Prabhjot K. Saini | Yunqing Zhu graduated with a masters degree in Material Science at Tongji University, in Shanghai, under the supervision of Prof. Jianzhong Du (2013). His research focused on the self-assembly behavior of amphiphilic polymers, their stimuli-responsive behaviour and the research of polymer vesicles with phase segregated membrane structure. He has been awarded an Imperial-CSC scholarship (2013–2016) and his PhD research focuses on the design and synthesis of functional block copolyesters, using a combination of ring-opening polymerization and ring-opening copolymerization methods. |
Dr Charles Romain graduated from the Université de Rennes 1 with an MSc in Chemistry and an MSc Engineering from Ecole Nationale Supérieure de Chimie de Rennes (ENSCR) (2008). Charles obtained a PhD from Université de Strasbourg, working under the supervision of Dr Samuel Dagorne and Dr Stéphane Bellemin-Laponnaz (2011). His research concerned the synthesis and reactivity of Group 4 metal complexes, bearing NHC ligands, for the ring-opening polymerization of cyclic polar monomers. He joined the Williams research group in 2012, working on the synthesis of new catalysts for ROCOP (CO2–epoxide and epoxide–anhydride) and developing the new switch catalysis. |
Rachel Brooks graduated with an MSci in Natural Sciences from Durham University (2012) and an MRes in Green Chemistry from Imperial College London (2013). Since 2013, she has been researching, for her PhD, the copolymerization of carbon dioxide and plant-based monomers. |
Shyeni Paul graduated with an MSci in Chemistry from Imperial College London (2013). Since 2013, she has been researching, for her PhD, new catalysts for the copolymerisation of epoxides and CO2. |
Prof. Charlotte K. Williams graduated with a BSc in Chemistry (1998) and a PhD (2001) from Imperial College London, working with Prof. Vernon Gibson and Prof. Nick Long on new catalysts for ethylene polymerization. She carried out postdoctoral research at the University of Minnesota, working with Professors William B. Tolman and Marc Hillmyer, on new catalysts for lactide polymerization (2001). She also worked as a postdoctoral researcher with Professors Andrew Holmes and Richard Friend, at Cambridge University, on new metallo-polymer composites for light emitting devices (2002). Charlotte returned to Imperial College London in 2003 and was awarded an EPSRC Advanced Research Fellowship (2005–2010). Her research interests are in polymerization catalysis and polymer chemistry. In particular, her group have investigated new catalysts for lactide polymerization, carbon dioxide copolymerization and combined switch catalysis. Her work has been recognised by awards from the RSC including the Laurie Vergnano Award, the Meldola Medal and the Energy, Environment and Sustainability Early Career Award. |
Dr Prabhjot K. Saini graduated with an MSci in Chemistry from Imperial College London (2011). Since 2011 she has been researching, for her PhD, new dinuclear metal catalysts for the synthesis of oxygenated polymers and just recently defended her thesis (2015). She is now working as a research associate within the Williams research group. |
1. Introduction
Polyesters and polycarbonates are amongst the most widely applied oxygenated polymers. The majority of commercial materials are prepared by condensation polymerizations and include well-known polymers such as poly(ethylene terephthalate) (PET) and polycarbonate (PC) (from bisphenol A). These materials are found in applications spanning packaging, fibres, rigid plastics and engineering materials.Currently, aliphatic polyesters and polycarbonates have fewer large scale commercial opportunities. This is partly due to their physical and chemical properties which are typically lower compared to materials containing aromatic/rigid functionalities in the polymer backbone. One interesting aliphatic polyester, produced on a several hundred thousand tonnes per annum scale, is poly(lactic acid) (PLA); this material is attracting considerable attention as a renewable and, in some cases, degradable alternative polymer for packaging, fibres, medical sutures/stents and in controlled release of active compounds.1 Another promising class of aliphatic polyesters are the poly(hydroxyl alkanoates) (PHAs), once again applied for both commodity and niche medical applications.2 Aliphatic polycarbonates prepared by the ring-opening copolymerization of epoxides and carbon dioxide are also attracting considerable attention, including commercialization ventures operating at pilot scale.3 The polymerization can be controlled to enable the production of either low molar mass polycarbonates or poly(ether carbonates), which are subsequently applied as the polyol portion in polyurethane synthesis.4 These materials are desirable as potential substitutes for conventional petrochemical polyols. Indeed, a recent detailed life cycle analysis shows that these materials consume ∼20% less petrochemical raw materials and reduce carbon dioxide emissions by approximately 20% compared to the use of conventional petrochemicals.5
Another topical area is the development of ‘controlled’ polymerizations, which in this context refers to polymerizations resulting in the precise control of polymer molar mass (typically with narrow dispersity), composition, architecture and end-group functionality.6 There is fundamental academic interest in understanding and developing such routes however, they are also essential for the production of (multi-) block copolymers and other sophisticated polymers.6b–k There are several controlled syntheses of aliphatic polyesters and polycarbonates, two of the most common are the ring-opening polymerization (ROP) of cyclic esters–carbonates and the ring-opening copolymerization (ROCOP) of epoxides and anhydride–carbon dioxide (Fig. 1). An alternative method, ring-opening polycondensation, to produce polyesters has been developed by Takasu and co-workers. However, the broad dispersity (ĐM > 2.0) indicates that the polymerization is not well controlled.7
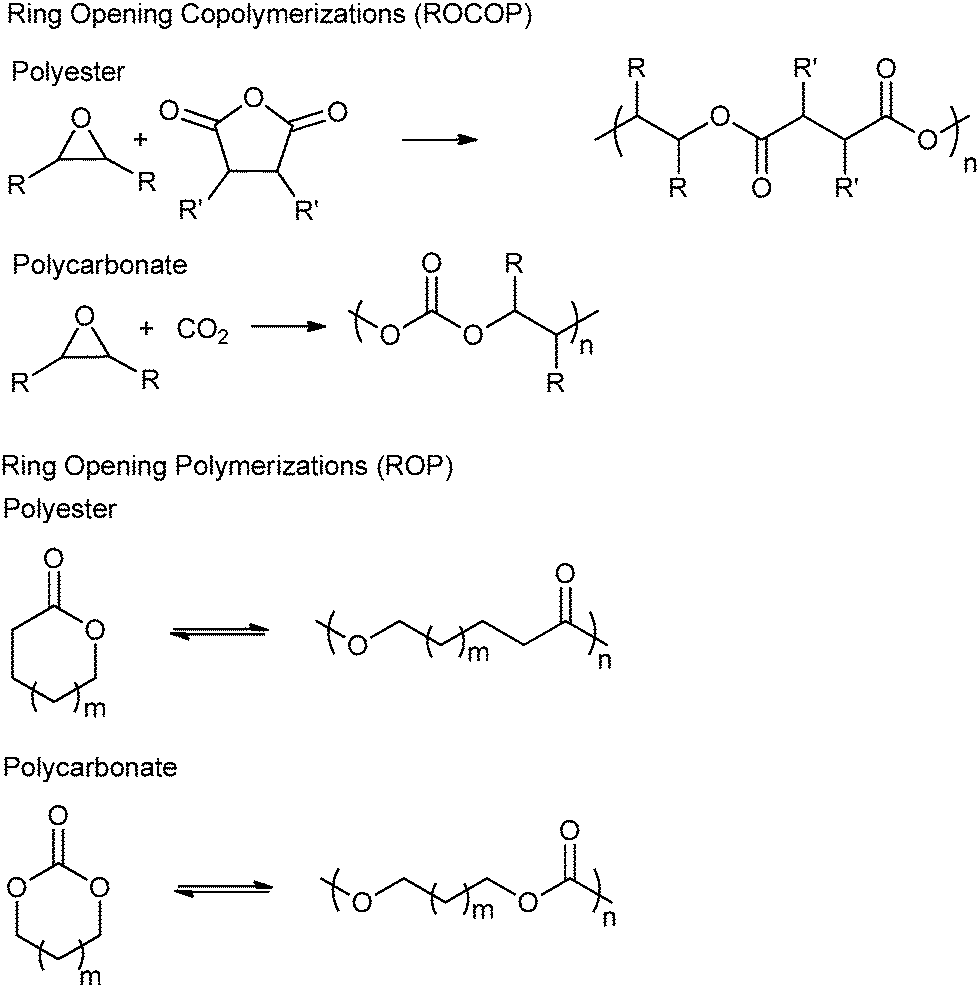 | ||
Fig. 1 Illustrates the generic reactions occurring during ring-opening copolymerization (ROCOP) and ring-opening polymerization (ROP) to prepare polyesters and polycarbonates. For the epoxides: where there is only one R group: R = alkyl, e.g. Me, Ph; CH2Cl, CH2OBn, and the other R group = H. Where both R groups are the same: R = cyclohexylene, cyclopentylene, naphthalene. For the anhydrides: where there is only one R′ group: R′ = Me, Ph, ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2, etc. and the other R′ = H. Where both R′ are the same: R′ = absent (maleic), H (succinic), phenylene, cyclohexylene. CH2, etc. and the other R′ = H. Where both R′ are the same: R′ = absent (maleic), H (succinic), phenylene, cyclohexylene. | ||
So far there have been a significant number of comprehensive reviews of ring-opening polymerization of lactones/cyclic carbonates, addressing areas spanning catalysis, new monomers, properties and applications for polyesters and polycarbonates.1,6d,8 Although it is an excellent controlled polymerization routes for controlled polymerization applications, there are opportunities to broaden the range materials produced, and to overcome some of the limitations of the ROP method. In contrast to the bright lights shining on ROP, the sister polymerization method of ROCOP has received far less attention. One attraction of ROCOP is that the properties of the resulting materials can be easily manipulated by facile substitution of just one of the monomers: for example, switching the epoxide from propylene oxide to cyclohexene oxide, using the same ROCOP catalysts, it is possible to moderate the polycarbonate glass transition temperatures over the range 33–123 °C.9 This facility to substitute different epoxides, using the same/very similar catalysts and conditions, stands in stark contrast to ROP methodologies, where changing the polymer repeating unit requires the preparation of new, functionalized lactones/cyclic carbonates.8b The synthesis of such functionalized monomers can be a very challenging undertaking, often requiring multiple steps.8b Furthermore, there are no guarantees that changing the lactone/cyclic carbonate will enable polymerization to occur at all, not least because this often reduces the ring strain, which is the thermodynamic driving force for polymerization.8b In addition, using ROP certain chemistries and repeating units are very difficult, and in some cases currently impossible, to incorporate into the polymer backbone. This is due to both thermodynamic limitations and the difficulty in preparing suitable lactones. One striking problem area for ROP is the production of polymers containing aromatic groups in the polymer backbone, a desirable goal to increase thermal and mechanical properties. ROCOP can overcome this limitation, in particular, it represents an attractive means to prepare semi-aromatic polyesters–polycarbonates.10 A further advantage of ROCOP is the common availability of the epoxide and anhydride (co)monomers. Many are commercially produced, some on a large scale, or they are relatively straightforward to prepare from olefins or di-carboxylic acids, respectively.
This review will focus on the application of ring-opening copolymerization (ROCOP) to prepare aliphatic polyesters and polycarbonates. The ring-opening copolymerization of epoxides–anhydrides will be examined and the range of different catalysts for this transformation highlighted. In the field of epoxide–CO2 copolymerization, there are already quite a range of reviews of different catalysts to which the reader is referred for more specific information.6a,9,11 Furthermore, a recent review of stereocontrolled epoxide polymerizations and copolymerizations provides a complementary perspective.12
The first part of this review focuses on describing catalysts for epoxide–anhydride ROCOP, an area not yet comprehensively reviewed, so as to enable efficient polyester synthesis and on recent discoveries enabling the efficient production of copoly(ester–carbonates). The second part, will describe the properties and performances of selected polyesters and polycarbonates, produced by ROCOP. This section seeks to illustrate both the range of materials already prepared, the possible chemistries and functionalities and to highlight areas in which future developments are expected. In particular, the emphasis will be on the influence that the polymer composition (epoxide–anhydride selection), tacticity and functional groups exert over the macroscopic properties. All the copolymerizations described in this review use epoxides as a co-monomer. Therefore, the reader may find Fig. 2 informative, as it illustrates the structures and abbreviations used for commonly applied epoxides and anhydrides.
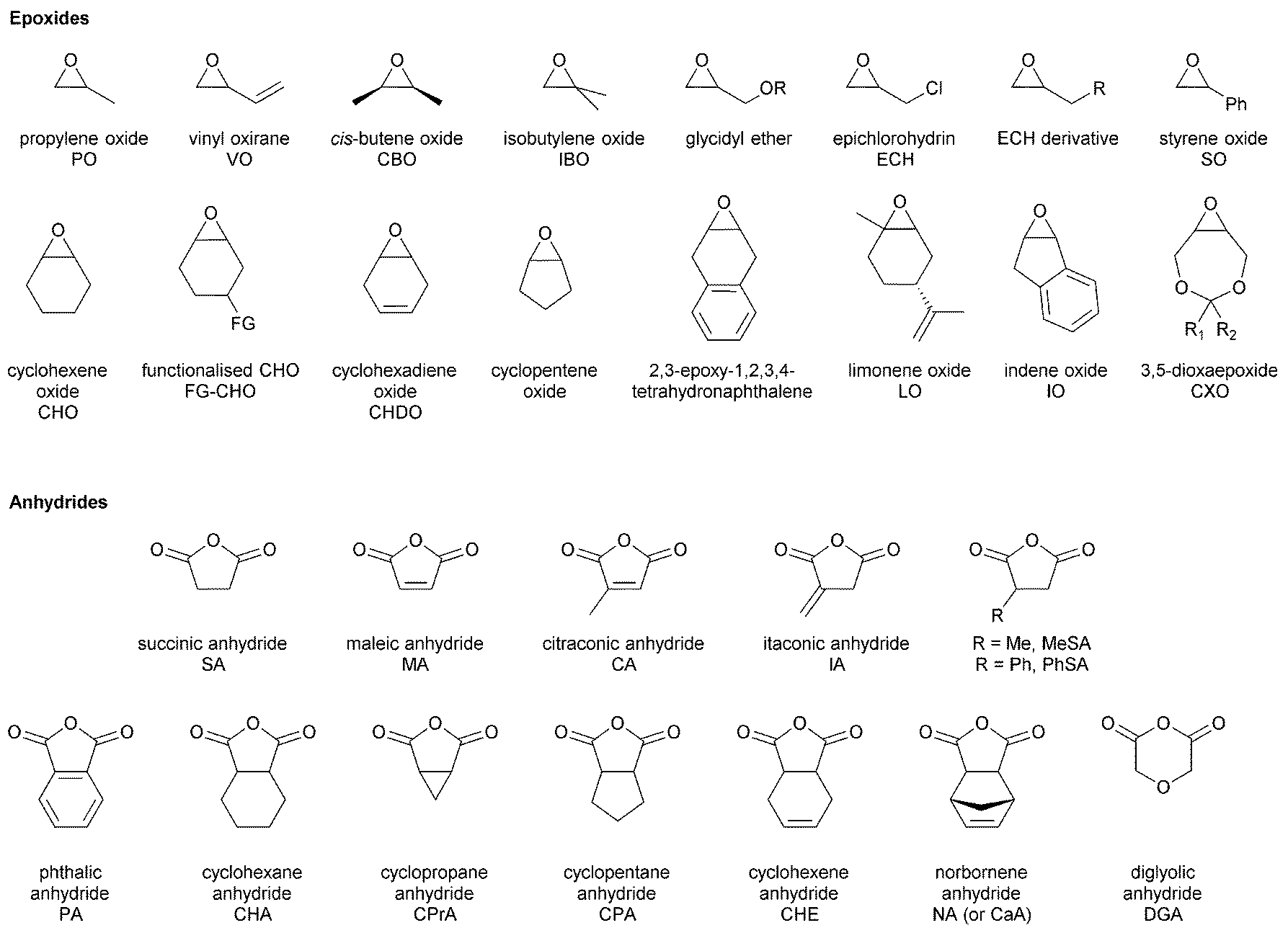 | ||
| Fig. 2 Shows the structures of commonly applied epoxides and anhydrides. | ||
2. Ring-opening copolymerization of epoxides and anhydrides: catalysis
2.1 Polymerization pathway
Ring-opening copolymerization reactions are used to produce polyesters and/or polycarbonates. Such polymerizations require the application of a ‘catalyst’ or, more accurately, an initiator. This species is often a single site metal complex of the form LMX, where L is an ancillary ligand, M is the metal site at which catalysis occurs and X is the initiating group and site at which propagation proceeds. Fig. 3 illustrates the generic elementary reactions which are proposed to occur during epoxide–anhydride ROCOP.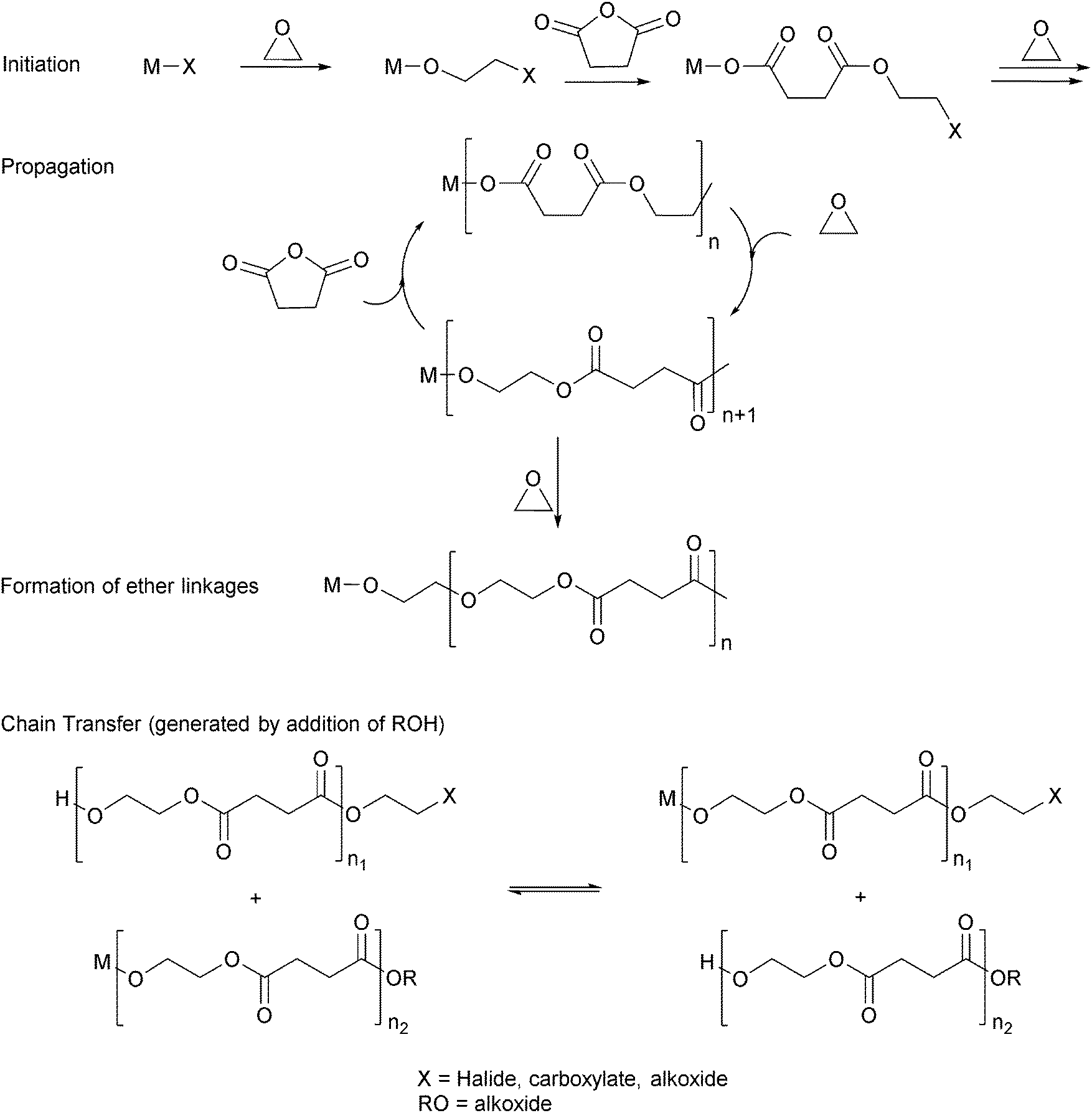 | ||
| Fig. 3 Illustrates the elementary steps occurring during epoxide–anhydride ring-opening copolymerizations (ROCOP). | ||
The initiation reactions involve a reaction between the MX initiator and the monomers, to generate metal alkoxide–carboxylate intermediates. The initiating group is commonly a carboxylate, alkoxide or halide group and, during controlled polymerization, this group becomes one of the chain end groups. The propagation reactions occur as monomers are sequentially enchained, this involves sequential formation of metal-alkoxide and carboxylate intermediates. The metal alkoxide intermediate attacks the anhydride to generate the metal carboxylate intermediate. The metal carboxylate intermediate attacks and ring-opens the epoxide co-monomer to (re-)generate the meal alkoxide. For this class of polymerizations, it is also important to consider chain transfer reactions. These are reactions in which the growing polymer chain equilibrates with added protic compounds, for example alcohols. The chain transfer reactions are typically assumed to occur more rapidly than propagation reactions; a hypothesis supported by the ability upon the addition of protic compounds, to control the molar mass and to narrow the dispersity of the polymers. Chain transfer reactions can be highly beneficial as they can be used to manipulate selectivity for a particular polymer end-group/molar mass. Other side reactions include sequential epoxide enchainment, leading to ether linkages. The termination of polymerization is frequently achieved by manipulating the conditions (reducing temperature, monomer removal) or by addition of water or acids.
Key parameters to consider when selecting a ROCOP catalyst include its productivity (often measured by its turn-over number, TON), activity (usually assessed as a turn over frequency, TOF), selectivity (against ether linkages), control of molar mass (and dispersity) and in some cases, control of regio- and stereochemistry during monomer enchainment.
2.2 Early catalyst discoveries
The first reports of the alternating copolymerization of epoxides and anhydrides were made in the 1960s.13 A range of catalyst systems including inorganic salts, tertiary amines or metal-alkyl initiators were reported.14 However, these copolymerizations were hampered by poor performance characteristics including low levels of polymerization control, poor activity and low molar mass products; additionally, in some cases, the catalysts exhibited poor selectivity resulting in significant quantities of ether linkages. In the mid-1960s, Inoue et al., investigated the preparation of polyesters using organometallic initiators.14b,c,f Thus, several metal alkyl initiators, together with added alcohol or water, were applied to epoxide (PO, ECH)–anhydride (PA, Carbic Anhydride) copolymerizations. An important mechanistic proposal, for PA–CHO ROCOP initiated by dialkyl zinc, was that polymerization proceeded by sequential formation of zinc-carboxylate and zinc-alkoxide species.14f In 1973, Hsieh et al. investigated the terpolymerization of epoxides, oxiranes and anhydrides, initiated by tri(alkyl)aluminium complexes.14g The resulting polymers consisted of alternating ABC blocks featuring ether–ester–ester linkages. In 1980, Kuran et al. reported MA–PO copolymerizations, using a series of organozinc catalysts of general formula RZnEt (R = alkoxide, aryloxide, carboxylate).14h Polyesters containing only 53 mol% incorporation of PO and of low molar mass (Mn < 2000 g mol−1) were produced. Although these early discoveries laid the ground work for future catalyst developments, the catalysts were lacked precise control and an inability to accurately define the catalyst structure.2.3 Well-defined metal catalysts
The next major series of catalyst developments involved the preparation of well-defined metal complexes, via the use of ancillary ligands to control/minimise aggregation reactions. These single site species include catalyst systems which comprise both a (transition) metal complex and added co-catalyst (typically at a loading of 1–5 eq. vs. catalyst). Common co-catalysts include inorganic salts, such as ammonium or phosphonium halides, or Lewis bases, such as methyl imidazole or dimethylaminopyridine (DMAP). The precise role for the co-catalyst remains rather complex, but it is generally proposed that either the Lewis base or the anion coordinates to the metal catalyst, resulting in the labilizing of the initiating group, X or the propagating polymer chain (which is coordinated in a trans-position) and accelerating polymerization (Fig. 3).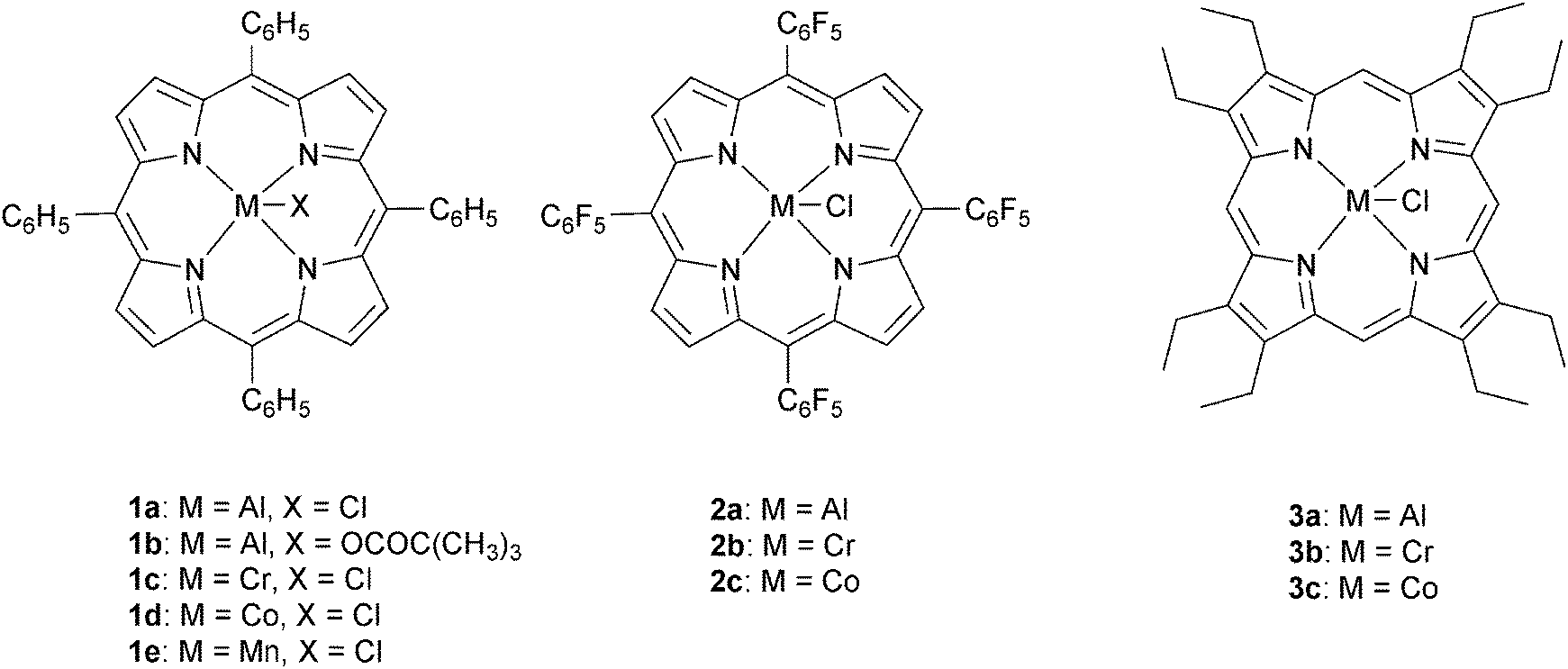 | ||
| Fig. 4 Illustrates the structures of various metalloporphyrin initiators. | ||
Recently, Chisholm and co-workers have extensively investigated a range of metal (Al(III), Cr(III), Co(III)) porphyrins for PO–anhydride (SA, MeSa, PhSA, MA, PA) copolymerizations.18 Comparing the metals, for MeSA–PO copolymerization, revealed that Cr(III) catalysts were significantly more active (TOF = 52 h−1) than Co(III) and Al(III) analogues (TOF = 19 h−1 and 25 h−1, respectively). Furthermore, the selectivity for ether–ester linkages was controlled by the quantity of co-catalyst and the ratio of monomers SO/MeSA. In addition, the chromium porphyrinato complexes were less affected by modifications to the porphyrin ligand than Al(III) and Co(III) counterparts, however in all cases the tetra(phenyl)porphyrin ligand was the best. The porphyrin substituents did affect the regioselectivity for PO–SA copolymerization, with the order being 1a > 2a > 3a and 2b > 3b > 1c for the aluminium and cobalt complexes, respectively.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1), perfectly alternating, polyesters.10g The ligands’ backbone and aryl substituents affected their stability and activity in polymerization, in common with previous observations for epoxide–CO2 copolymerization.19e Complexes with R3 = CN (4b, 4d, 4e in Fig. 5) were among the fastest and most tolerant.10g The use of 4d for CHO–DGA copolymerization (TOF = 79 h−1) afforded alternating polyesters with high molar masses (Mn = 23000 g mol−1) and narrow distributions (ĐM = 1.2). In addition, a wide range of epoxides–anhydrides were investigated leading, in some cases, to unsaturated polyesters. These polyesters showed some promising features, including relatively high decomposition temperatures (∼290 °C) and moderate glass transition temperatures (50–60 °C). The (BDI)ZnOAc complex shows low activity (TOF < 1 h−1) for MA–PO copolymerization to afford polymers with high ether linkages (86%).17
000 g mol−1), perfectly alternating, polyesters.10g The ligands’ backbone and aryl substituents affected their stability and activity in polymerization, in common with previous observations for epoxide–CO2 copolymerization.19e Complexes with R3 = CN (4b, 4d, 4e in Fig. 5) were among the fastest and most tolerant.10g The use of 4d for CHO–DGA copolymerization (TOF = 79 h−1) afforded alternating polyesters with high molar masses (Mn = 23000 g mol−1) and narrow distributions (ĐM = 1.2). In addition, a wide range of epoxides–anhydrides were investigated leading, in some cases, to unsaturated polyesters. These polyesters showed some promising features, including relatively high decomposition temperatures (∼290 °C) and moderate glass transition temperatures (50–60 °C). The (BDI)ZnOAc complex shows low activity (TOF < 1 h−1) for MA–PO copolymerization to afford polymers with high ether linkages (86%).17
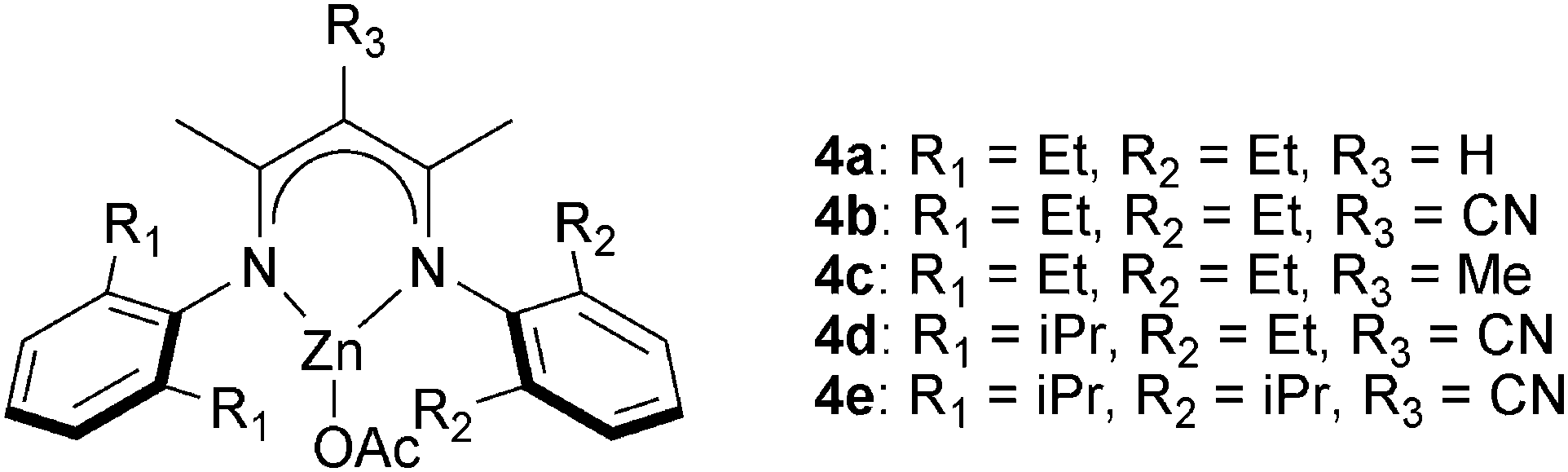 | ||
| Fig. 5 Illustrates the structures of β-diiminate zinc catalysts.10g | ||
000 g mol−1, 6a and 6b, respectively). The PPM was then successfully converted to poly(propylene fumarate) PPF by controlled cis–trans isomerization in the presence of dimethylamine. Complex 6b was applied to a range of epoxide–MA copolymerizations (Fig. 6), always affording perfectly alternating polyesters (<1% ether linkages) with high molar masses (21000 < Mn < 31000 g mol−1), reasonably narrow distributions (ĐM < 1.7) and moderate activities (11 h−1 < TOF < 50 h−1). By applying a range of epoxides, the polyester glass transition temperatures could be controlled from −29 to 50 °C.
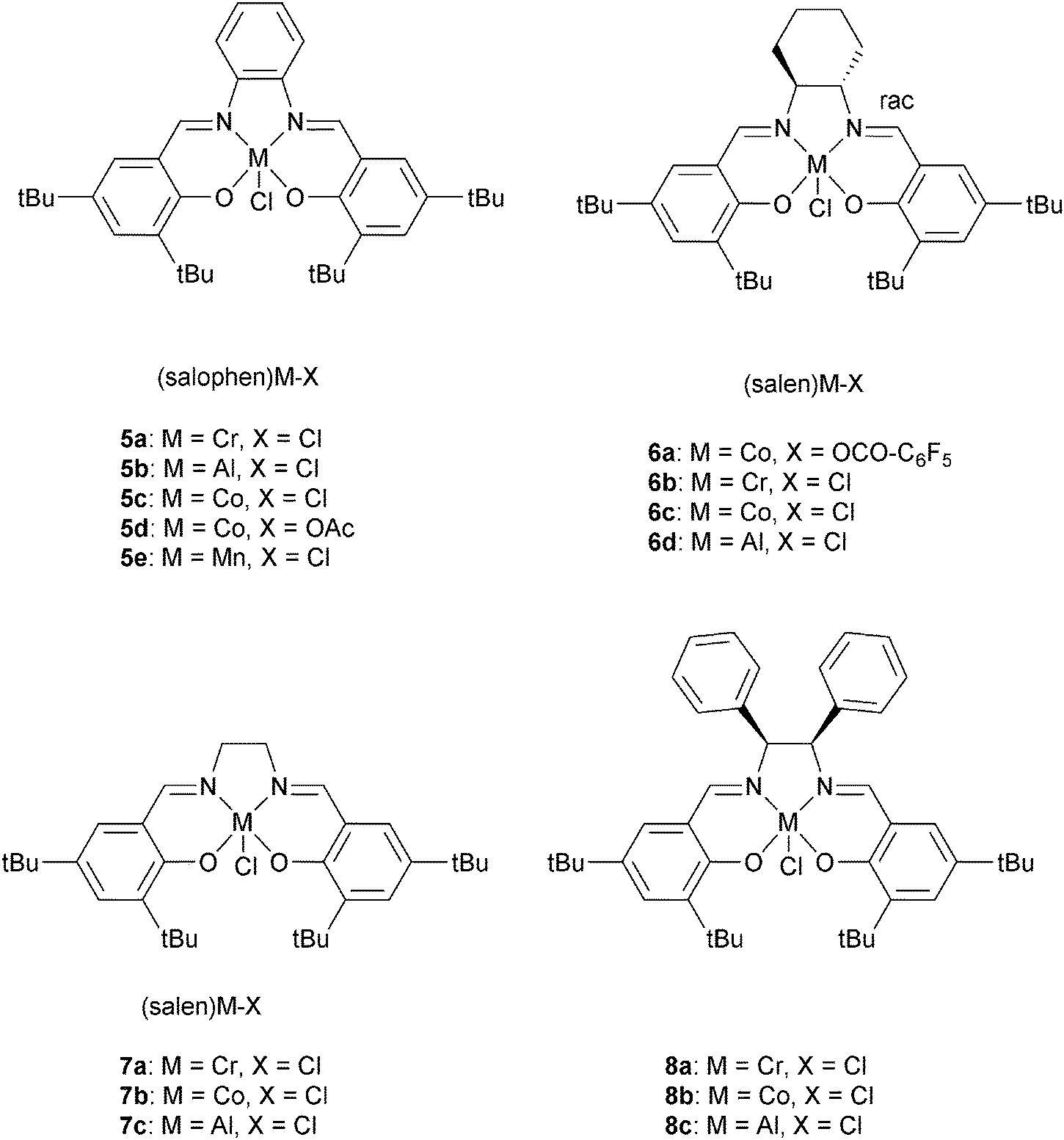 | ||
| Fig. 6 Shows the structures of various metal salen and salophen complexes. | ||
Darensbourg and co-workers also investigated 6a, with various onium salts as the co-catalysts, for epoxide–anhydride copolymerization.21 They found that 6b on its own is inactive for CHO–PA copolymerization. Using 6b, PPN+X− (X = Cl−, N3−, DNP−) salts were slightly better co-catalysts than NBu4+ X− (X = Br−, I−) salts. The relative reactivity of various anhydrides, with CHO as co-monomer, were found to follow the order: CHA > PA > CHE. If CHA was applied as the anhydride, the relative order in epoxides was PO > CHO = SO. Once again, the most active metal was chromium, with 5a/DMAP being significantly more active (bulk: TOF = 150 h−1) than analogous systems of Al(III), Mn(III) or Co(III). Duchateau and co-workers also reported an extensive study of metal salen/co-catalyst combinations for CHO–anhydride copolymerization.22 Again, the chromium complexes outperformed cobalt and aluminium analogues. Duchateau and co-workers also investigated limonene oxide (LO)–PA copolymerization, catalyzed by metal salophen complexes (5a, 5b, 5c, 5e), to yield partially renewable polyesters.10a
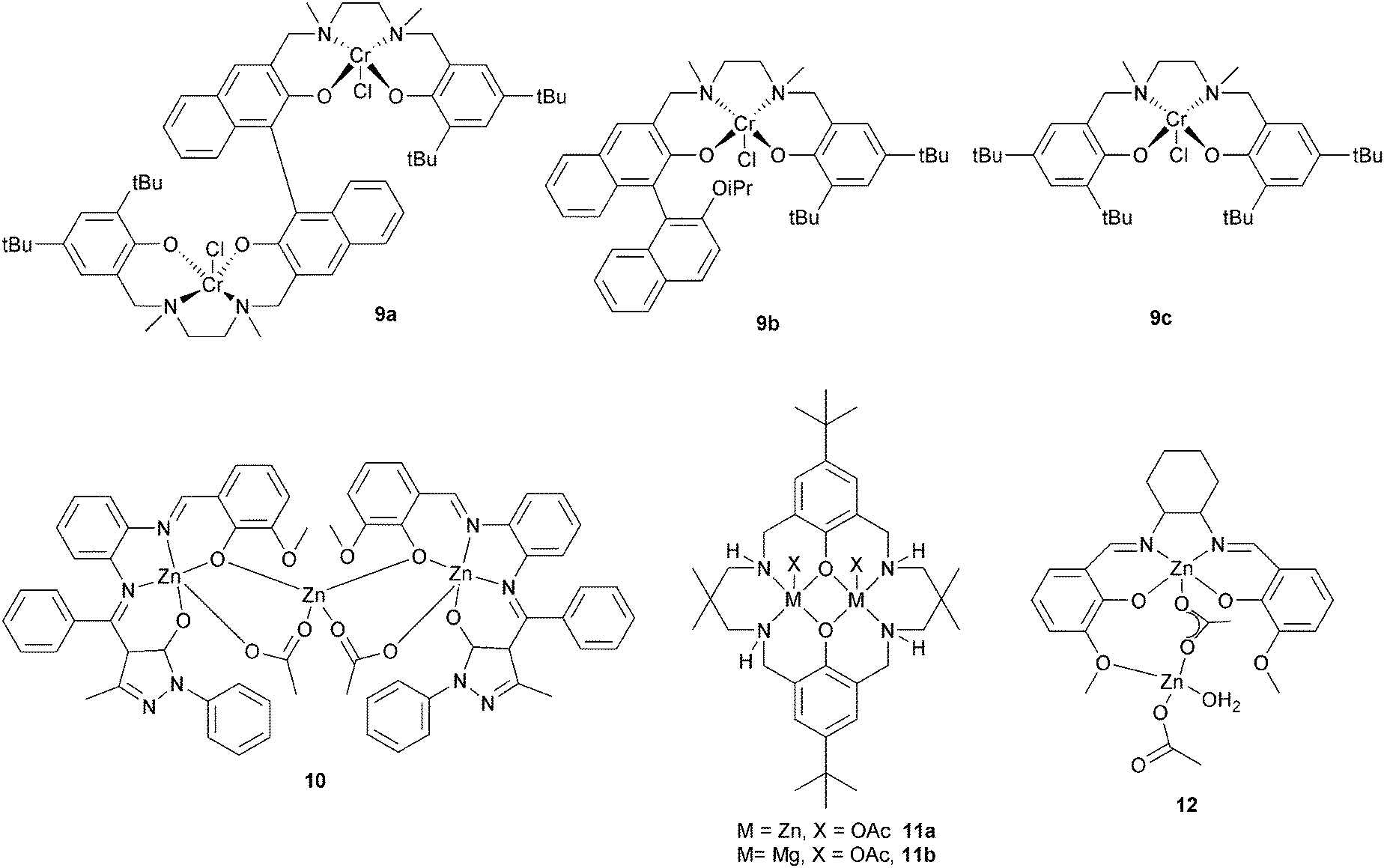 | ||
| Fig. 7 Illustrates the structures of various bimetallic catalysts. | ||
Lu et al. compared a dinuclear chromium salan catalyst, 9a, with two mononuclear analogues 9b and 9c.24 The dinuclear catalyst, 9a, was approximately four times faster for epichlorohydrin–MA (TOF = 7.8 h−1vs. 0.9 h−1) and approximately seven times faster for glycidyl phenyl ether–MA compared to the mononuclear analogue 9c (TOF = 6.0 h−1vs. 0.4 h−1). Furthermore, the polyesters showed perfectly alternating structures and were of high molar masses (Mn = 32500 g mol−1, ĐM < 1.6). The hydrolysis of the polymer obtained by (S)-GO–MA copolymerization shows an ee of 98.5% suggesting that ring-opening predominantly occurs at the methylene carbon. A novel trinuclear zinc complex, 10, with DMAP as co-catalyst, showed good activity for CHO–MA co-polymerisation (TOF = 116 h−1 at 110 °C in toluene).25 However, the polymerization conditions were important: in bulk there were ether linkages (>29%), these could be reduced using a solvent (toluene or DMF). Our group have reported di-zinc and di-magnesium catalysts for CO2–epoxide copolymerizations.9,23a–h The di-zinc and magnesium catalyst, 11a and b, are also effective for CHO–PA copolymerizations; representing the first example of a well-defined magnesium catalyst for this polymerization.26 The optimum performance was in bulk, where the complexes show moderate/good activities (11b, TOF = 97 h−1, 100 °C) and produced perfectly alternating semi-aromatic polyesters. Dizinc complex, 12 with DMAP as a cocatalyst, was shown to selectively copolymerise CHO and MA at 110 °C.27 The polymer produced is a perfectly alternating polyester (<1% ether linkages). Polymerization was controlled and yielded low molar mass polyester (Mn = 4000 g mol−1) with a narrow distribution (ĐM < 1.1). The activity was good (TOF = 130 h−1), but longer reaction times lead to broader distributions and increased amounts of ether linkages.
Very recently, Nozaki and co-workers have reported metal–corrole complexes 13a–e (Fig. 8), some of which are dimeric, for the homopolymerization of epoxides, as well as epoxide–anhydride or epoxide–carbon dioxide ROCOP.23j Manganese (13a–b) and iron corrole complexes (13d–e), with the co-catalyst (PPN)OCO–C6F5, were slow systems for PO–GA copolymerization (13e, TOF = 3 h−1, in bulk, at 30 °C) affording perfectly alternating polyesters (5700 < Mn < 8000 g mol−1 and ĐM ≤ 1.2). Interestingly, in toluene solutions poly(ester-co-ether) products formed, whereas no activity was observed in THF. Furthermore, the same catalyst system was suitable for use with other epoxides, including PO or ECH, and anhydrides, including GA.
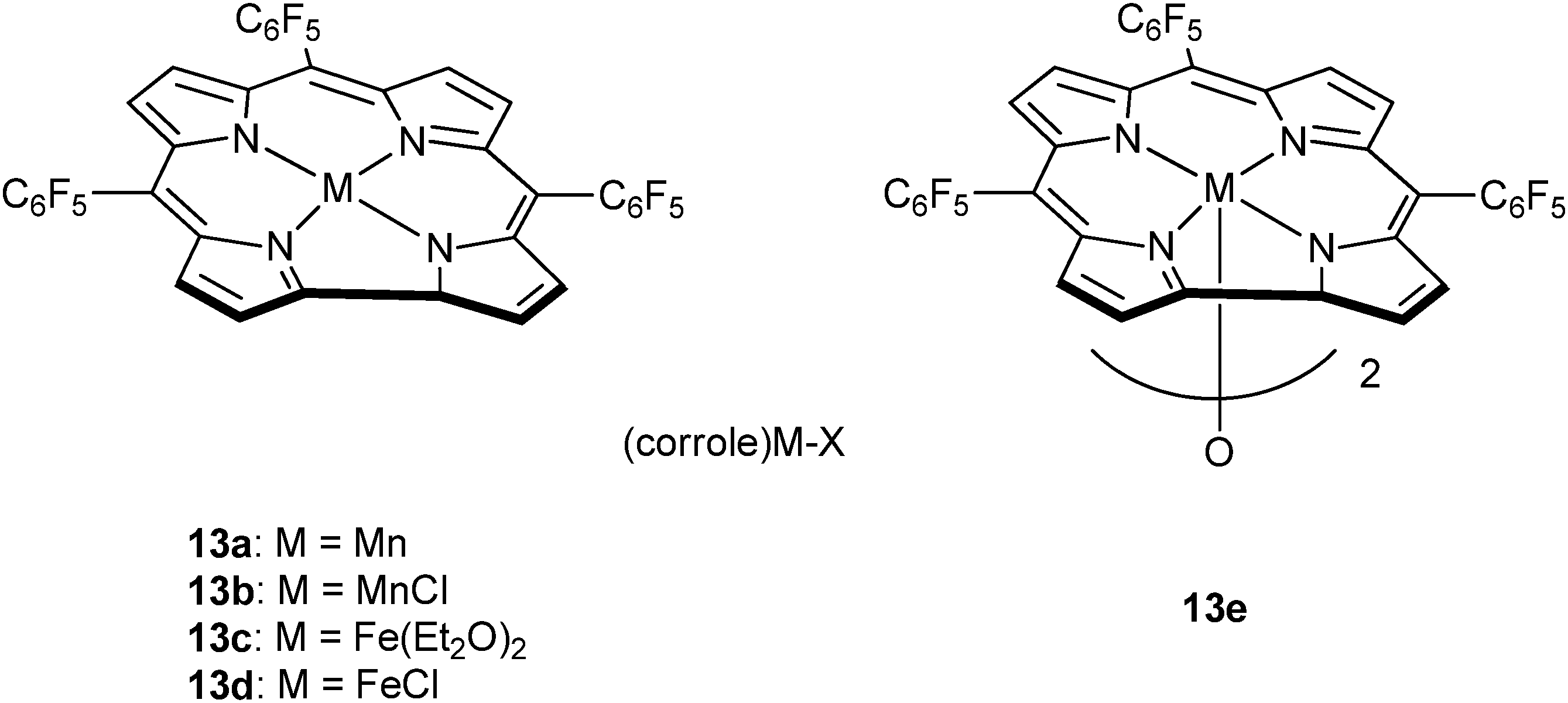 | ||
| Fig. 8 Illustrates the structures of various metal corrole catalysts.23j | ||
2.4 Copoly(ester–carbonate): combining ROCOP and ROP processes
Recently, interest in copoly(ester–carbonates) has intensified, driven by the opportunities to modify and improve upon the polymer properties. A range of different copolymer microstructures/architectures have been investigated, including block, graft and multi-branched/star copolymers. The syntheses applied depend on the desired microstructures and include sequential monomer addition (for block copolymers), the use of macro-initiators (for graft/star polymers) and the application of various multi-functional or polymeric chain transfer agents. There has been a particular focus on the development of so-called ‘one-pot’ methods, i.e. where all monomers are combined and copolymer selectivity results from catalysts/polymerization control.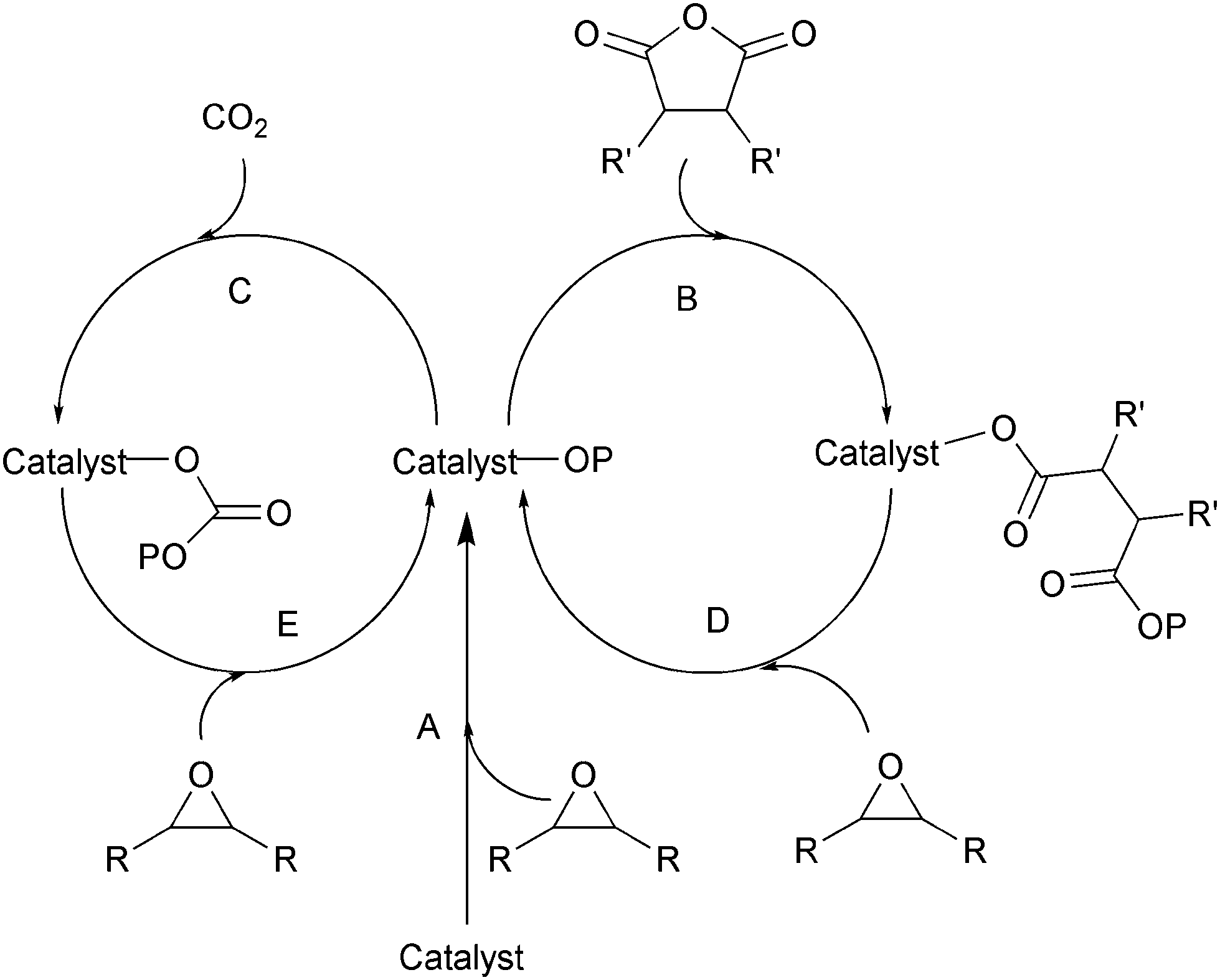 | ||
| Fig. 9 Shows the proposed mechanism for the formation of copoly(ester-b-carbonate), as reported by Coates and co-workers.29 | ||
Subsequently, other catalysts have also been shown to exhibit the same selectivity and produce copoly(ester-b-carbonates) group.10b,d,21,26,28,30 Duchateau and co-workers used 5a with DMAP as the co-catalyst for the terpolymerisation of CHO–anhydrides (SA–CPrA–CPA or PA) and CO2.10b,d By monitoring the reaction they found that ∼90% of the anhydride is converted before any carbonate functionalities are formed. It was also found that the presence of CO2, suppresses the formation of ether linkages, even in the ester blocks. It was suggested that the coordination of CO2 to the metal, may reduce its Lewis acidity and thereby quench sequential epoxide enchainment. The resulting copolymers show a single glass transition temperature, proposed to be due to block miscibility. Darensbourg and co-workers also prepared block copolymers from CHO–PA–CO2, using a chromium salen catalyst 6b with PPNCl/N3 as the co-catalyst, however, in this case two glass transition temperatures were observed, consistent with phase separation of the blocks (Tg = 48 °C and 115 °C).21 A Co(III)salen catalyst with a tethered ammonium co-catalyst, was also successfully applied, to copolymerize PO/NA/CO2.30a Duchateau and co-workers reported that metal porphyrin catalyst 1c with DMAP (as co-catalyst), was effective for the terpolymerisation of CHO, anhydrides (SA–CPrA–CPA or PA) and CO2.10d In this case however, there was concurrent carbonate linkage formation during the enchainment of ester linkages, leading to tapered block structures. This was attributed to relatively similar rates of anhydride and CO2 insertion into the metal-alkoxide bonds. In contrast, Chisholm et al. also used the same catalyst, 1c with PPNCl as co-catalyst, for the terpolymerization of PO–SA–CO2 and reported the formation of diblock copoly(ester–carbonates).18 Our group have reported that the di-zinc and di-magensium catalysts, 11a/b, are also selective in the terpolymerizations of CHO, PA and CO2 producing block copoly(ester–carbonates).26 Heterogeneous catalysts such as zinc glutarate or double metal cyanides (DMC) have been investigated. Using zinc glutarate for PO–MA–CO2 copolymerizaiton produced tapered block copoly(ester–carbonates).30b This is proposed to be due to similarities in the rate of insertion of the anhydride and CO2 co-monomers. A similar result occurs using double metal cyanide catalysts.30c The polymerisation of CHO–MA–CO2 gave a sequence where initially polyester forms, together with the random insertion of carbon dioxide. Once the MA is mostly consumed (>90%), the carbonate block forms. In contrast, the polymer supported double metal cyanide showed a different selectivity.28 During the polymerisation of PO–MA–CO2, only polycarbonate formed, with occasional, random insertion of MA, there was no formation of polyester blocks.
Earlier this year, our group reported a novel ‘Switch’ catalysis which enabled the two polymerization cycles to be combined and controlled.34 Using the di-zinc catalyst, 11a, for the terpolymerization of CHO, CO2 and ε-caprolactone (CL) led to the selective formation of block copoly(ester–carbonates) (Fig. 10).35 The reactions were monitored using in situ ATR-IR spectroscopy which showed that firstly the ROCOP reaction occurred leading to formation of only polycarbonate, subsequently, after carbon dioxide removal, the lactone ROP occurred leading to a block copoly(carbonate ester). The selectivity was interesting and unexpected because the rate of ROCOP was significantly slower than the rate of ROP, however, it was controlled by the pre-rate determining step (carbon dioxide insertion into the metal alkoxide bond). This is comparable to the earlier findings on epoxide–anhydride–CO2 copolymerisation. It was discovered that the catalyst selectivity could be easily manipulated, thus under a nitrogen atmosphere the combination of CHO and CL led to the exclusive formation of polyester. When the gas atmosphere was changed to CO2, the only product was the polycarbonate. The polymerizations were easily controlled, in one-pot, by adding different monomers at selected time points and this enabled the production of block copolymers.
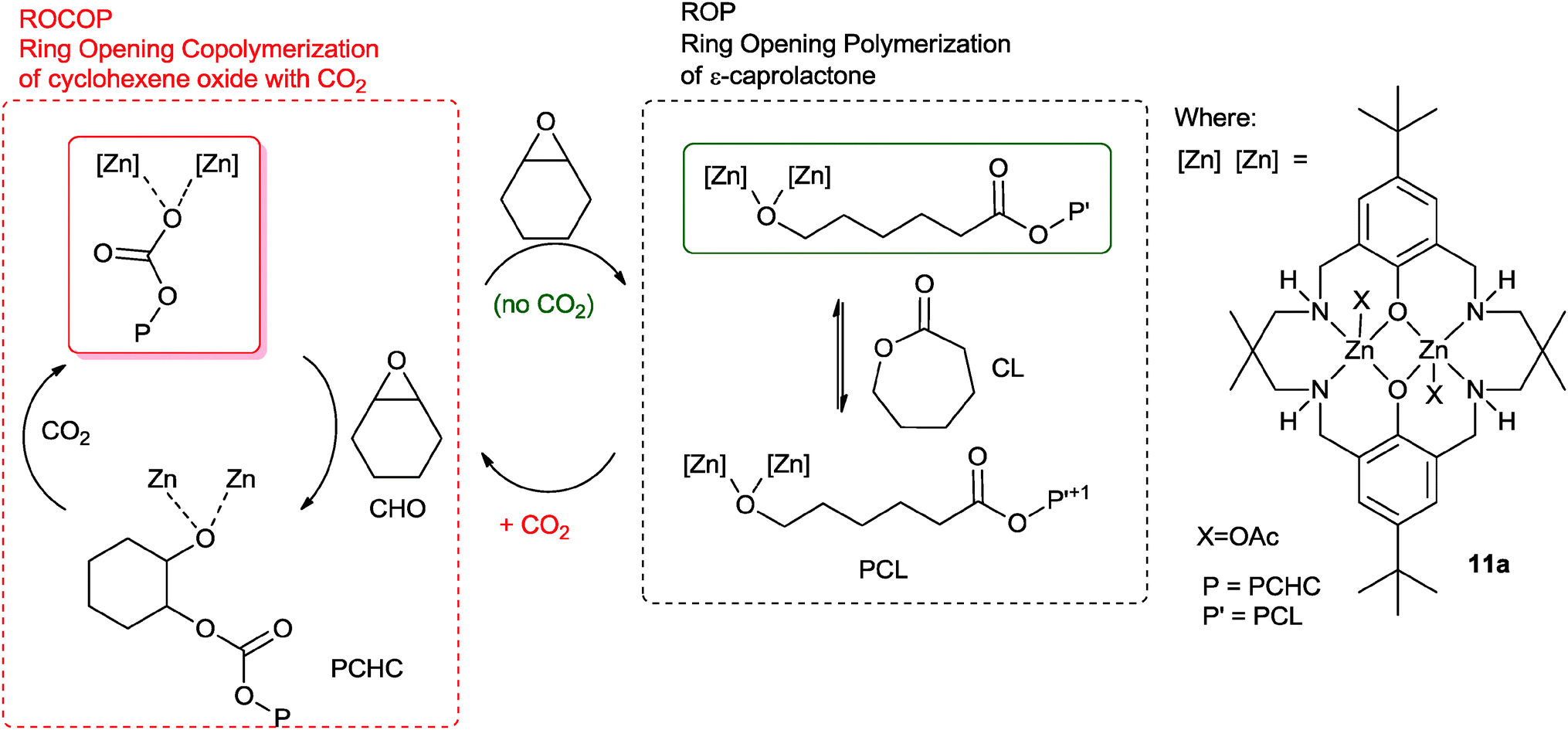 | ||
| Fig. 10 Illustrates the combination of ROCOP and ROP reactions, using a single catalyst as reported by Williams and co-workers.34 | ||
The polymerization is initiated by reaction between the metal carboxylate species and cyclohexene oxide, resulting in the formation of a metal alkoxide species. This intermediate can enter into either catalytic cycle (ROP or ROCOP). Despite the fact that the overall rate for ROP is significantly faster than ROCOP, in the presence of all three monomers the catalyst selectively enchains carbonate units. This is because the fastest reaction for the alkoxide intermediate is with carbon dioxide (1 bar pressure), leading to the formation of a zinc carbonate intermediate. The zinc carbonate species cannot react with lactones, thus it can only react with epoxide to (re)-generate the metal alkoxide species. The polymerization progresses through the ROCOP cycle. Upon removal or consumption of the carbon dioxide, in the presence of excess cyclohexene oxide, the catalyst can switch to ring-opening polymerization and selectively produce copoly(carbonate-b-ester). An attraction of this one-pot, single catalyst, switch catalysis is the potential to use it to prepare multi-block materials.
An alternative route to such copolymers is by carrying out the CO2–epoxide ROCOP in the presence of (di)hydroxyl terminated polymeric chain transfer agents. In 2010, Lee and co-workers carried out an extensive study of CO2–epoxide ROCOP, using cobalt salen catalysts, and the in presence of various macromolecular chain transfer agents.36 Hydroxyl terminated polymers, including PCL and PEG, were used to prepare di or triblock co-polymers (AB or ABA type). An indication of the promising properties for such terpolymers is that the copoly(carbonate–ether) PPC–PEG block copolymer was less brittle than the homopolycarbonate (PPC).
In an alternative strategy, our group and the Darensbourg group have both applied polycarbonate macro-initiators in the ring-opening polymerization of lactones.23c,37 In both cases, two catalysts were required and it was important to ensure compatibility between the different species and cycles. In 2011, we reported a di-zinc catalyst which was highly selective for the preparation of telechelic dihyroxyl terminated polycyclohexene carbonate (PCHC).23c The telechelic PCHC was subsequently applied, together with an yttrium initiator, for the ring-opening polymerization of S- or rac-lactide, leading to the formation of ABA block copoly(lactide-b-PCHC-b-lactide). Subsequently, Darensbourg and co-workers applied a cobalt(III) salen catalyst, featuring a tethered ammonium co-catalyst, for the ROCOP of SO–CO2.37 Once the ROCOP was complete, water was added to quench the ROCOP reaction and form, in situ, the hydroxyl terminated polymer. This addition was followed by the addition of LA, and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) as the ROP catalyst; in the second phase a block copoly(carbonate–ester) was produced. This initial report was followed by the discovery that a related cobalt(III) salen catalyst, featuring a tethered co-catalyst, could catalyse the polymerisation of PO and CO2 in the presence of water to give dihydroxyl terminated PPC.38 This telechelic polymer was subsequently applied as a macroinitiator for LA polymerisation using the same sequence of additions as previously. This year, Coates and co-workers reported the copolymerization of various epoxides, including PO, CHO, CPO and various glycidyl ethers, with dihydrocoumarin.39 This novel copolymerization produced perfectly alternating, atactic polyesters; it was surprising that two of these polyesters showed significant crystallinity.
3. ROCOP: polymer properties
The recent focus and development of ring-opening copolymerization (ROCOP) catalysts is significant because the product polyesters differ considerably in backbone structure and functionality compared to those accessible by ROP. Most importantly, simply by changing the epoxide or cyclic anhydride, the properties of the resulting material can be easily controlled, including the thermal properties (glass transition temperature (Tg), thermal decomposition temperature) as well as properties such as the lower critical solution temperature (LCST) and UV-stability.2c,10a,g,17,40 In 2014, Coates and co-workers communicated the first example of a polyester stereocomplex, prepared by catalytic and regio-selective ROCOP.41 In the following sections, the structures/properties of this novel class of polyesters and polycarbonates will be outlined.3.1 Polyesters
The nature of the polymer backbone linkages exerts a significant influence, also, the presence of ether linkages is often proposed to introduce backbone flexibility leading to a lower glass transition temperature. Nishimura and co-workers, reported that for poly(1,2-butylene itaconate), the Tg decreased upon the increasing the content of ether linkages (from 11 to −9 °C).43 It is notable that although isomerization from the itaconic to citraconic configuration occurred during the polymerization, the molar ratio of the two configurations hardly affected the Tg. In addition, it was noted that the Tg of the polyester could be further increased by the crosslinking of the polymer through the itaconic units.
ROCOP using maleic anhydride has been quite widely investigated (vide supra) and is attractive from the property perspective because moderation of the maleic group enables control of the Tg.44 Poly(propylene maleate) had only Z-configuration CC bonds (Fig. 11a). However, these can be isomerized to the E-configuration, using morpholine (Fig. 11b). In addition, by varying the isomerization reaction time, the ratio of Z- to E- was easily controlled. The Tg increased from −14 °C to 4 °C with the increasing content of the E-configuration (from 0 to 92 mol%).
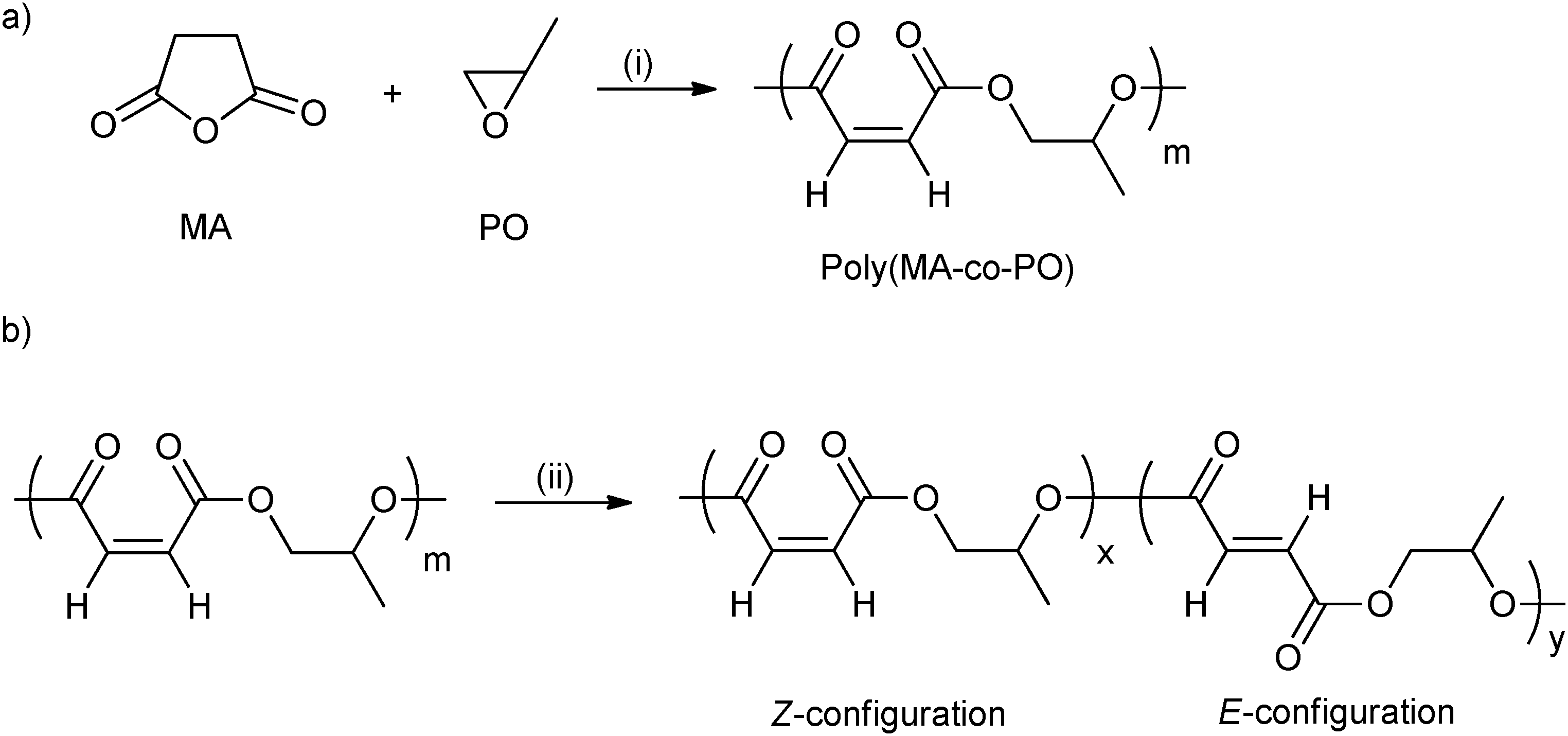 | ||
| Fig. 11 (a) Ring-opening copolymerization of maleic anhydride with propylene oxide; (b) isomerization of poly(maleic anhydride-co-propylene oxide) catalyzed by morpholine.44 Reagents and conditions: (i) Mg(OEt)2, toluene; (ii) morpholine, dichloroethane. | ||
As mentioned, Coates and co-workers reported a range of polyesters synthesized from maleic anhydride and various epoxides, including PO, epichlorohydrin, glycidyl ethers, perfluoroalkyl substituted epoxides, via ROCOP.17 The Tg of the poly(alkylene maleate) was tuned from −26 °C to 41 °C by simply changing the epoxide co-monomer. The highest Tg was observed using perfluoro alkyl substituted epoxides, whilst epoxides with diethylene glycol substituents afforded materials with the lowest Tg. Furthermore, the isomerization of poly(propylene maleate) to poly(propylene fumarate) slightly increased the Tg of the polyesters.
Since the choice of epoxide affects the Tg of the resulting polyester, several epoxides with a range of anhydrides were selected for investigation by Darensbourg and co-workers.21 They pointed out that for a particular anhydride, the Tg of the resultant polyesters increases in the order PO < SO < CHO, whilst for a particular epoxide, the Tg increases in the order SA < MA < CHA ≤ PA < CHE. Thus, as expected, the steric effect of the pendant groups and the rigidity of the repeating unit exert a significant influence on the Tg of polyesters [from −39 °C (SA–PO) to 95 °C (CHE–CHO)]. Moreover, two different Tg values were obtained for MA–PO copolymer due to photo-isomerization reactions.
A further influence on the polyesters Tg is the molar mass of the polymer. Duchateau and co-workers, reported that the Tg of poly(styrene phthalate) increases from 43 to 73 °C as the molar mass increases from 4500 to 9000 g mol−1.10c
In 2014, Coates and co-workers reported the first example of a polyester stereocomplex, prepared by the ROCOP of enantiopure epoxides–anhydrides (Fig. 12).41 Using highly regio-selective chiral cobalt salen catalysts, with ionic co-catalysts, the ROCOP of R- or S-propylene oxide, with succinic anhydride enabled preparation of highly regio-regular, isotactic poly(propylene succinate). The thermal properties of the two enantiopure polymers showed very slow crystallization (Tm: 50 °C, with a smaller amount of high melting polymorph 70 °C). However, by mixing the two polymers a stereocomplex or co-crystallite between the two isotactic (enantiomeric) polymers was formed. The stereocomplex showed a higher melting temperature (120 °C) and the t1/2 of recrystallization was three orders of magnitude faster than for the separate enantiomers. This very promising result shows the potential to manipulate the stereo- and regiochemistry of ROCOP to enable the preparation of polyesters with a melting temperature approaching that of low density polyethylene.
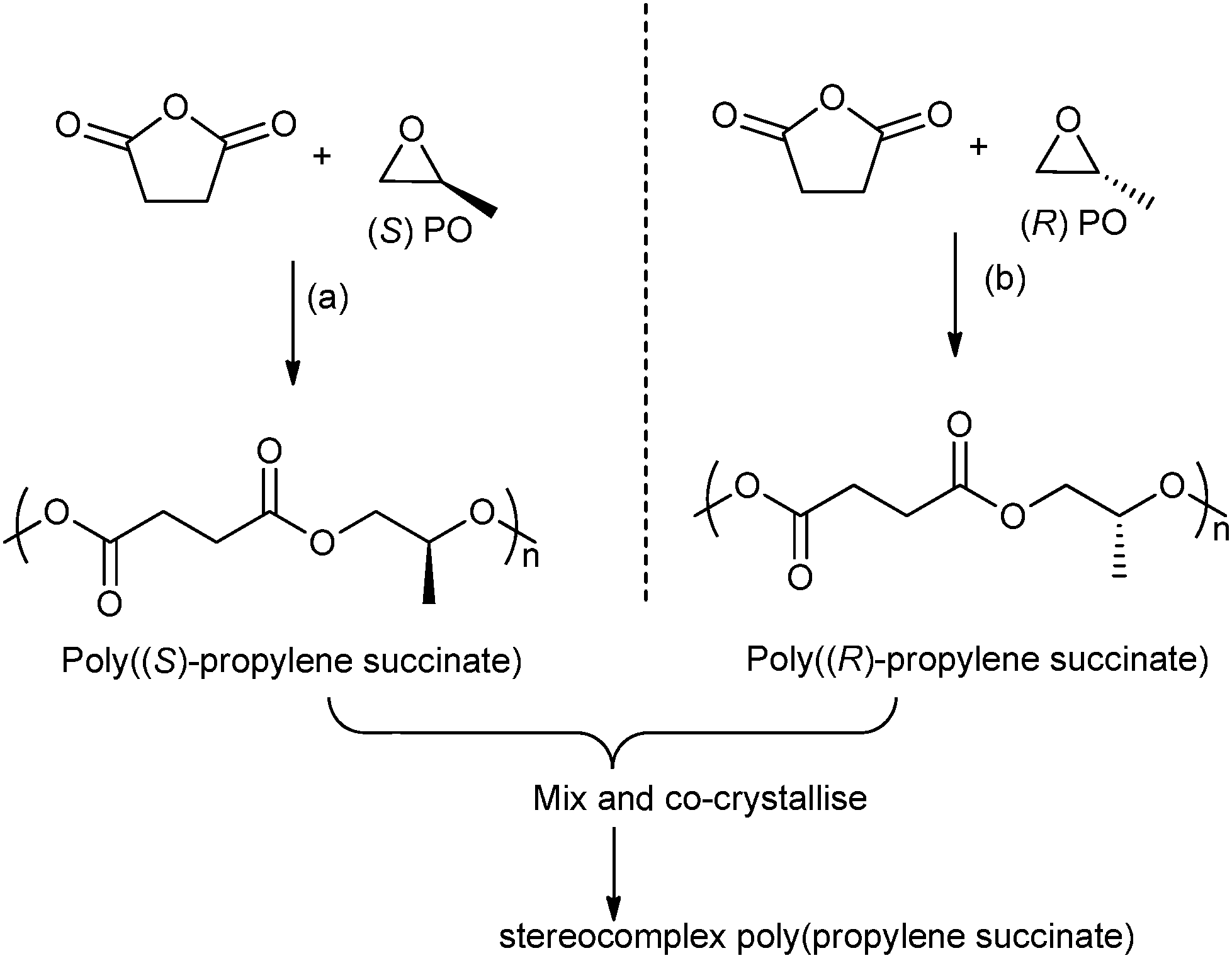 | ||
| Fig. 12 Illustrates the formation of stereocomplex poly(propylene succinate) by co-crystallisation of poly((S)-propylene succinate) and poly((R)-propylene succinate). Reagents and conditions: (a) [(R,R)-(Cl-salcy)CoNO3], [PPN][NO3], 30 °C, 36 h. (b) [(S,S)-(Cl-salcy)CoNO3], [PPN][NO3], 30 °C, 36 h.41 | ||
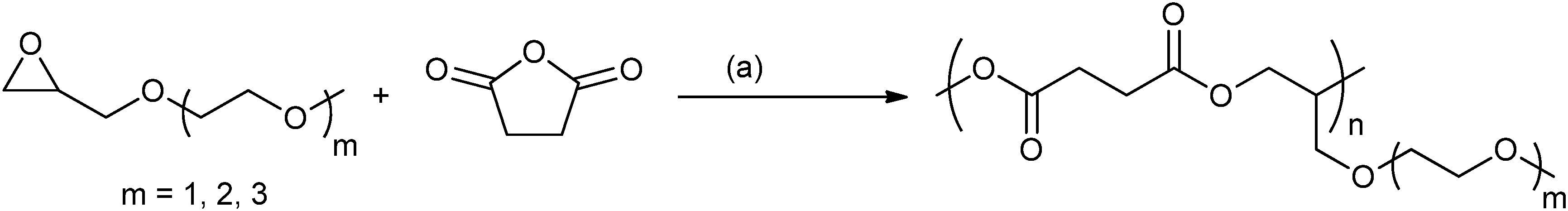 | ||
| Fig. 13 Shows the synthesis of the thermo-response polyesters prepared by ROCOP. Reagents and Conditions: (a) Al(OiPr)3.47 | ||
Although these initial results are promising, the successive tuning of the LCST cannot be easily achieved. In order to achieve finer degrees of control, a mixture of MEMO–ME2MO epoxides were copolymerized with SA.40 By changing the molar ratio of MEMO/ME2MO (from 0 to 100 mol%), fine control of the LCST of the polyester was possible over the range 17.8 to 49.2 °C. Importantly, a linear increase of LCST was observed with the increasing ME2MO content in the initial feed.
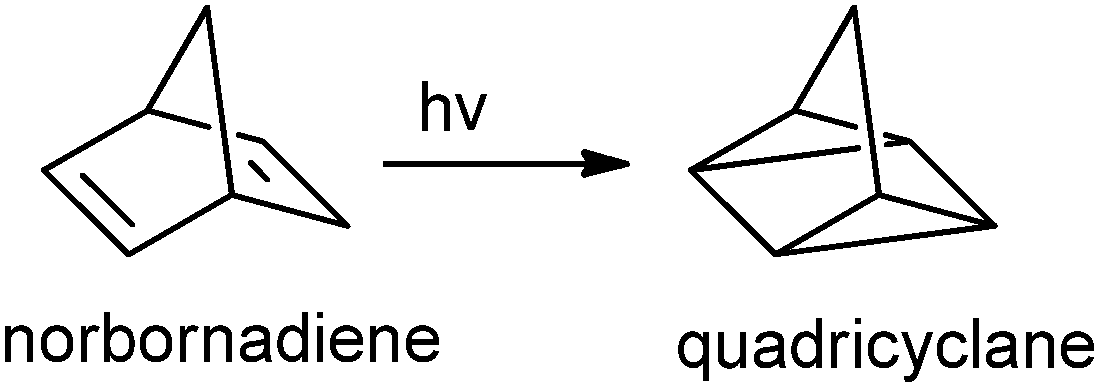 | ||
| Fig. 14 Shows the photoisomerization process from norbornadiene to quadricyclane. | ||
A photosensitizer addition was required to achieve the conversion, however, Nishikubo et al. successfully synthesized a self-photosensitizing polyester using a mixture of epoxides, with norbornadiene (NBD) and benzophenone substituents, copolymerized with PA.50 The reactivity of photoisomerization was increased, while the energy storage capacity remains the same (90 kJ mol−1). Nevertheless these polymers are hampered by relatively low photoreactivity and material fatigue. To solve this problem, polyesters bearing donor–acceptor norbornadiene (NBD) moieties in both the main chain and the side chain were prepared via ROCOP.51 The rate of photoisomerization using these donor–acceptor NBD derivatives is increased 5–8 times compared to earlier generations of materials. They also demonstrate the highest storage capacity ∼150–190 J g−1.
3.2 Polycarbonate and terpolymer properties
As mentioned above, there is a plethora of comprehensive reviews of catalysts for CO2–epoxide ROCOP.6a,9,11 Rather than any further catalysts review, this section will review the material properties of these aliphatic polycarbonates, prepared by ROCOP, as well as the potential for copoly(ester–carbonates). In our 2011 review of catalysts for CO2–epoxide copolymerisation, we wrote that “so far, polycarbonates produced from CO2 cannot match the properties of conventional polycarbonate (from bisphenol A).”9 Three years on, the outlook is markedly different.It has been known since the initial discovery of ROCOP, by Inoue, that various epoxides, including styrene oxide (SO),53 epichlorohydrin,53 3-phenyl-1,2-epoxypropane,54 cyclohexylepoxyethane,55 1,2- and 2,3-epoxybutane,56 isobutylene oxide,14f and various glycidyl ethers, bearing functional moieties including methyl, ethyl, benzoate and cholestryl,57 could be copolymerised (Fig. 2). Inoue and co-workers also showed that ROCOP using trimethylsilyl protected glycidyl ethers, with aluminium porphyrin systems (1a–e), could furnish hydroxyl-functionalised polycarbonates after deprotection.58 Inoue's approach showed great foresight, marking an important milestone and enabling further functionalization (post-polymerization) of the hydroxyl groups to give a greatly expanded range of polycarbonates. Other epoxides that have been investigated for ROCOP include cyclopentene oxide,23o,59 2,3-epoxy-1,2,3,4-tetrahydronaphthalene,60 limonene oxide,61 indene oxide,10f isomers of butene oxide and functionalized 3,5-dioxaepoxides (Fig. 2).62
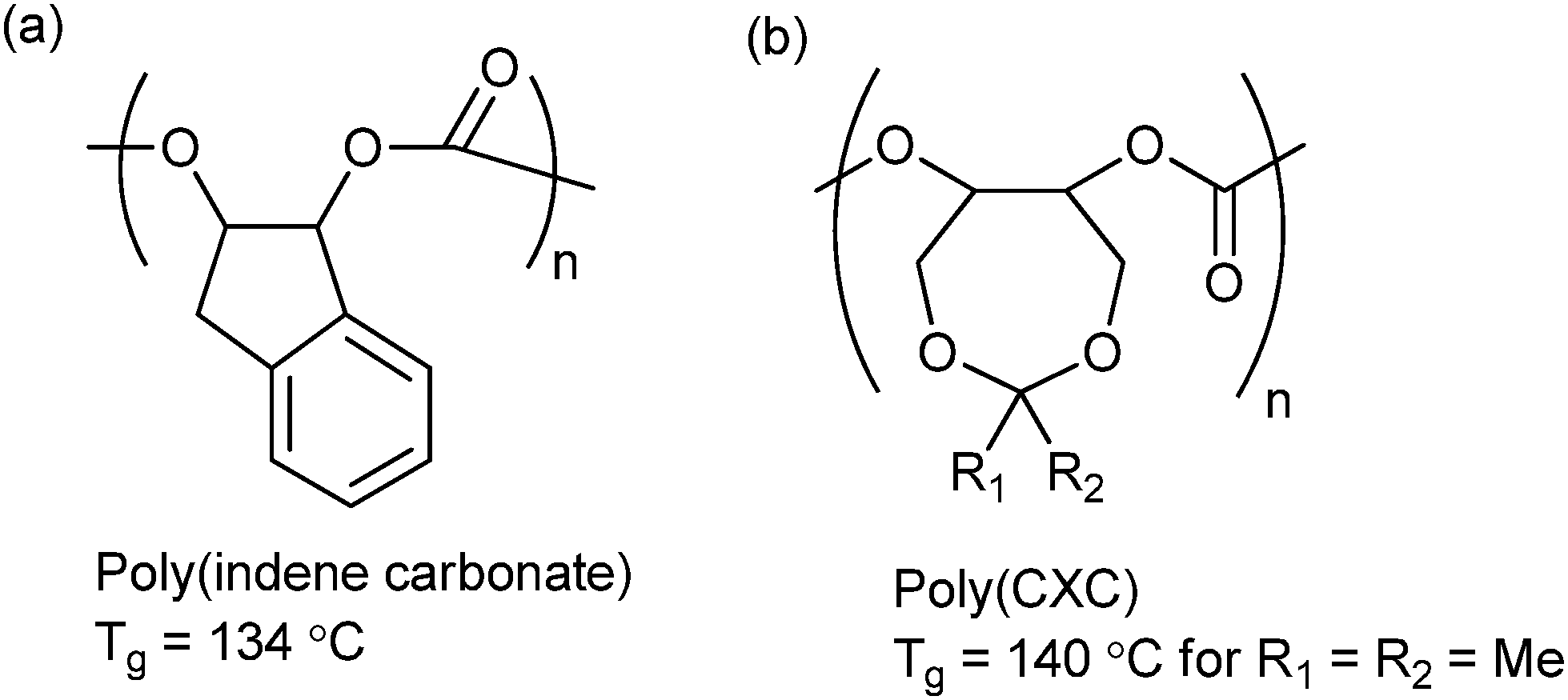 | ||
| Fig. 15 Highest Tg polycarbonates reported to date from ROCOP of CO2 and (a) indene oxide (IO)10f and (b) 4,4-dimethyl-3,5,8-trioxabicyclo[5.1.0]octane (CXO).62 | ||
Lu and co-workers synthesized a range of 3,5-dioxaepoxides (Fig. 15b) which were copolymerized with CO2, using a range of enantioselective dinuclear catalysts.23o,62 For the dimethyl substituted dioxaepoxide, an isotactic polymer (PCXC) with >99% enantioselectivity and high crystallinity was prepared, exhibiting a melting temperature of 242 °C. When the achiral analogue of the dinuclear cobalt salen catalyst was used, the polymer was atactic and notably, demonstrated the highest Tg yet reported (140 °C). This study once again highlights the importance of both tacticity control and polymer backbone rigidity in achieving the optimum thermal properties. The polymers are also highly suited to post-polymerization modification producing polycarbonates with pendant hydroxyl groups; which can subsequently be applied as macro-initiators for the ring-opening polymerization of lactide.62 The number of branch points, and therefore the thermal and mechanical properties of the polymer, could be altered by simple variation of the feed ratio of dioxaepoxides, CHO and CO2. This strategy could be of interest in biomedical applications, since the PLA regions introduce degradability and biocompatibility into the polymer.
Isotactic PCHC has been synthesised by several groups,23i,o,63f,i,65 usually with chiral catalysts such as Co/Cr salen catalysts and ZnBDI catalysts (Fig. 16).23i,o,63i,k,66 The degree of isoselectivity is usually determined by comparing the integrals of various tetrad/carbonyl signals in the 1H{1H} and 13C{1H} NMR spectra or by polymer hydrolysis and analysis of the chirality of the diol degradation products. An elegant study by Lu and co-workers23o demonstrated the highest ee values (>99%) for isotactic PCHC through careful modification of an asymmetric cobalt salen catalyst system. For iso-enriched PCHC (ee ≥ 92%), a Tg of 124 °C was observed. For isotactic PCHC (ee > 99%), there was no amorphous region and a Tm value of 272 °C was observed. Equivalent degrees of iso-selectivity were also recently demonstrated by Coates and co-workers by systematic modification of the (BDI)Zn catalysts, particularly applying C1 symmetric complexes.23i Very recently Guillaume, Carpentier and co-workers have demonstrated that the ring-opening polymerization of trans-cyclohexene carbonate, using a range of metal/organo-catalysts, and provides an alternative route to prepare isotactic PCHC, showing a similarly high degree of stereoselectivity and melting temperature.67 This latter result is particularly important as it had previously been assumed to be thermodynamically unfeasible for five-membered ring cyclic carbonates to undergo ring opening polymerization.
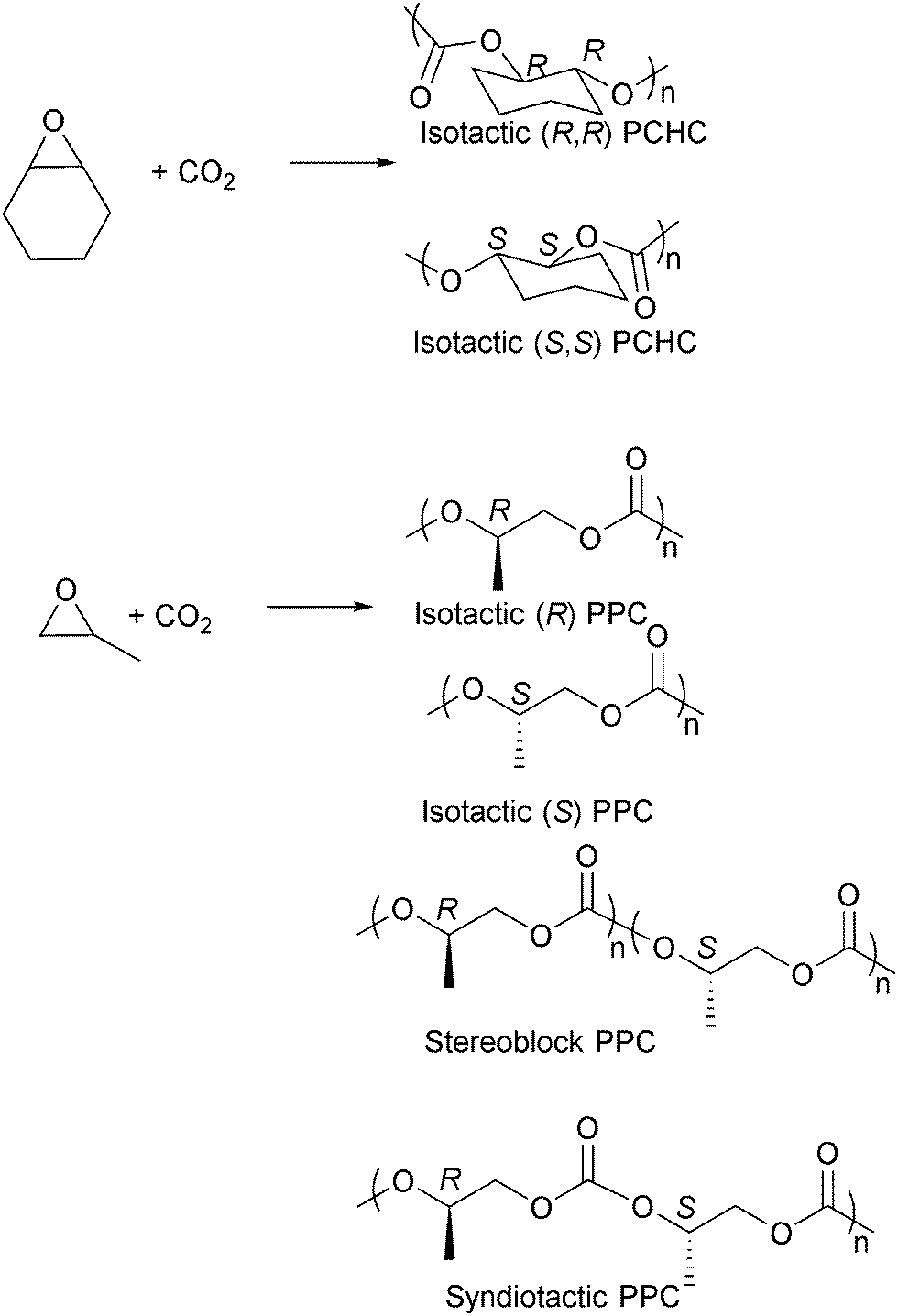 | ||
| Fig. 16 Selected structures of stereocontrolled polycarbonates (considering head-to-tail enchainment for PPC). | ||
The preparation of isotactic PPC has also been widely investigated and various chiral cobalt and chromium salen catalysts have been developed (Fig. 16).23m,63j,66b,68 One very interesting result was achieved by Nozaki and co-workers who used chiral cobalt salen catalysts to prepare a tapered stereoblock (isotactic) poly(propylene carbonate) from rac-PO (Fig. 16).63h The finding was particularly significant as it had previously not been known that PPC could form stereoblock structures and it offers the intriguing potential to prepare a stereocomplex PPC in the future. The stereoselective PPCs prepared by Nozaki and co-workers were semi-crystalline, showing Tg of 33 °C (Mn = 15000 g mol−1), and increased the thermal decomposition temperatures. Syndiotactic PPC, having 79–96% of head-to-tail linkages, was also synthesised by Coates and co-workers.68b Other epoxides have been copolymerised stereoselectively with CO2, including 1,2-hexene oxide,68g 1,2-butene oxide,68g styrene oxide,63c,69 phenyl glycidyl ether63a,b and epichlorohydrin.70
When the functional groups are terminal this leads to block copolymers. Terminal hydroxyl groups can be obtained by use of specific catalysts or the addition of chain transfer agents.23b,71 The hydroxyl terminated polymers (polyols) can then be used as chain transfer agents for further polymerisations (see 2.4.2). Polyols can also be reacted with isocyanates to synthesise polyurethanes.4,52,73 This is proposed as a readily accessible way to displace a fraction of conventional petrochemically derived polymer with renewable CO2 based polymers. CO2 based polyols have been shown to perform favourably on a life cycle basis (relative to conventional polyols),5 with production now reaching industrial scales (Fig. 17).3
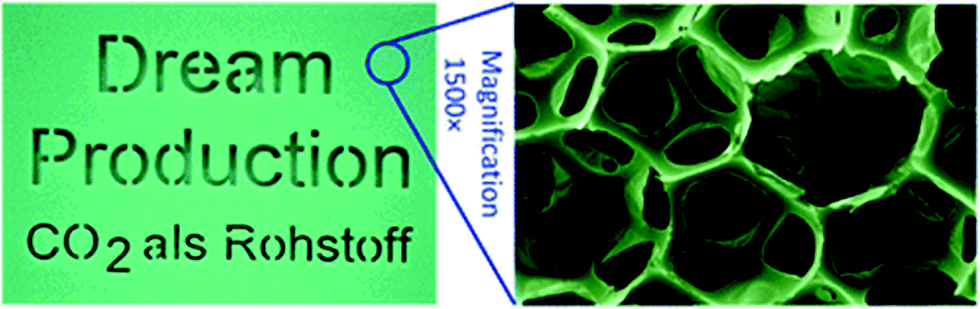 | ||
| Fig. 17 Shows an image of a polyurethane foam and micrograph showing the foam structure. The foam was prepared by the reaction between poly(ether carbonate) polyol (10.5 wt% CO2) and toluene di-isocyanate. The images are reproduced from ref. 4. | ||
When the functional groups are pendant or ‘mid chain’, further modification leads to graft or brush copolymers. Pendant functional groups occur from the ROCOP of functionalised monomers. The main challenge is ensuring that the functional group does not interfere with the polymerization. This becomes a particular problem with hydroxyl groups which are highly effective chain transfer agents and thus should be considered as reactive functionalities. Instead, functionalized monomers that can be modified after polymerization to give hydroxyl groups are used.
A range of different glycidyl ethers have been copolymerized with CO2 to give various poly(1,2-glycerol carbonates).57,63a,74 These polymers, which are often modified post polymerization to produce hydroxyl functionalized polycarbonates, are reported to be biodegradable and biocompatible – contrasting with poly(propylene carbonate), which has been shown to be resistant to enzymatic attack.75 Ren and co-workers have prepared regio-regular (HT), isotactic poly(phenyl glycerol carbonate) which is a semi-crystalline material with a melting temperature of 75 °C. In contrast, its atactic counterpart exhibited a Tg of 50 °C.63a Deng and co-workers used epichlorohydrin47 to produce a series of epoxides that gave polycarbonates with oligo(ethylene glycol) side chains which were thermo-responsive materials.74i,j A common strategy has been to use various protecting groups to enable efficient polymerization and after polymerization to effect deprotection without degrading the polymer backbone, for example by the hydrogenation of benzyl groups or acid hydrolysis of alkyl groups (Fig. 18). The nature of the protecting group is important and a reactivity series for the production of polymer using various protecting groups has been established: allyl > butyl > isopropyl.74h Using glycidyl ethers to prepare polycarbonates, followed by deprotection produces poly(1,2-glycerol carbonate), which is substituted with primary hydroxyl groups, in contrast to the ROP of cyclic carbonate which affords poly(1,3-glycerol carbonate) that contains only secondary hydroxyl groups. Poly(1,2-glycerol carbonate) was shown to be ‘highly degradable with a t1/2 of 3 days by Grinstaff and co-workers.74d Luinstra, Theato and co-workers applied a 2-nitrobenzyl glycidyl ether in copolymerization, the resulting polycarbonates were deprotected using ultra-violet light, without any backbone degradation, to prepare poly(1,2-glycerol carbonates).76
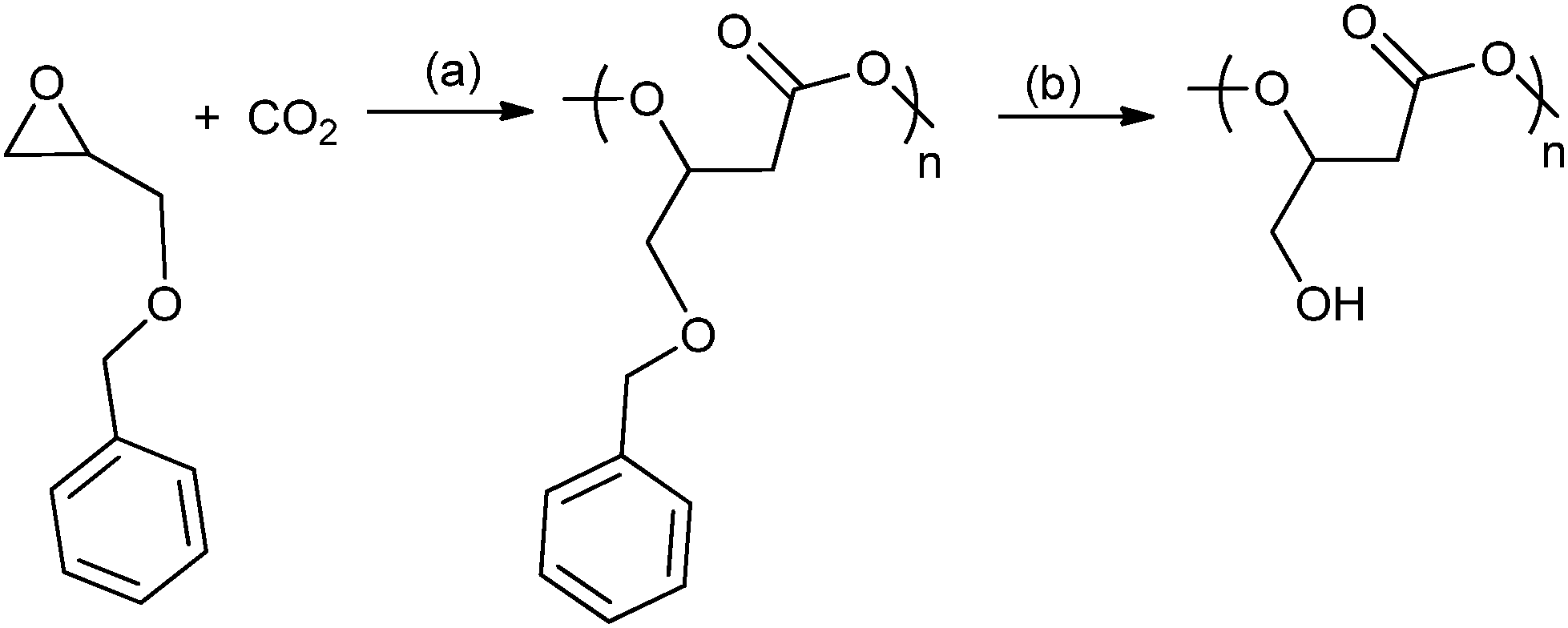 | ||
| Fig. 18 Shows the preparation of poly(1,2-glycerol carbonate) as reported, independently, by Frey and co-workers and Grinstaff and co-workers.74a,b,d Reagents and conditions: (a) zinc pyrogallol catalyst (Et2Zn:pyrogallol, 2:1), 20 bar, 40 h or [Co(salcy)(O2CCCl3)]·[PPN][Cl], 40 bar, 22 °C, 4 h. (b) H2 (40 bar), Pd/C, ethyl acetate, 24 h. | ||
Using other functionalized glycidyl ether monomers allows access to polycarbonates with controllable reactivity. Frey and co-workers copolymerized propargyl glycidyl ether with CO2 to produce polycarbonates.74l The alkyne functional groups on the polymer were reacted with benzyl azide, using the copper-catalyzed Huisgen-1,3-dipolar addition, to produce functionalized polycarbonates.74e,l The same group also prepared various random copolycarbonates containing different ratios of glycidyl methyl ether and 1,2-isopropylidene glycidyl ether units. The acetal functional group was deprotected, under acidic conditions, to yield polycarbonates functionalized with side-chain diol groups.74e,l
Ester functionalization of epoxides while potentially useful for post polymerisation modification, is rare for these polymerizations as the ester functional group can react by transesterification processes. The copolymerisation of glycidyl esters–CO2, to give acrylate functionalized PC was achieved using a heterogeneous zinc–cobalt double metal cyanide catalyst.77 Although epoxide homopolymerization also occurred, resulting in formation of a poly(carbonate-co-ether). Duchateau and co-workers copolymerised an ester functionalised CHO using a (BDI)ZnOEt catalyst, however significant amounts of transesterification occurred both from the pendant ester and the carbonate unit.78
As discussed previously, 3,5-dioxaepoxides (CXO) can be copolymerised with CO2 to give isotactic and semicrystalline polycarbonate (Fig. 15). The polymer can be easily deprotected to produce a polymer with pendant hydroxyl groups. This was used as a macro-initiator in the ring-opening polymerisation of lactide to give graft copolymers.62
One potential draw-back of using protecting groups is the need for deprotection strategies which are compatible with the polymer backbone. Alternatively, the copolymerization of vinyl functionalized epoxides enables alternative post-polymerization functionalization. Coates and co-workers carried out a comprehensive investigation of the copolymerisation of CO2 with 4-vinyl cyclohex-1,2-ene oxide (VCHO), and other functionalised CHOs with vinyl, triethylsiloxy, PEG, ketal, alkyl and fluorophilic substituents at the 4 position.79 They employed a [(BDI)ZnOAc]complex that was both tolerant of the functional groups, and enabled production of high molar mass polymers with narrow distributions (ĐM < 1.1). A range of multiblock copolymers, were prepared by addition of different functionalized –CHOs upon full consumption of the previous monomer. It was possible to prepare multi-block copolycarbonates featuring lipophilic, hydrophilic and fluorophilic units in the same linear chain. The authors highlight that this method could provide a ready means for the systematic study on the effect of block miscibility on polymer nanostructure.78,80
Frey and co-workers, copolymerized various epoxides possessing terminal vinyl groups, e.g. 1,2-epoxy-5-hexene (EH) and 1,2-epoxy-9-decene (ED), with PO and CO2.72a The frequency of pendant vinyl groups to the PC backbone could be controlled through adjusting the proportion of PO in the feed ratio, although neither ED nor EH could be directly copolymerised with CO2. Carboxyl and hydroxyl moieties were introduced using the thiol–ene reaction. The hydroxyl functionalised polycarbonates were used as macro-initiators for lactide ROP, giving graft copolymers with thermal properties linked to the PLA branch length.72a
In 2014, Darensbourg and co-workers reported ROCOP using vinyl-oxirane, PO and CO2, with cobalt(III) salen catalysts to produce a range of polycarbonates (Fig. 19).80c These were modified using post-polymerization thiol–ene ‘Click’ reactions to produce amphiphilic and water soluble polycarbonates with multiple hydroxyl or carboxyl functionalities.80c
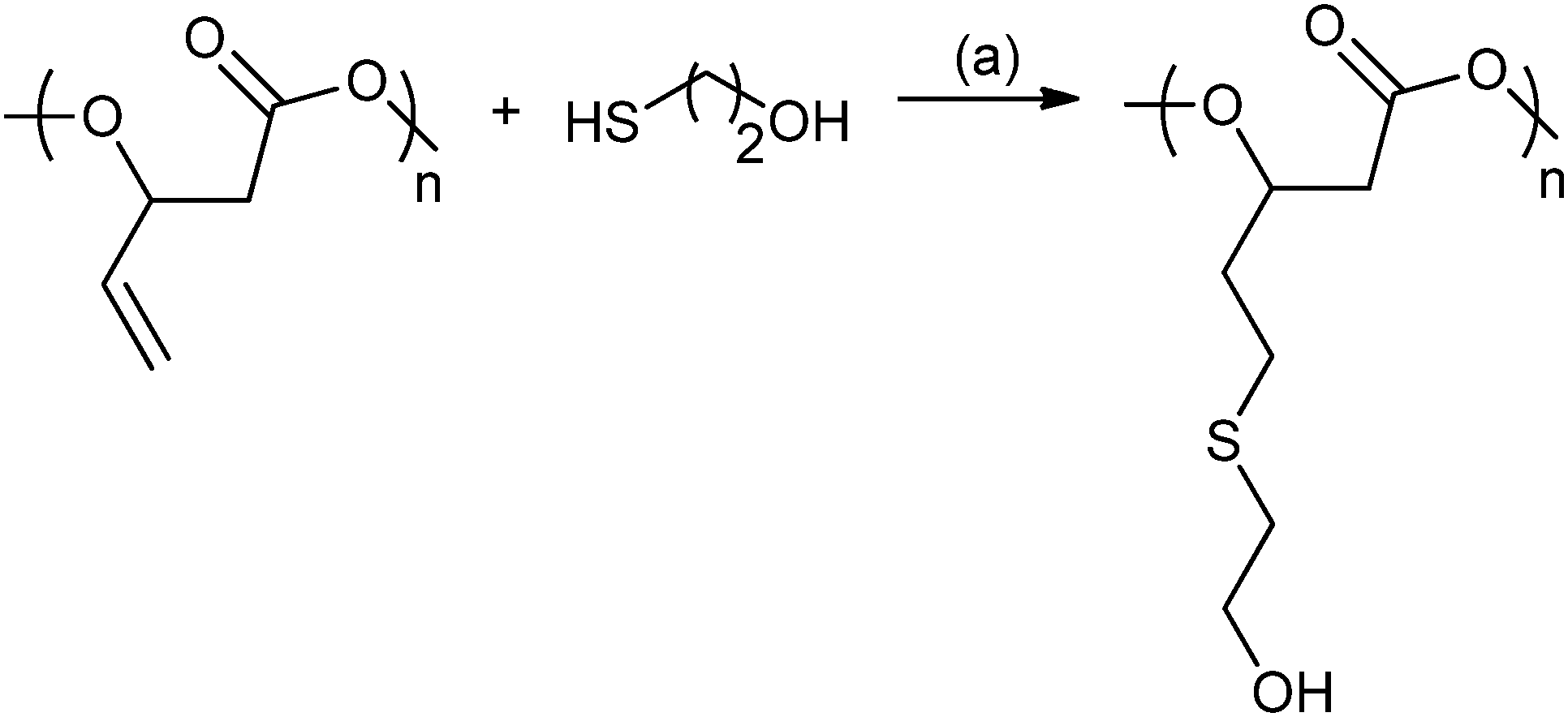 | ||
| Fig. 19 Illustrates the functionalization of poly(2-vinylpropylene carbonate) using the thiol–ene reaction. Reagents and conditions: (a) AIBN, THF, 24 h, 70 °C.80c | ||
Zhang and co-workers, also investigated the post-modification of poly(VCHC) via the thiol–ene reaction to generate OH groups. These were subsequently used as initiators in the ROP of ε-caprolactone to give graft copolymers.72c
An interesting possible alternative to the ROCOP between epoxides and carbon dioxide was presented in 2014 by Nozaki and co-workers who reported the copolymerization of carbon dioxide and butadiene via a lactone intermediate.81 They applied a palladium catalyst to prepare a metastable lactone intermediate, in addition to various side-products, via the condensation of carbon dioxide and buta-1,4-diene. The lactone was reacted by free radical polymerization to afford polymers. So far, this reaction represents a proof of principle, with a range of different repeating units present in the polymer backbone, but further research may enable its application to prepare functionalized polycarbonates.
Terpenes contain double bonds which can be epoxidised, such epoxides include limonene oxide and pinene oxide. The first success of terpene derived epoxide–CO2 ROCOP came from Coates and co-workers, in 2004, who reported the successful copolymerization of limonene oxide–CO2, using a (BDI)ZnOAc catalyst.61 The polymer was highly regio- and stereoregular, and moderate–good activity was possible (TOF = 37 h−1). The catalyst is highly selective for the ROCOP of the trans-epoxide diastereoisomers (leaving the cis-epoxides unreacted). In 2014, Coates and co-workers reported on the application of closely related catalysts to prepare enantiopure isotactic poly(limonene carbonates) (Fig. 20).82 These were prepared by reacting cis/trans mixture of (R)- or (S)-limonene oxide with CO2. The catalyst was found to only polymerize the trans diastereoisomer letting the cis isomer unreacted. It results in highly regio-, diastereo-, and enantiopure polymers which were found to have amorphous structures. However, mixing an equal proportion of the two enantiomers (i.e. the racemic mixture of poly(R)- and poly(S)-limonene carbonate) enabled the preparation of a co-crystallite or stereocomplex poly(limonene carbonate) which had a crystalline structure (as determined by powder XRD measurements).
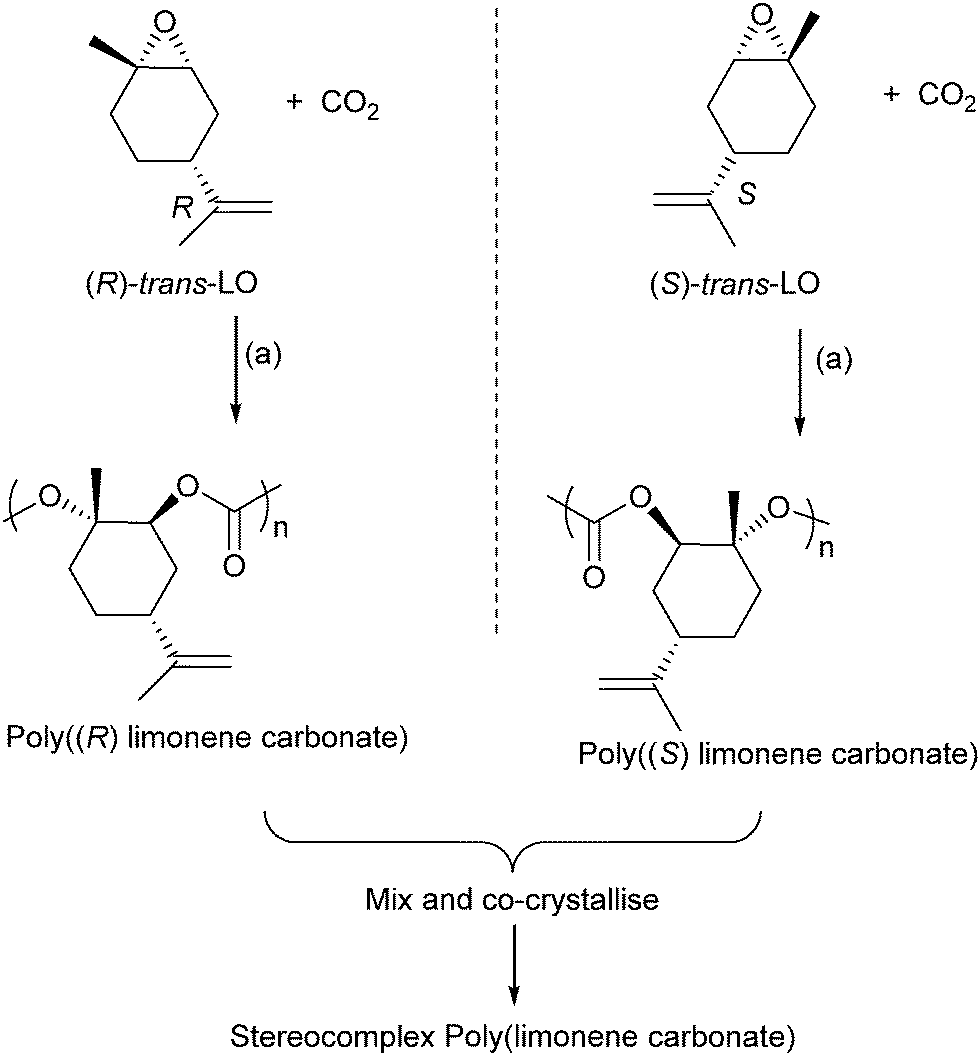 | ||
| Fig. 20 Shows the preparation of stereocomplex poly(limonene carbonate). Reagents and conditions: (a) [(BDI)Zn(N(SiMe3)2)], 22 °C, 7 bar pressure CO2.82 | ||
Thomas and co-workers used a tandem catalysis method to form polyesters from a range of diacids, produced from renewable resources. Dicarboxylic acids were cyclized to give anhydrides, which then underwent ROCOP with epoxides. In the cases were the epoxides are limonene oxide and pinene oxide (anhydrides are CA and GA respectively) then the polyester is fully renewable.83
Fatty acids, derived from vegetable oils, contain unsaturated groups which can be epoxidised. Recently, our group in collaboration with the group of Meier, have reported the preparation of various cyclohexadiene oxides derived from fatty acids (Fig. 21).84 This provides a bio-based route to prepare PCHC, as well as functionalized polycarbonates.
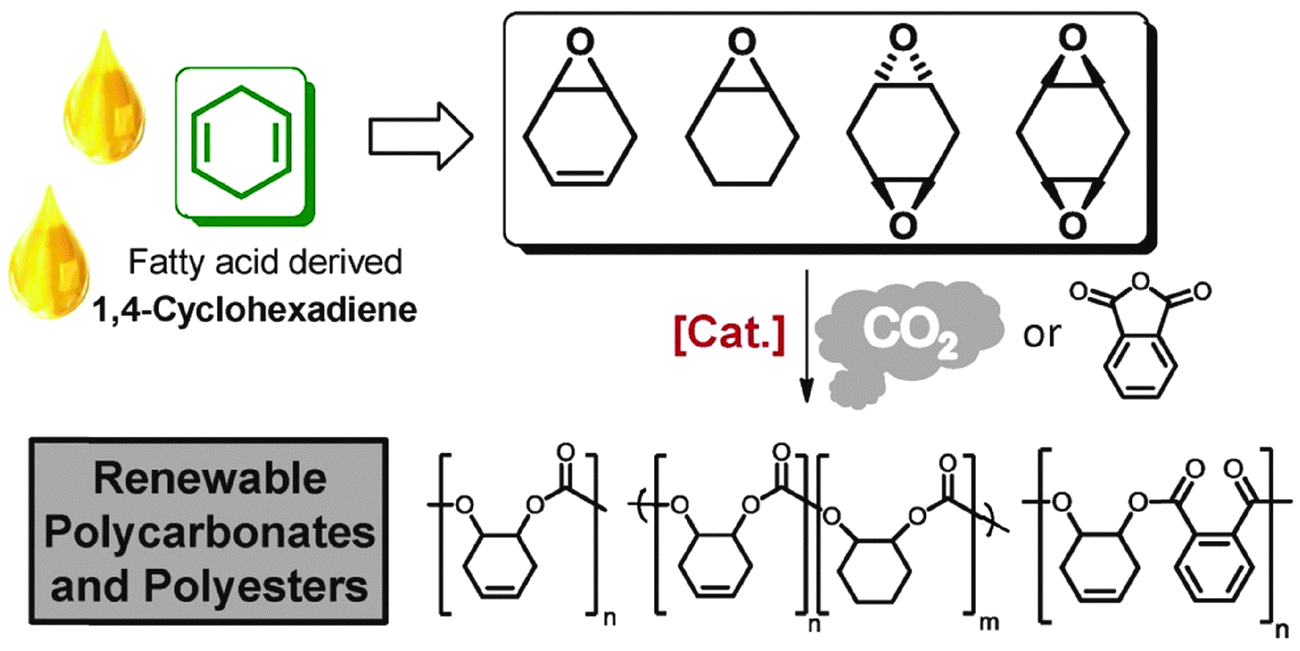 | ||
| Fig. 21 Illustrates the structures of various polycarbonates and polyesters which can be prepared from fatty acids, via ROCOP reactions, where [Cat.] represents the di-zinc and di-magnesium catalysts (11/b) illustrated in Fig. 7.84 | ||
Carbohydrates can be transformed into furfuraldehyde by a range of acidic treatments. Wang and co-workers reported the preparation of furfuryl glycidyl ether by reaction between epichlorohydrin and furfuryl alcohol.85 The same group investigated the copolymerization of furfuryl glycidyl ether with CO2 to produce a carbohydrate derived polycarbonate. The post-polymerization functionalization was possible by a Diels–Alder (DA) reaction between the furan and N-phenylmaleimide; it resulted in a polycarbonate with higher Tg (from 6.8 °C to 40.3 °C) and decreased the rate of polymer degradation. Maleic anhydride can be synthesised from furfuraldehyde using VOx/Al2O3 catalysts.86 Furthermore, phthalic anhydride can be synthesised by the Diels–Alder reaction of furan, derived from carbohydrates, and maleic anhydride, followed by dehydration.87 Many different anhydrides can be synthesised by cyclisation of dicarboxylic acids, available from carbohydrates, including SA, GA, IA, MA, and PA.88
Epichlorohydrin, is produced on a 1.8 million tons per year scale worldwide from petrochemicals, however it can be derived from glycerin,89 and therefore polymers derived from epichlorohydrin have the potential to be renewable in the future. Epichlorohydrin, can be directly copolymerized with CO2,70,74m,90 or used as a monomer precursor to other epoxides – in particular glycidyl ethers.74a,e,j–m Darensbourg and co-workers used ROCOP to producing crystalline and perfectly alternated epichlorohydrin–CO2 copolymers.70,90
4. Conclusions
Amongst the approaches to prepare polyesters and polycarbonates, ring-opening copolymerization (ROCOP) provides a large scope to modify the properties of the materials by facile substitution of at least one of the monomers (epoxide–anhydride–CO2). The polymerizations require the application of catalysts; various homogeneous and heterogeneous catalysts have been reported. Although the field of CO2–epoxide catalysis is well developed, and has not been reviewed here, the contrasting area of epoxide–anhydride catalysis remains under-developed and there is much scope to increase activity, selectivity and molar mass of the target polyesters. The polymerizations catalyzed using single site homogeneous metal complexes are generally controllable, yielding polymers of predictable molar masses, with narrow dispersities. There remains a need for fundamental understanding of the polymerization kinetics and the elementary steps occurring during these polymerization pathways so as to enable the preparation of more highly active and selective catalysts.In terms of the product properties, there are indications of the significant potential for this class of polymer with superior thermal properties. Although the range of materials explored is still a fraction of those which could be prepared from available epoxide–anhydride–heterocumulene precursors, there have already been some promising properties. For example, in the field of polyester synthesis, the application of ROCOP may prove an attractive means to prepare semi-aromatic materials which are otherwise very difficult to synthesise (e.g. using ring-opening polymerization). Such polymers, together with those with rigid polymer repeat units, show higher glass transition temperatures, than aliphatic polyesters prepared by ROP. It is possible to control aspects such as amphiphilicity, biodegradability, thermal-response and, in future, it is anticipated that more detailed understanding and study of the polymer properties will result from the advances in catalyst control.
Following from a period of intense catalyst development in the field of CO2–epoxide ROCOP, there is now significant interest and scope for exploring the limits of polycarbonate properties. Recent highlights include the potential to access glass transition temperatures well above 100 °C, indeed maximum values of 140 °C are close to those for some commercial polycarbonate materials (although other material property aspects are not yet optimised). In the area of tacticity control, the combination of advances in organometallic chemistry and the application to polymerization have enabled the production of isotactic polycarbonates, some of which are crystalline materials. As an example of the potential to moderate properties, isotactic poly(cyclohexene carbonate) has shown a melting temperature in excess of 272 °C. Such a high melting temperature, combined with high degrees of stereocontrol (>99% ee) offer much promise in the quest for the next generation of polycarbonate materials. In the field of functionalized epoxides and bioderived epoxides there has been significant recent activity and it is feasible, by a number of approaches, to prepare polycarbonates with pendant hydroxyl groups (via post-polymerization modifications). Such polymers are proposed as interesting materials for bio-medicine or as macro-initiators for further functionalization, for example by ring-opening polymerization with cyclic esters to produce partially degradable poly(ester carbonates).
There are many opportunities for the future development of aliphatic polycarbonates, including the expansion of the range of epoxides, the application of tacticity control to moderate crystallinity and the ability to control polymer properties either by post-polymerization modification or by copolymerization. The next chapter in the quest for advanced CO2-based polycarbonates will require improved control over the polymerization of epoxides, in order that high degrees of polymerization may be reached and the true thermal and mechanical properties of these materials may be evaluated. It is clear that a number of synthetic challenges need to overcome so that the engineering of advanced polymer architectures can be realised.
Acknowledgements
The Engineering and Physical Sciences Research Council (EPSRC, EP/K035274/1, EP/K014070/1, EP/L017393/1), the Grantham Institute for Climate Change and Climate KIC (scholarship to PS) and Imperial College London-CSC Scholarship (YZ) are acknowledged for research funding.References
- (a) S. Inkinen, M. Hakkarainen, A. C. Albertsson and A. Sodergard, Biomacromolecules, 2011, 12, 523–532 CrossRef CAS PubMed; (b) R. H. Platel, L. M. Hodgson and C. K. Williams, Polym. Rev., 2008, 48, 11–63 CrossRef CAS; (c) M. J. L. Tschan, E. Brule, P. Haquette and C. M. Thomas, Polym. Chem., 2012, 3, 836–851 RSC; (d) R. Auras, B. Harte and S. Selke, Macromol. Biosci., 2004, 4, 835–864 CrossRef CAS PubMed.
- (a) K. Sudesh, H. Abe and Y. Doi, Prog. Polym. Sci., 2000, 25, 1503–1555 CrossRef CAS; (b) H. M. Muller and D. Seebach, Angew. Chem., Int. Ed., 1993, 32, 477–502 CrossRef; (c) R. W. Lenz and R. H. Marchessault, Biomacromolecules, 2005, 6, 1–8 CrossRef CAS PubMed.
- http://www.empowermaterials.com/, http://www.novomer.com/, http://www.materialscience.bayer.de/Projects-and-Cooperations/CO2-Project.aspx and http://www.econic-technologies.com/, accessed 23/09/14.
- J. Langanke, A. Wolf, J. Hofmann, K. Bohm, M. A. Subhani, T. E. Muller, W. Leitner and C. Gurtler, Green Chem., 2014, 16, 1865–1870 RSC.
- N. v. d. Assen and A. Bardow, Green Chem., 2014, 16, 3272–3280 RSC.
- (a) S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2861–2871 CrossRef CAS; (b) C. J. Hawker, Acc. Chem. Res., 1997, 30, 373–382 CrossRef CAS; (c) G. W. Coates, Chem. Rev., 2000, 100, 1223–1252 CrossRef CAS PubMed; (d) M. Okada, Prog. Polym. Sci., 2002, 27, 87–133 CrossRef CAS; (e) T. P. Lodge, Macromol. Chem. Phys., 2003, 204, 265–273 CrossRef CAS; (f) N. Hadjichristidis, H. Iatrou, M. Pitsikalis and J. Mays, Prog. Polym. Sci., 2006, 31, 1068–1132 CrossRef CAS PubMed; (g) W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS PubMed; (h) G. J. Domski, J. M. Rose, G. W. Coates, A. D. Bolig and M. Brookhart, Prog. Polym. Sci., 2007, 32, 30–92 CrossRef CAS PubMed; (i) N. V. Tsarevsky and K. Matyjaszewski, Chem. Rev., 2007, 107, 2270–2299 CrossRef CAS PubMed; (j) G. Moad, E. Rizzardo and S. H. Thang, Acc. Chem. Res., 2008, 41, 1133–1142 CrossRef CAS PubMed; (k) R. R. Schrock, Acc. Chem. Res., 2014, 47, 2457–2466 CrossRef CAS PubMed.
- (a) T. Tang, M. Oshimura, S. Yamada, A. Takasu, X. Yang and Q. Cai, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3171–3183 CrossRef CAS; (b) T. Tang, T. Moyori and A. Takasu, Macromolecules, 2013, 46, 5464–5472 CrossRef CAS.
- (a) M. Helou, O. Miserque, J.-M. Brusson, J.-F. Carpentier and S. M. Guillaume, Adv. Synth. Catal., 2009, 351, 1312–1324 CrossRef CAS; (b) C. K. Williams, Chem. Soc. Rev., 2007, 36, 1573–1580 RSC; (c) C. M. Thomas and J. F. Lutz, Angew. Chem., Int. Ed., 2011, 50, 9244–9246 CrossRef CAS PubMed; (d) M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486–494 RSC; (e) N. Ajellal, J. F. Carpentier, C. Guillaume, S. M. Guillaume, M. Helou, V. Poirier, Y. Sarazin and A. Trifonov, Dalton Trans., 2010, 39, 8363–8376 RSC; (f) O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed; (g) B. J. O’Keefe, M. A. Hillmyer and W. B. Tolman, Dalton Trans., 2001, 2215–2224 RSC; (h) P. J. Dijkstra, H. Z. Du and J. Feijen, Polym. Chem., 2011, 2, 520–527 RSC; (i) C. G. Jaffredo and S. M. Guillaume, Polym. Chem., 2014, 5, 4168–4194 RSC; (j) S. Tempelaar, L. Mespouille, O. Coulembier, P. Dubois and A. P. Dove, Chem. Soc. Rev., 2013, 42, 1312–1336 RSC; (k) S. Dagorne, M. Normand, E. Kirillov and J.-F. Carpentier, Coord. Chem. Rev., 2013, 257, 1869–1886 CrossRef CAS PubMed; (l) H. A. Brown and R. M. Waymouth, Acc. Chem. Res., 2013, 46, 2585–2596 CrossRef CAS PubMed; (m) P. Lecomte and C. Jerome, in Synthetic Biodegradable Polymers, ed. B. Rieger, A. Kunkel, G. W. Coates, R. Reichardt, E. Dinjus and T. A. Zevaco, 2012, pp. 173–217 Search PubMed; (n) S. M. Guillaume and J.-F. Carpentier, Catal. Sci. Technol., 2012, 2, 898–906 RSC; (o) S. Dutta, W.-C. Hung, B.-H. Huang and C.-C. Lin, in Synthetic Biodegradable Polymers, ed. B. Rieger, A. Kunkel, G. W. Coates, R. Reichardt, E. Dinjus and T. A. Zevaco, 2012, pp. 219–283 Search PubMed; (p) L. Mespouille, O. Coulembier, M. Kawalec, A. P. Dove and P. Dubois, Prog. Polym. Sci., 2014, 39, 1144–1164 CrossRef CAS PubMed; (q) M. A. Hillmyer and W. B. Tolman, Acc. Chem. Res., 2014, 47, 2390–2396 CrossRef CAS PubMed; (r) K. Yao and C. Tang, Macromolecules, 2013, 46, 1689–1712 CrossRef CAS; (s) M. Labet and W. Thielemans, Chem. Soc. Rev., 2009, 38, 3484–3504 RSC; (t) A. Arbaoui and C. Redshaw, Polym. Chem., 2010, 1, 801–826 RSC.
- M. R. Kember, A. Buchard and C. K. Williams, Chem. Commun., 2011, 47, 141–163 RSC.
- (a) E. H. Nejad, A. Paoniasari, C. G. W. van Melis, C. E. Koning and R. Duchateau, Macromolecules, 2013, 46, 631–637 CrossRef CAS; (b) C. E. Koning, R. J. Sablong, E. H. Nejad, R. Duchateau and P. Buijsen, Prog. Org. Coat., 2013, 76, 1704–1711 CrossRef CAS PubMed; (c) E. H. Nejad, A. Paoniasari, C. E. Koning and R. Duchateau, Polym. Chem., 2012, 3, 1308–1313 RSC; (d) S. Huijser, E. HosseiniNejad, R. l. Sablong, C. D. Jong, C. E. Koning and R. Duchateau, Macromolecules, 2011, 44, 1132–1139 CrossRef CAS; (e) D. J. Darensbourg and S. J. Wilson, Macromolecules, 2013, 46, 5929–5934 CrossRef CAS; (f) D. J. Darensbourg and S. J. Wilson, J. Am. Chem. Soc., 2011, 133, 18610–18613 CrossRef CAS PubMed; (g) R. C. Jeske, A. M. DiCiccio and G. W. Coates, J. Am. Chem. Soc., 2007, 129, 11330–11331 CrossRef CAS PubMed.
- (a) D. J. Darensbourg and A. D. Yeung, Polym. Chem., 2014, 5, 3949–3962 RSC; (b) D. J. Darensbourg, in CO2 Chemistry, ed. M. Aresta and R. V. Eldik, 2014, pp. 1–23 Search PubMed; (c) D. J. Darensbourg and S. J. Wilson, Green Chem., 2012, 14, 2665–2671 RSC; (d) D. J. Darensbourg, Inorg. Chem., 2010, 49, 10765–10780 CrossRef CAS PubMed; (e) D. J. Darensbourg, Chem. Rev., 2007, 107, 2388–2410 CrossRef CAS PubMed; (f) D. J. Darensbourg and M. W. Holtcamp, Coord. Chem. Rev., 1996, 153, 155–174 CrossRef CAS; (g) D. J. Darensbourg, in Synthetic Biodegradable Polymers, ed. B. Rieger, A. Kunkel, G. W. Coates, R. Reichardt, E. Dinjus and T. A. Zevaco, 2012, pp. 1–27 Search PubMed; (h) S. D. Allen, C. M. Byrne and G. W. Coates, in Feedstocks for the Future: Renewables for the Production of Chemicals and Materials, ed. J. J. Bozell and M. K. Patel, 2006, pp. 116–129 Search PubMed; (i) E. Brule, J. Guo, G. W. Coates and C. M. Thomas, Macromol. Rapid Commun., 2011, 32, 169–185 CrossRef CAS PubMed; (j) G. W. Coates and D. R. Moore, Angew. Chem., Int. Ed., 2004, 43, 6618–6639 CrossRef CAS PubMed; (k) S. Klaus, M. W. Lehenmeier, C. E. Anderson and B. Rieger, Coord. Chem. Rev., 2011, 255, 1460–1479 CrossRef CAS PubMed; (l) X.-B. Lu, W.-M. Ren and G.-P. Wu, Acc. Chem. Res., 2012, 45, 1721–1735 CrossRef CAS PubMed; (m) W. Kuran, Prog. Polym. Sci., 1998, 23, 919–992 CrossRef CAS; (n) H. Sugimoto and S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5561–5573 CrossRef CAS; (o) G. A. Luinstra, Polym. Rev., 2008, 48, 192–219 CrossRef CAS; (p) R. Korashvili, B. Noernberg, N. Bornholdt, E. Borchardt and G. A. Luinstra, Chem. Ing. Tech., 2013, 85, 437–446 CrossRef CAS.
- M. I. Childers, J. M. Longo, N. J. Van Zee, A. M. LaPointe and G. W. Coates, Chem. Rev., 2014, 114, 8129–8152 CrossRef CAS PubMed.
- R. F. Fischer, J. Polym. Sci., 1960, 44, 155–172 CrossRef CAS.
- (a) V. E. Schwenk, K. Gulbins, M. Roth, G. Benzing, R. Maysenhölder and K. Hamann, Macromol. Chem. Phys., 1962, 51, 53–69 CrossRef; (b) T. Tsuruta, K. Matsuura and S. Inoue, Macromol. Chem. Phys., 1964, 75, 211–214 CrossRef CAS; (c) K. Matsuura, T. Tsuruta, Y. Terada and S. Inoue, Macromol. Chem. Phys., 1965, 81, 258–260 CrossRef CAS; (d) V. A. Hilt, K. H. Reichert and K. Hamann, Macromol. Chem. Phys., 1967, 101, 246–270 CrossRef; (e) J. Schaefer, R. J. Katnik and R. J. Kern, J. Am. Chem. Soc., 1968, 90, 2476–2480 CrossRef CAS; (f) S. Inoue, K. Kitamura and T. Tsuruta, Macromol. Chem. Phys., 1969, 126, 250–265 CrossRef CAS; (g) H. L. Hsieh, J. Macromol. Sci., Part A, 1973, 7, 1525–1535 CrossRef; (h) W. Kuran and A. Niestochowski, Polym. Bull., 1980, 2, 411–416 CrossRef CAS.
- (a) T. Aida and S. Inoue, J. Am. Chem. Soc., 1985, 107, 1358–1364 CrossRef CAS; (b) T. Aida, K. Sanuki and S. Inoue, Macromolecules, 1985, 18, 1049–1055 CrossRef CAS.
- (a) N. D. Harrold, Y. Li and M. H. Chisholm, Macromolecules, 2013, 46, 692–698 CrossRef CAS; (b) C. Chatterjee, M. H. Chisholm, A. El-Khaldy, R. D. McIntosh, J. T. Miller and T. Wu, Inorg. Chem., 2013, 52, 4547–4553 CrossRef CAS PubMed; (c) C. Chatterjee and M. H. Chisholm, Inorg. Chem., 2012, 51, 12041–12052 CrossRef CAS PubMed; (d) C. Chatterjee and M. H. Chisholm, Inorg. Chem., 2011, 50, 4481–4492 CrossRef CAS PubMed; (e) P. Chen, M. H. Chisholm, J. C. Gallucci, X. Zhang and Z. Zhou, Inorg. Chem., 2005, 44, 2588–2595 CrossRef CAS PubMed; (f) C. Chatterjee and M. H. Chisholm, Chem. Rec., 2013, 13, 549–560 CrossRef CAS PubMed; (g) H. Sugimoto, H. Ohshima and S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3549–3555 CrossRef CAS; (h) N. Takeda and S. Inoue, Macromol. Chem. Phys., 1978, 179, 1377–1381 CrossRef CAS; (i) S. Mang, A. I. Cooper, M. E. Colclough, N. Chauhan and A. B. Holmes, Macromolecules, 2000, 33, 303–308 CrossRef CAS.
- A. M. DiCiccio and G. W. Coates, J. Am. Chem. Soc., 2011, 133, 10724–10727 CrossRef CAS PubMed.
- A. Bernard, C. Chatterjee and M. H. Chisholm, Polymer, 2013, 54, 2639–2646 CrossRef CAS PubMed.
- (a) M. Cheng, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 1998, 120, 11018–11019 CrossRef CAS; (b) M. Cheng, D. R. Moore, J. J. Reczek, B. M. Chamberlain, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 8738–8749 CrossRef CAS PubMed; (c) S. D. Allen, D. R. Moore, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 14284–14285 CrossRef CAS PubMed; (d) D. R. Moore, M. Cheng, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2003, 125, 11911–11924 CrossRef CAS PubMed; (e) D. R. Moore, M. Cheng, E. B. Lobkovsky and G. W. Coates, Angew. Chem., Int. Ed., 2002, 41, 2599–2602 CrossRef CAS.
- (a) M. Cheng, A. B. Attygalle, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 1999, 121, 11583–11584 CrossRef CAS; (b) B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 3229–3238 CrossRef CAS PubMed; (c) L. R. Rieth, D. R. Moore, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 15239–15248 CrossRef CAS PubMed.
- D. J. Darensbourg, R. R. Poland and C. Escobedo, Macromolecules, 2012, 45, 2242–2248 CrossRef CAS.
- E. Hosseini Nejad, C. G. W. van Melis, T. J. Vermeer, C. E. Koning and R. Duchateau, Macromolecules, 2012, 45, 1770–1776 CrossRef.
- (a) P. K. Saini, C. Romain and C. K. Williams, Chem. Commun., 2014, 50, 4164–4167 RSC; (b) M. R. Kember and C. K. Williams, J. Am. Chem. Soc., 2012, 134, 15676–15679 CrossRef CAS PubMed; (c) M. R. Kember, F. Jutz, A. Buchard, A. J. P. White and C. K. Williams, Chem. Sci., 2012, 3, 1245–1255 RSC; (d) A. Buchard, F. Jutz, M. R. Kember, A. J. P. White, H. S. Rzepa and C. K. Williams, Macromolecules, 2012, 45, 6781–6795 CrossRef CAS; (e) F. Jutz, A. Buchard, M. R. Kember, S. B. Fredrickson and C. K. Williams, J. Am. Chem. Soc., 2011, 133, 17395–17405 CrossRef CAS PubMed; (f) A. Buchard, M. R. Kember, K. G. Sandeman and C. K. Williams, Chem. Commun., 2011, 47, 212–214 RSC; (g) M. R. Kember, P. D. Knight, P. T. R. Reung and C. K. Williams, Angew. Chem., Int. Ed., 2009, 48, 931–933 CrossRef CAS PubMed; (h) M. R. Kember, A. J. P. White and C. K. Williams, Inorg. Chem., 2009, 48, 9535–9542 CrossRef CAS PubMed; (i) W. C. Ellis, Y. Jung, M. Mulzer, R. Di Girolamo, E. B. Lobkovsky and G. W. Coates, Chem. Sci., 2014, 5, 4004–4011 RSC; (j) C. Robert, T. Ohkawara and K. Nozaki, Chem. – Eur. J., 2014, 4789–4795 CrossRef CAS PubMed; (k) T. Ohkawara, K. Suzuki, K. Nakano, S. Mori and K. Nozaki, J. Am. Chem. Soc., 2014, 136, 10728–10735 CrossRef CAS PubMed; (l) K. Nakano, S. Hashimoto and K. Nozaki, Chem. Sci., 2010, 1, 369–373 RSC; (m) E. K. Noh, S. J. Na, S. Sujith, S.-W. Kim and B. Y. Lee, J. Am. Chem. Soc., 2007, 129, 8082–8083 CrossRef CAS PubMed; (n) B. Y. Lee, H. Y. Kwon, S. Y. Lee, S. J. Na, S.-i. Han, H. Yun, H. Lee and Y.-W. Park, J. Am. Chem. Soc., 2005, 127, 3031–3037 CrossRef CAS PubMed; (o) Y. Liu, W.-M. Ren, J. Liu and X.-B. Lu, Angew. Chem., Int. Ed., 2013, 52, 11594–11598 CrossRef CAS PubMed; (p) C. E. Anderson, S. I. Vagin, M. Hammann, L. Zimmermann and B. Rieger, ChemCatChem, 2013, 5, 3269–3280 CrossRef CAS; (q) S. Klaus, S. I. Vagin, M. W. Lehenmeier, P. Deglmann, A. K. Brym and B. Rieger, Macromolecules, 2011, 44, 9508–9516 CrossRef CAS; (r) S. I. Vagin, R. Reichardt, S. Klaus and B. Rieger, J. Am. Chem. Soc., 2010, 132, 14367–14369 CrossRef CAS PubMed.
- J. Liu, Y.-Y. Bao, Y. Liu, W.-M. Ren and X.-B. Lu, Polym. Chem., 2013, 4, 1439–1444 RSC.
- (a) D.-F. Liu, L.-Y. Wu, W.-X. Feng, X.-M. Zhang, J. Wu, L.-Q. Zhu, D.-D. Fan, X.-Q. Lü and Q. Shi, J. Mol. Catal. A: Chem., 2014, 382, 136–145 CrossRef CAS PubMed; (b) L. Zhu, D. Liu, L. Wu, W. Feng, X. Zhang, J. Wu, D. Fan, X. Lü, R. Lu and Q. Shi, Inorg. Chem. Commun., 2013, 37, 182–185 CrossRef CAS PubMed.
- P. K. Saini, C. Romain, Y. Zhu and C. K. Williams, Polym. Chem., 2014, 5, 6068–6075 RSC.
- L. Y. Wu, D. D. Fan, X.-Q. Lü and R. Lu, Chin. J. Polym. Sci., 2014, 32, 768–777 CrossRef CAS.
- Y. Liu, K. Huang, D. Peng and H. Wu, Polymer, 2006, 47, 8453–8461 CrossRef CAS PubMed.
- R. C. Jeske, J. M. Rowley and G. W. Coates, Angew. Chem., Int. Ed., 2008, 47, 6041–6044 CrossRef CAS PubMed.
- (a) Z. Duan, X. Wang, Q. Gao, L. Zhang, B. Liu and I. Kim, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 789–795 CrossRef CAS; (b) Y. Liu, M. Xiao, S. Wang, L. Xia, D. Hang, G. Cui and Y. Meng, RSC Adv., 2014, 4, 9503–9508 RSC; (c) X.-K. Sun, X.-H. Zhang, S. Chen, B.-Y. Du, Q. Wang, Z.-Q. Fan and G.-R. Qi, Polymer, 2010, 51, 5719–5725 CrossRef CAS PubMed.
- L. Gu, Y. Qin, Y. Gao, X. Wang and F. Wang, Chin. J. Chem., 2012, 30, 2121–2125 CrossRef CAS.
- (a) Y. Hwang, J. Jung, M. Ree and H. Kim, Macromolecules, 2003, 36, 8210–8212 CrossRef CAS; (b) Y. Hwang, H. Kim and M. Ree, Macromol. Symp., 2005, 224, 227–238 CrossRef CAS; (c) S. Liu, J. Wang, K. Huang, Y. Liu and W. Wu, Polym. Bull., 2011, 66, 327–340 CrossRef CAS.
- M. Kroger, C. Folli, O. Walter and M. Doring, Adv. Synth. Catal., 2006, 348, 1908–1918 CrossRef.
- P. K. Saini, C. Romain, Y. Zhu and C. K. Williams, Polym. Chem., 2014, 5, 6068–6075 RSC.
- C. Romain and C. K. Williams, Angew. Chem., Int. Ed., 2014, 53, 1607–1610 CrossRef CAS PubMed.
- A. Cyriac, S. H. Lee, J. K. Varghese, E. S. Park, J. H. Park and B. Y. Lee, Macromolecules, 2010, 43, 7398–7401 CrossRef CAS.
- G.-P. Wu, D. J. Darensbourg and X.-B. Lu, J. Am. Chem. Soc., 2012, 134, 17739–17745 CrossRef CAS PubMed.
- D. J. Darensbourg and G.-P. Wu, Angew. Chem., Int. Ed., 2013, 52, 10602–10606 CrossRef CAS PubMed.
- N. J. Van Zee and G. W. Coates, Chem. Commun., 2014, 50, 6322–6325 RSC.
- L. Feng, Y. Liu, J. Hao, C. Xiong and X. Deng, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1812–1818 CrossRef CAS.
- J. M. Longo, A. M. DiCiccio and G. W. Coates, J. Am. Chem. Soc., 2014, 136, 15897–15900 CrossRef CAS PubMed.
- G. L. Baker, E. B. Vogel and M. R. Smith, Polym. Rev., 2008, 48, 64–84 CrossRef CAS.
- A. Takasu, M. Ito, Y. Inai, T. Hirabayashi and Y. Nishimura, Polym. J., 1999, 31, 961–969 CrossRef CAS.
- S. Takenouchi, A. Takasu, Y. Inai and T. Hirabayashi, Polym. J., 2002, 34, 36–42 CrossRef CAS.
- (a) E. S. Gil and S. M. Hudson, Prog. Polym. Sci., 2004, 29, 1173–1222 CrossRef CAS PubMed; (b) X. J. Loh, J. del Barrio, P. P. C. Toh, T. C. Lee, D. Z. Jiao, U. Rauwald, E. A. Appel and O. A. Scherman, Biomacromolecules, 2012, 13, 84–91 CrossRef CAS PubMed; (c) T. Rossow, S. Hackelbusch, P. van Assenbergh and S. Seiffert, Polym. Chem., 2013, 4, 2515–2527 RSC; (d) Z. X. Zhang, K. L. Liu and J. Li, Macromolecules, 2011, 44, 1182–1193 CrossRef CAS.
- H. Vihola, A. Laukkanen, L. Valtola, H. Tenhu and J. Hirvonen, Biomaterials, 2005, 26, 3055–3064 CrossRef CAS PubMed.
- L. Feng, Y. Liu, J. Hao, X. Li, C. Xiong and X. Deng, Macromol. Chem. Phys., 2011, 212, 2626–2632 CrossRef CAS.
- R. Hautala, R. B. King and C. Kutal, in Solar Energy, ed. R. Hautala, R. B. King and C. Kutal, Humana Press, 1979, pp. 333–369 Search PubMed.
- I. Nishimura, A. Kameyama, T. Sakurai and T. Nishikubo, Macromolecules, 1996, 29, 3818–3825 CrossRef CAS.
- I. Nishimura, A. Kameyama and T. Nishikubo, Macromolecules, 1998, 31, 2789–2796 CrossRef CAS.
- Y. Ono, N. Kawashima, H. Kudo, T. Nishikubo and T. Nagai, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4412–4421 CrossRef CAS.
- D. X. Wang, G. J. Zhang, Y. C. Zhang, Y. J. Gao, Y. H. Zhao, C. Y. Zhou, Q. Y. Zhang and X. K. Wang, J. Appl. Polym. Sci., 2007, 103, 417–424 CrossRef CAS.
- S. Inoue, J. Polym. Sci., Part B: Polym. Lett., 1969, 7, 287–292 CrossRef CAS.
- T. Hirano, S. Inoue and T. Tsuruta, Makromol. Chem., 1976, 177, 3245–3253 CrossRef CAS.
- T. Hirano, S. Inoue and T. Tsuruta, Makromol. Chem., 1976, 177, 3237–3243 CrossRef CAS.
- S. Inoue, K. Matsumoto and Y. Yoshida, Makromol. Chem., 1980, 181, 2287–2292 CrossRef CAS.
- M. Takanashi, Y. Nomura, Y. Yoshida and S. Inoue, Makromol. Chem., 1982, 183, 2085–2092 CrossRef CAS.
- (a) S. Inoue, J. Macromol. Sci., Chem., 1979, 13, 651–664 CrossRef; (b) H. Koinuma, S. Inoue and T. Tsuruta, Makromol. Chem., 1970, 136, 65–71 CrossRef CAS.
- D. J. Darensbourg, W.-C. Chung and S. J. Wilson, ACS Catal., 2013, 3, 3050–3057 CrossRef CAS.
- D. J. Darensbourg, C. C. Fang and J. L. Rodgers, Organometallics, 2004, 23, 924–927 CrossRef CAS.
- C. M. Byrne, S. D. Allen, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2004, 126, 11404–11405 CrossRef CAS PubMed.
- Y. Liu, M. Wang, W.-M. Ren, K.-K. He, Y.-C. Xu, J. Liu and X.-B. Lu, Macromolecules, 2014, 47, 1269–1276 CrossRef CAS.
- (a) W.-M. Ren, M.-W. Liang, Y.-C. Xu and X.-B. Lu, Polym. Chem., 2013, 4, 4425–4433 RSC; (b) K. Nakano, K. Kobayashi, T. Ohkawara, H. Imoto and K. Nozaki, J. Am. Chem. Soc., 2013, 135, 8456–8459 CrossRef CAS PubMed; (c) R.-J. Wei, X.-H. Zhang, B.-Y. Du, X.-K. Sun, Z.-Q. Fan and G.-R. Qi, Macromolecules, 2013, 46, 3693–3697 CrossRef CAS; (d) R. M. Thomas, P. C. B. Widger, S. M. Ahmed, R. C. Jeske, W. Hirahata, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2010, 132, 16520–16525 CrossRef CAS PubMed; (e) S. M. Ahmed, A. Poater, M. I. Childers, P. C. B. Widger, A. M. LaPointe, E. B. Lobkovsky, G. W. Coates and L. Cavallo, J. Am. Chem. Soc., 2013, 135, 18901–18911 CrossRef CAS PubMed; (f) Y.-Z. Hua, L.-J. Lu, P.-J. Huang, D.-H. Wei, M.-S. Tang, M.-C. Wang and J.-B. Chang, Chem. – Eur. J., 2014, 20, 12394–12398 CrossRef CAS PubMed; (g) G.-p. Wu, S.-d. Jiang, X.-b. Lu, W.-m. Ren and S.-k. Yan, Chin. J. Polym. Sci., 2012, 30, 487–492 CrossRef CAS PubMed; (h) K. Nakano, S. Hashimoto, M. Nakamura, T. Kamada and K. Nozaki, Angew. Chem., Int. Ed., 2011, 50, 4868–4871 CrossRef CAS PubMed; (i) L. Shi, X.-B. Lu, R. Zhang, X.-J. Peng, C.-Q. Zhang, J.-F. Li and X.-M. Peng, Macromolecules, 2006, 39, 5679–5685 CrossRef CAS; (j) B. Li, G. P. Wu, W. M. Ren, Y. M. Wang, D. Y. Rao and X. B. Lu, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6102–6113 CrossRef CAS; (k) G.-P. Wu, W.-M. Ren, Y. Luo, B. Li, W.-Z. Zhang and X.-B. Lu, J. Am. Chem. Soc., 2012, 134, 5682–5688 CrossRef CAS PubMed; (l) M. Luo, X. H. Zhang, B. Y. Du, Q. Wang and Z. Q. Fan, Macromolecules, 2013, 46, 5899–5904 CrossRef CAS.
- (a) X.-B. Lu and D. J. Darensbourg, Chem. Soc. Rev., 2012, 41, 1462–1484 RSC; (b) N. Kielland, C. J. Whiteoak and A. W. Kleij, Adv. Synth. Catal., 2013, 355, 2115–2138 CrossRef CAS.
- (a) K. Nozaki, K. Nakano and T. Hiyama, J. Am. Chem. Soc., 1999, 121, 11008–11009 CrossRef CAS; (b) K. Nakano, K. Nozaki and T. Hiyama, J. Am. Chem. Soc., 2003, 125, 5501–5510 CrossRef CAS PubMed; (c) M. Cheng, N. A. Darling, E. B. Lobkovsky and G. W. Coates, Chem. Commun., 2000, 2007–2008 RSC; (d) Y. Xiao, Z. Wang and K. Ding, Chem. – Eur. J., 2005, 11, 3668–3678 CrossRef CAS PubMed.
- (a) C. T. Cohen, C. M. Thomas, K. L. Peretti, E. B. Lobkovsky and G. W. Coates, Dalton Trans., 2006, 237–249 RSC; (b) B. Li, R. Zhang and X.-B. Lu, Macromolecules, 2007, 40, 2303–2307 CrossRef CAS; (c) K. Nakano, M. Nakamura and K. Nozaki, Macromolecules, 2009, 42, 6972–6980 CrossRef CAS.
- W. Guerin, A. K. Diallo, E. Kirilov, M. Helou, M. Slawinski, J.-M. Brusson, J.-F. Carpentier and S. M. Guillaume, Macromolecules, 2014, 47, 4230–4235 CrossRef CAS.
- (a) X.-B. Lu, B. Liang, Y.-J. Zhang, Y.-Z. Tian, Y.-M. Wang, C.-X. Bai, H. Wang and R. Zhang, J. Am. Chem. Soc., 2004, 126, 3732–3733 CAS; (b) C. T. Cohen, T. Chu and G. W. Coates, J. Am. Chem. Soc., 2005, 127, 10869–10878 CrossRef CAS PubMed; (c) C. T. Cohen and G. W. Coates, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5182–5191 CrossRef CAS; (d) X.-B. Lu, L. Shi, Y.-M. Wang, R. Zhang, Y.-J. Zhang, X.-J. Peng, Z.-C. Zhang and B. Li, J. Am. Chem. Soc., 2006, 128, 1664–1674 CrossRef CAS PubMed; (e) R. L. Paddock and S. T. Nguyen, Macromolecules, 2005, 38, 6251–6253 CrossRef CAS; (f) W. M. Ren, W. Z. Zhang and X. B. Lu, Sci. China: Chem., 2010, 53, 1646–1652 CrossRef CAS; (g) W. M. Ren, Y. Liu, G. P. Wu, J. Liu and X. B. Lu, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4894–4901 CrossRef CAS; (h) Z. Qin, C. M. Thomas, S. Lee and G. W. Coates, Angew. Chem., Int. Ed., 2003, 115, 5642–5645 CrossRef; (i) X.-B. Lu and Y. Wang, Angew. Chem., Int. Ed., 2004, 43, 3574–3577 CrossRef CAS PubMed; (j) H. Sugimoto and K. Kuroda, Macromolecules, 2007, 41, 312–317 CrossRef.
- G.-P. Wu, S.-H. Wei, W.-M. Ren, X.-B. Lu, B. Li, Y.-P. Zu and D. J. Darensbourg, Energy Environ. Sci., 2011, 4, 5084–5092 CAS.
- G.-P. Wu, P.-X. Xu, X.-B. Lu, Y.-P. Zu, S.-H. Wei, W.-M. Ren and D. J. Darensbourg, Macromolecules, 2013, 46, 2128–2133 CrossRef CAS.
- M. R. Kember, J. Copley, A. Buchard and C. K. Williams, Polym. Chem., 2012, 3, 1196–1201 RSC.
- (a) J. Geschwind, F. Wurm and H. Frey, Macromol. Chem. Phys., 2013, 214, 892–901 CrossRef CAS; (b) A. Dag, M. Aydin, H. Durmaz, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4476–4483 CrossRef CAS; (c) J.-F. Zhang, W.-M. Ren, X.-K. Sun, Y. Meng, B.-Y. Du and X.-H. Zhang, Macromolecules, 2011, 44, 9882–9886 CrossRef CAS.
- (a) H. Blattmann, M. Fleischer, M. Bähr and R. Mülhaupt, Macromol. Rapid Commun., 2014, 35, 1238–1254 CrossRef CAS PubMed; (b) L. Chen, Y. Qin, X. Wang, X. Zhao and F. Wang, Polymer, 2011, 52, 4873–4880 CrossRef CAS PubMed; (c) G. Ren, X. Sheng, Y. Qin, X. Chen, X. Wang and F. Wang, Polymer, 2014, 55, 5460–5468 CrossRef CAS PubMed; (d) Y. Hao, H. Ge, L. Han, H. Liang, H. Zhang and L. Dong, Polym. Bull., 2013, 70, 1991–2003 CrossRef CAS.
- (a) J. Geschwind and H. Frey, Macromolecules, 2013, 46, 3280–3287 CrossRef CAS; (b) H. Zhang and M. W. Grinstaff, J. Appl. Polym. Sci., 2014, 131, 39893–39899 Search PubMed; (c) D. J. Darensbourg and A. D. Yeung, Green Chem., 2014, 16, 247–252 RSC; (d) H. Zhang and M. W. Grinstaff, J. Am. Chem. Soc., 2013, 135, 6806–6809 CrossRef CAS PubMed; (e) J. Geschwind and H. Frey, Macromol. Rapid Commun., 2013, 34, 150–155 CrossRef CAS PubMed; (f) J. Łukaszczyk, K. Jaszcz, W. Kuran and T. Listos, Macromol. Rapid Commun., 2000, 21, 754–757 CrossRef; (g) R. Kudashev, E. Glukhov, E. Kuramshina, Y. Monakov, I. Gailyunas and S. Rafikov, Dokl. Phys. Chem., 1987, 292, 4–6 Search PubMed; (h) J. Łukaszczyk, K. Jaszcz, W. Kuran and T. Listoś, Macromol. Biosci., 2001, 1, 282–289 CrossRef; (i) Q. Zhou, L. Gu, Y. Gao, Y. Qin, X. Wang and F. Wang, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1893–1898 CrossRef CAS; (j) L. Gu, Y. S. Qin, Y. G. Gao, X. H. Wang and F. S. Wang, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2834–2840 CrossRef CAS; (k) J. Hilf, A. Phillips and H. Frey, Polym. Chem., 2014, 5, 814–818 RSC; (l) J. Hilf and H. Frey, Macromol. Rapid Commun., 2013, 34, 1395–1400 CrossRef CAS PubMed; (m) J. T. Guo, X. Y. Wang, Y. S. Xu and J. W. Sun, J. Appl. Polym. Sci., 2003, 87, 2356–2359 CrossRef CAS; (n) Y. Tominaga, T. Shimomura and M. Nakamura, Polymer, 2010, 51, 4295–4298 CrossRef CAS PubMed; (o) J. Hilf, P. Schulze and H. Frey, Macromol. Chem. Phys., 2013, 214, 2848–2855 CrossRef CAS; (p) J. Hilf, P. Schulze, J. Seiwert and H. Frey, Macromol. Rapid Commun., 2014, 35, 198–203 CrossRef CAS PubMed.
- M. Zhou, M. Takayanagi, Y. Yoshida, S. Ishii and H. Noguchi, Polym. Bull., 1999, 42, 419–424 CrossRef CAS.
- X. Wu, H. Zhao, B. Nörnberg, P. Theato and G. A. Luinstra, Macromolecules, 2014, 47, 492–497 CrossRef CAS.
- R.-J. Wei, X.-H. Zhang, Y.-Y. Zhang, B.-Y. Du, Z.-Q. Fan and G.-R. Qi, RSC Adv., 2014, 4, 3188–3194 RSC.
- R. Duchateau, W. J. van Meerendonk, L. Yajjou, B. B. P. Staal, C. E. Koning and G.-J. M. Gruter, Macromolecules, 2006, 39, 7900–7908 CrossRef CAS.
- J. G. Kim, C. D. Cowman, A. M. LaPointe, U. Wiesner and G. W. Coates, Macromolecules, 2011, 44, 1110–1113 CrossRef CAS.
- (a) T. J. Hsu and C. S. Tan, Polymer, 2002, 43, 4535–4543 CrossRef CAS; (b) D. J. Darensbourg, J. L. Rodgers and C. C. Fang, Inorg. Chem., 2003, 42, 4498–4500 CrossRef CAS PubMed; (c) D. J. Darensbourg and F.-T. Tsai, Macromolecules, 2014, 47, 3806–3813 CrossRef CAS; (d) D. J. Darensbourg, R. R. Poland and A. L. Strickland, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 127–133 CrossRef CAS.
- R. Nakano, S. Ito and K. Nozaki, Nat. Chem., 2014, 6, 325–331 CrossRef CAS PubMed.
- F. Auriemma, C. De Rosa, M. R. Di Caprio, R. Di Girolamo, W. C. Ellis and G. W. Coates, Angew. Chem., Int. Ed., 2015, 54, 1215–1218 CrossRef CAS PubMed.
- C. Robert, F. de Montigny and C. M. Thomas, Nat. Commun., 2011, 2, 586 CrossRef PubMed.
- M. Winkler, C. Romain, M. A. R. Meier and C. K. Williams, Green Chem., 2015, 17, 300–306 RSC.
- Y. Hu, L. Qiao, Y. Qin, X. Zhao, X. Chen, X. Wang and F. Wang, Macromolecules, 2009, 42, 9251–9254 CrossRef CAS.
- N. Alonso-Fagúndez, M. L. Granados, R. Mariscal and M. Ojeda, ChemSusChem, 2012, 5, 1984–1990 CrossRef PubMed.
- E. Mahmoud, D. A. Watson and R. F. Lobo, Green Chem., 2014, 16, 167–175 RSC.
- C. Robert, F. de Montigny and C. M. Thomas, ACS Catal., 2014, 4, 3586–3589 CrossRef CAS.
- B. M. Bell, J. R. Briggs, R. M. Campbell, S. M. Chambers, P. D. Gaarenstroom, J. G. Hippler, B. D. Hook, K. Kearns, J. M. Kenney, W. J. Kruper, D. J. Schreck, C. N. Theriault and C. P. Wolfe, Clean: Soil, Air, Water, 2008, 36, 657–661 CrossRef CAS.
- G.-P. Wu, S.-H. Wei, W.-M. Ren, X.-B. Lu, T.-Q. Xu and D. J. Darensbourg, J. Am. Chem. Soc., 2011, 133, 15191–15199 CrossRef CAS PubMed.
Footnote |
| † SP and YZ are equal first authors. |
| This journal is © The Royal Society of Chemistry 2015 |