
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric hydroamination catalyzed by a new chiral zirconium system: reaction scope and mechanism†
Xiaoguang
Zhou
ab,
Bing
Wei
ab,
Xiu-Li
Sun
a,
Yong
Tang
*a and
Zuowei
Xie
*bc
aState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry (SIOC), Chinese Academy of Sciences, 345 Lingling Lu, Shanghai 200032, P. R. China. E-mail: tangy@sioc.ac.cn
bShanghai-Hong Kong Joint Laboratory in Chemical Synthesis, SIOC, P. R. China
cDepartment of Chemistry, The Chinese University of Hong Kong, Shatin, New Territories, Hong Kong, P. R. China. E-mail: zxie@cuhk.edu.hk
First published on 19th February 2015
Abstract
A new class of chiral zirconium complexes supported by chiral tridentate [O−NO−]-type of ligands derived from amino acids were synthesized and structurally characterized. They catalyzed asymmetric hydroamination/cyclization of primary aminoalkenes to give five- and six-membered N-heterocyclic amines with up to 94% ee.
Hydroamination as a highly atom-economic reaction for efficient synthesis of nitrogen containing compounds has received great attention. In particular, chiral intramolecular hydroamination provides a convenient way to obtain chiral N-heterocycles, which plays an important role in pharmaceuticals.1 Since the first report on asymmetric hydroamination in 1992 based on chiral ansa-lanthanocenes,2 several chiral catalyst systems have been developed, including group 4 metals,3 alkaline earth metals,4 alkali metals,5 late transition metals,6 rare earth metals2,7–12 and chiral Brønsted acids.13 Though significant progress has been made in the past decade in this field, the reported enantioselective systems remain very limited. In view of the recent progress in asymmetric catalysis,14 it is envisioned that new ligand design, new catalyst synthesis, and mechanism tests may offer strategies for addressing challenging issues in asymmetric olefin hydroamination, thus further advancing this field.
Very recently, we reported a highly active cationic zirconium system based on a tridentate [O−N−S] ligand for catalytic hydroamination with a broad substrate scope from primary amines to secondary amines, and from terminal alkenes to much less reactive internal alkenes.15 This system also catalyzed tandem intramolecular hydroamination of primary aminoalkenes to give bicyclic amines, and was tolerant of many functional groups. We wondered if chiral ligands derived from such a tridentate backbone system would offer both high catalytic activity and enantioselectivity in asymmetric hydroamination. Here we report the new chiral catalyst systems for asymmetric hydroamination of aminoalkenes with up to 94% ee and ≥95% conversion. The reaction mechanism is also discussed.
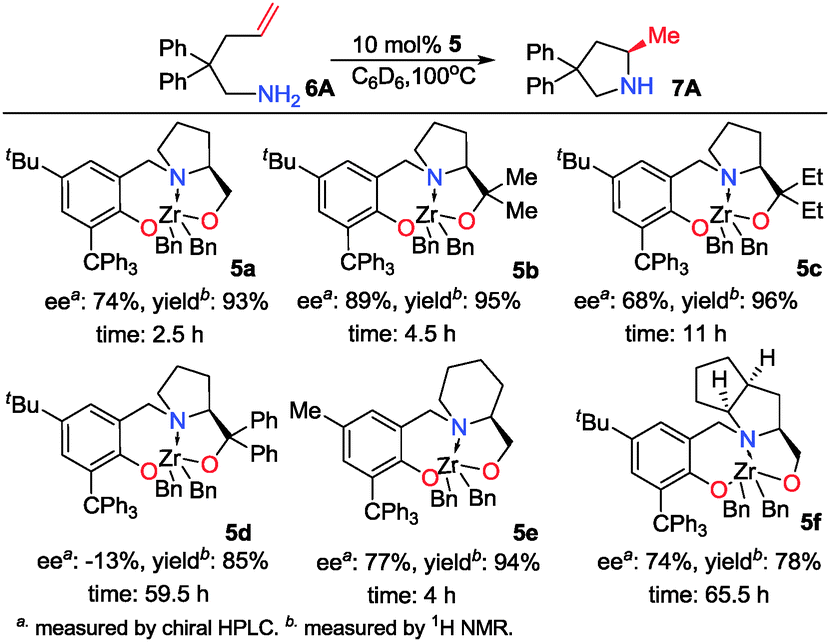
Initially, a series of new chiral auxiliary [O−N−O] ligands were synthesized via the condensation of salicylaldehyde with O-alkylated chiral aminoalcohol, followed by reduction with LiAlH4 (see ESI†). They were treated with 1 equiv. of tetrabenzyl-zirconium in toluene to give new chiral dibenzyl zirconium complexes 1–4 (Chart 1). Screening results on catalytic hydroamination of 2,2-diphenyl-4-pentenamine (6A) indicated that 4b offered the best ee value and the lability of the OR2 group disfavored the chiral induction (Table S3 in ESI†). To fix this problem, a covalent bond between the Zr atom and O(2) was desirable, which led to the design of a new ligand backbone. Subsequently, a series of new ligands with a [O−NO−] backbone were prepared. Their Zr complexes 5a–f were also synthesized and characterized by various spectroscopic data and single-crystal X-ray analyses (Fig. S8–S11 in ESI†). Scheme 1 shows the screening results of 5a–f. The enantioselectivity of the reaction was greatly improved, 74% ee for 5a, 89% ee for 5b and 68% ee for 5c. It was clear that gem-disubstituents had a large impact on ee values with gem-dimethyl being the best choice. Replacement of a five-membered N-heterocycle by a six-membered one (5e) or a fused five-membered ring (5f) offered similar enantioselectivity. After identifying 5b as the best catalyst for hydroamination of 6A, the effect of reaction temperature on enantioselectivity was examined and the results are listed in Table S4 (ESI†).
 | ||
| Chart 1 Chiral Zr complexes 1–4. | ||
 | ||
| Scheme 1 Screening results of asymmetric hydroamination catalyzed by 5a–f. | ||
Under the optimal reaction conditions, several substrates were examined, and the results are summarized in Table 1. Both five- and six-membered N-heterocyclic ring products were generated with up to 94% ee. The substrate 6B gave spiro[4,5] product 7B in 93% ee (entry 2), and the substrate 6C offered spiro[4,4] product 7C in 87% ee (entry 3). Desymmetrization with the two allyl groups in aminodiene 6F produced both diastereomers with 88% ee and 92% ee, respectively, whereas the diastereoselectivity was low (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.7) (entry 6). Upon replacing phenyl in 6F with methyl, even higher ee values of 90% and 93% were achieved (entry 5). The gem-dialkyl effect was also observed. As anticipated, replacement of gem-diphenyl substitutes in 6A by gem-dimethyl-ones resulted in a much lower activity (entry 4). Complex 5e was found to give 7G in 66% ee, which represents the best enantioselectivity so far observed for the hydroamination of 6G (entry 7).
:1.7) (entry 6). Upon replacing phenyl in 6F with methyl, even higher ee values of 90% and 93% were achieved (entry 5). The gem-dialkyl effect was also observed. As anticipated, replacement of gem-diphenyl substitutes in 6A by gem-dimethyl-ones resulted in a much lower activity (entry 4). Complex 5e was found to give 7G in 66% ee, which represents the best enantioselectivity so far observed for the hydroamination of 6G (entry 7).
| Entry | Substrate | Product | Time (h) | Yieldf/eed (%) |
|---|---|---|---|---|
| a Temp. = 85 °C. b Using 10 mol% 5e at 130 °C. c Temp. = 70 °C. d The ee value measured by chiral HPLC. e Products were converted to N-Ts compounds and the ee value was measured by chiral HPLC, the dr value was determined by the analyses of the 1H NMR spectrum of the crude product. f Isolated yield. | ||||
| 1c |

|

|
19 | 84/94 |
| 2a,e |
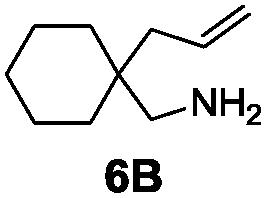
|

|
21 | 97/93 |
| 3a,e |

|

|
72 | 91/87 |
| 4a,e |

|

|
120 | 96/89 |
| 5a,e |

|

|
89 | 89/90, 93 |
| dr = 1:1.2 |
||||
| 6a,e |

|

|
24 | 91/88, 92 |
| dr = 1:1.7 |
||||
| 7b |
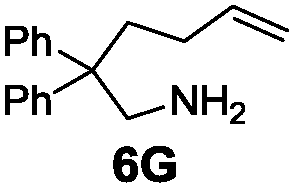
|
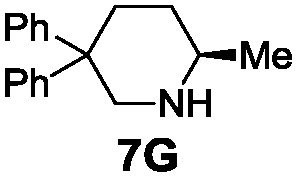
|
2 | 91/66 |
Since 5b did not show catalytic activity toward secondary aminoalkenes, it seemed that the above [ONO]Zr(CH2Ph)2 system might proceed via a Zr-imido intermediate. Many attempts to isolate the reaction intermediate from the stoichiometric reaction of 5b with 6A were unsuccessful. We then prepared an achiral Zr complex 8 with the same [O−NO−] backbone. It could catalyze the hydroamination of 6A to a racemic mixture of 7A with a comparable activity to 5b.
Reaction of 8 with an excess amount of t-BuNH2 in toluene at room temperature gave an imido-bridged zirconium amide complex 9 in 44% yield. Complex 9 also catalyzed the hydroamination of 6A to offer (±)7A in a similar activity to that of 8. Single-crystal X-ray analyses reveal that 9 is a dimer with bridging imido tBuN and alkoxy units, respectively (Scheme 2). One of the Zr atoms is five coordinated, and the other is six coordinated. In addition, each Zr atom is σ-bonded to one amido unit tBuNH, and the two tBuNH moieties are oriented in trans positions (Fig. S13 in ESI†).
 | ||
| Scheme 2 Reaction of 8 with t-BuNH2. | ||
To gain further insight into the reaction mechanism, a kinetic analysis of hydroamination/cyclization of aminoalkene 6A catalyzed by 5b was performed for the determination of an empirical rate law using in situ1H NMR monitoring. Ferrocene was employed as an internal 1H NMR integration standard, as it was readily differentiated from the substrate, product, and catalyst resonances in deuterated toluene. The appearance of the product 7ACH2CH signal was monitored as a function of time and normalized versus the internal standard.16
The results show that there is a first-order dependence on both catalyst and substrate concentration, giving the empirical rate law in the following equation on the basis of initial rate measurements: rate = kobs[catalyst][substrate].
To gauge the effect of N-deuteration on the reaction catalyzed by 5b, the conversion of d2-6A was studied (Scheme 3). It was found that the cyclization of N-deuterated d2-6A was much slower than for the proteo counterpart 6A, giving kH/kD = 5.2 at 60 °C (see Fig. S19 in ESI†).
 | ||
| Scheme 3 Primary kinetic isotopic effect observed in the cyclization of 6A (E = H/D). | ||
To further probe the nature of the turnover-limiting step, the temperature dependence of the cyclization rate of 6A was investigated by determining the kobs values at 50 °C, 60 °C, 70 °C and 80 °C (see Fig. S20 in ESI†). The Eyring plot generated from these data is shown in Fig. S21 in ESI.† The calculated thermodynamic parameters are ΔH≠ = 9.9 ± 0.3 kcal mol−1 and ΔS≠ = −53.4 ± 0.9 cal K−1 mol−1, which are very comparable to ΔH≠ = 6.7 ± 0.2 kcal mol−1 and ΔS≠ = −43 ± 7 cal K−1 mol−1 observed for the [PhB(C6H4)(OX)]Zr(NMe2)2 system.3f These values may also be compared to ΔH≠ = 17 to 21 kcal mol−1 and ΔS≠ = −13 to −23 cal K−1 mol−1 in the salicyloxazoline zirconium system17 and ΔH≠ = 20.1 ± 0.5 kcal mol−1 and ΔS≠ = −21 ± 1 cal K−1 mol−1 in the tethered bis(ureate)zirconium system.18 The parameters obtained in the current study show a highly organized transition state.
Though 9 displayed a similar catalytic activity to that of 8 and 5b, it was not clear whether a dinuclear species was involved in the turnover-limiting step of the catalytic cycle as the first-order dependence on precatalyst concentration was observed. Accordingly, experiments on the non-linear effect were carried out. A strict linear relationship between the ee values of 7A and those of precatalyst 5b was observed (see Fig. S22 in the ESI†), as would be expected for the exclusively monomeric mechanism. In addition, the 1H NMR spectrum showed that 9 dissociated into the monomeric form [ONO]Zr(NHBut)2 in the presence of excess t-BuNH2 (see Schemes S1 and S2 in ESI†). These results suggest that the reaction may proceed via a mononuclear intermediate.
In addition, the isotopic substitution significantly affects the reaction's enantioselectivity, where the % ee's for deutero-pyrrolidines are systematically and significantly higher than the values for the corresponding proteo-pyrrolidines (see Table S7 in ESI†). Such effects show that an N–H (or N–D) bond is involved in the stereochemistry-determining step, supporting a concerted C–H/C–N bond forming mechanism.3d
On the basis of aforementioned experimental data, a plausible catalytic mechanism is proposed in Scheme 4. Initial aminolysis of the precatalyst 5b by excess substrates liberates toluene to give a monomeric species I. The third substrate binds reversibly to the Zr atom to give the species II. Intramolecular hydroamination takes place via irreversible C–H and C–N bond formation through a highly ordered transition state. This is supported by a very large and negative value of ΔS≠ (−53.4(9) cal K−1 mol−1) as well as a large KIE value of 5.2. The dissociation of neutral pyrrolidine regenerates the active catalyst 1 to complete the catalytic cycle. Such a mechanism is similar to those reported in the literature.3d,18
 | ||
| Scheme 4 Proposed catalytic cycle for intramolecular hydroamination. | ||
In summary, this work describes our journey of exploring new chiral zirconium catalysts for asymmetric hydroamination/cyclization. Through systematic studies, we have developed a class of new pincer-like [O−NO−]Zr systems that can efficiently catalyze the hydroamination of primary aminoalkenes with up to 94% ee and ≥95% conversion. The catalyst structure–enantioselectivity relationships have also been addressed, which may shed some light on the ligand design of new catalyst systems. A mechanism has also been proposed to account for all of the experimental observations, which involves a highly ordered transition state and a concerted bond formation pathway.
We are grateful for the financial support from the National Natural Science Foundation of China (21121062 to YT) and the CAS-Croucher Funding Scheme.
Notes and references
- (a) T. C. Nugent, Chiral Amine Synthesis: Methods, Developments and Applications, Wiley-VCH, Weinheim, Germany, 2010 Search PubMed; (b) A. Ricci, Modern Amination Methods, Wiley-VCH, Weinheim, Germany, 2000 Search PubMed.
- (a) M. R. Gagné, L. Brard, V. P. Conticello, M. A. Giardello, C. L. Stern and T. J. Marks, Organometallics, 1992, 11, 2003 CrossRef; (b) M. A. Giardello, V. P. Conticello, L. Brard, M. Sabat, A. L. Rheingold, C. L. Stern and T. J. Marks, J. Am. Chem. Soc., 1994, 116, 10212 CrossRef CAS; (c) K. Manna, M. L. Kruse and A. D. Sadow, ACS Catal., 2011, 1, 1637 CrossRef CAS; (d) M. R. Gagne, C. L. Stern and T. J. Marks, J. Am. Chem. Soc., 1992, 114, 275 CrossRef CAS.
- (a) P. D. Knight, I. Munslow, P. N. O'Shaughnessy and P. Scott, Chem. Commun., 2004, 894 RSC; (b) M. C. Wood, D. C. Leitch, C. S. Yeung, J. A. Kozak and L. L. Schafer, Angew. Chem., Int. Ed., 2007, 46, 354 CrossRef CAS PubMed; (c) A. L. Gott, A. J. Clarke, G. J. Clarkson and P. Scott, Chem. Commun., 2008, 1422 RSC; (d) K. Manna, S. Xu and A. D. Sadow, Angew. Chem., Int. Ed., 2011, 50, 1865 CrossRef CAS PubMed; (e) G. Zi, X. Liu, L. Xiang and H. Song, Organometallics, 2009, 28, 1127 CrossRef CAS; (f) K. Manna, W. C. Everett, G. Schoendorff, A. Ellern, T. L. Windus and A. D. Sadow, J. Am. Chem. Soc., 2013, 135, 7235 CrossRef CAS PubMed; (g) M. C. Hansen, C. A. Heusser, T. C. Narayan, K. E. Fong, N. Hara, A. W. Kohn, A. R. Venning, A. L. Rheingold and A. R. Johnson, Organometallics, 2011, 30, 4616 CrossRef CAS; (h) J. M. Hoover, J. R. Petersen, J. H. Pikul and A. R. Johnson, Organometallics, 2004, 23, 4614 CrossRef CAS.
- (a) P. Horrillo-Martínez and K. C. Hultzsch, Tetrahedron Lett., 2009, 50, 2054 CrossRef PubMed; (b) J. S. Wixey and B. D. Ward, Chem. Commun., 2011, 47, 5449 RSC; (c) X. Zhang, T. J. Emge and K. C. Hultzsch, Angew. Chem., Int. Ed., 2012, 51, 394 CrossRef CAS.
- (a) P. H. Martinez, K. C. Hultzsch and F. Hampel, Chem. Commun., 2006, 2221 RSC; (b) T. Ogata, A. Ujihara, S. Tsuchida, T. Shimizu, A. Kaneshige and K. Tomioka, Tetrahedron Lett., 2007, 48, 6648 CrossRef CAS PubMed; (c) J. Deschamp, J. Collin, J. Hannedouche and E. Schulz, Eur. J. Org. Chem., 2011, 3329 CrossRef CAS.
- (a) X. Shen and S. L. Buchwald, Angew. Chem., Int. Ed., 2010, 49, 564 CrossRef CAS PubMed; (b) B. W. Turnpenny, K. L. Hyman and S. R. Chemler, Organometallics, 2012, 31, 7819 CrossRef CAS PubMed; (c) Z. Zhang and R. A. Widenhoefer, Angew. Chem., Int. Ed., 2007, 46, 283 CrossRef CAS PubMed; (d) Z. Zhang, S. D. Lee and R. A. Widenhoefer, J. Am. Chem. Soc., 2009, 131, 5373 Search PubMed; (e) G. L. Hamilton, E. J. Kang, M. Mba and F. D. Toste, Science, 2007, 317, 496 CrossRef CAS PubMed.
- (a) S. Hong, S. Tian, M. V. Metz and T. J. Marks, J. Am. Chem. Soc., 2003, 125, 14768 CrossRef CAS PubMed; (b) X. Yu and T. J. Marks, Organometallics, 2007, 26, 365 CrossRef CAS.
- P. N. O'Shaughnessy, P. D. Knight, C. Morton, K. M. Gillespie and P. Scott, Chem. Commun., 2003, 1770 RSC.
- (a) J. Collin, J.-C. Daran, E. Schulz and A. Trifonov, Chem. Commun., 2003, 3048 RSC; (b) J. Hannedouche, I. Aillaud, J. Collin, E. Schulz and A. Trifonov, Chem. Commun., 2008, 3552 RSC; (c) I. Aillaud, K. Wright, J. Collin, E. Schulz and J. P. Mazaleyrat, Tetrahedron: Asymmetry, 2008, 19, 82 CrossRef CAS PubMed; (d) Y. Chapurina, J. Hannedouche, J. Collin, R. Guillot, E. Schulz and A. Trifonov, Chem. Commun., 2010, 46, 6918 RSC; (e) Y. Chapurina, H. Ibrahim, R. Guillot, E. Kolodziej, J. Collin, A. Trifonov, E. Schulz and J. Hannedouche, J. Org. Chem., 2011, 76, 10163 CrossRef CAS PubMed.
- (a) D. V. Gribkov, K. C. Hultzsch and F. Hampel, Chem. – Eur. J., 2003, 9, 4796 CrossRef CAS PubMed; (b) D. V. Gribkov, K. C. Hultzsch and F. Hampel, J. Am. Chem. Soc., 2006, 128, 3748 CrossRef CAS PubMed; (c) A. L. Reznichenko, H. N. Nguyen and K. C. Hultzsch, Angew. Chem., Int. Ed., 2010, 49, 8984 CrossRef CAS PubMed.
- (a) J. Y. Kim and T. Livinghouse, Org. Lett., 2005, 7, 1737 CrossRef CAS PubMed; (b) N. Meyer, A. Zulys and P. W. Roesky, Organometallics, 2006, 25, 4179 CrossRef CAS; (c) P. Benndorf, J. Jenter, L. Zielke and P. W. Roesky, Chem. Commun., 2011, 47, 2574 RSC.
- (a) L. Xiang, Q. Wang, H. Song and G. Zi, Organometallics, 2007, 26, 5323 CrossRef CAS; (b) G. Zi, L. Xiang and H. Song, Organometallics, 2008, 27, 1242 CrossRef CAS.
- N. D. Shapiro, V. Rauniyar, G. L. Hamilton, J. Wu and F. D. Toste, Nature, 2011, 470, 245 CrossRef CAS PubMed.
- (a) K. Mikami and M. Lautens, New Frontiers in Asymmetric Catalysis, J. Wiley and Sons, Inc., Hoboken, NJ, 2007 Search PubMed; (b) B. List, Asymmetric Organocatalysis, Springer, New York, 2010 Search PubMed.
- X. Wang, Z. Chen, X.-L. Sun, Y. Tang and Z. Xie, Org. Lett., 2011, 13, 4758 CrossRef CAS PubMed.
- C. He, J. Ke, H. Xu and A. Lei, Angew. Chem., Int. Ed., 2013, 125, 1527 CrossRef PubMed.
- L. E. N. Allan, G. J. Clarkson, D. J. Fox, A. L. Gott and P. Scott, J. Am. Chem. Soc., 2010, 132, 15308 CrossRef CAS PubMed.
- D. C. Leitch, R. H. Platel and L. L. Schafer, J. Am. Chem. Soc., 2011, 133, 15453 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Complete experimental details, characterization data, kinetic plots, and crystallographic data. CCDC 1016330–1016342 for 1–3, 4a,b,c,d, 5a,b,c,d, 8, and 9. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc10032h |
| This journal is © The Royal Society of Chemistry 2015 |