
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Targeting G-quadruplex structures with extrinsic fluorogenic dyes: promising fluorescence sensors
Achikanath C.
Bhasikuttan
* and
Jyotirmayee
Mohanty
*
Radiation & Photochemistry Division, Bhabha Atomic Research Centre, Mumbai 400 085, India. E-mail: bkac@barc.gov.in; jyotim@barc.gov.in; Fax: +91-22-25505331/25519613; Tel: +91-22-25593771
First published on 13th February 2015
Abstract
The research on the G-quadruplex DNAs has received much attention in recent years and numerous reports appeared probing their detection, structure, stability, reactivity, selectivity, etc. for the chemical intervention of their biological activity or sensor applications. This feature article provides an account of the recent reports from different research groups on the intriguing fluorescence properties showcased by certain fluorogenic dyes upon their binding to the G-quadruplex DNAs. Aptly, these selective and sensitive emission features demonstrated with structure specific G-quadruplex DNAs have been turned into label-free fluorescence-based detection methods for various metal ions and small biomolecules, down to the pico molar range, having promising bio-analytical applications. While the in vivo formation of G-quadruplexes is dynamically sensitive to the cell cycle, in tandem with the in vitro applications, it is essential to understand the factors that affect chemical, biological and genetic roles of the G-quadruplex structures plausible along the human genome. Towards this, the recent findings on the quantitative visualization of the quadruplex structures in the human cells using immunofluorescent probes open up avenues to explore highly specific quadruplex responsive agents for diagnostic and therapeutic applications, especially to develop a clinically viable method for cancer treatment.
 A. C. Bhasikuttan | Achikanath C. Bhasikuttan obtained his MSc in Chemistry from University of Calicut, Kerala, in 1989 and joined Bhabha Atomic Research Centre, Mumbai, India, in 1991 after a one year advanced orientation course conducted by the institute. After his PhD from the University of Mumbai in 1998 he joined as a JSPS postdoctoral fellow at Osaka University, Japan, 1999–2001. His research interests include the excited state molecular dynamics and to probe the intricacies of non-covalent interactions in supra-biomolecular systems. He is a recipient of Scientific & Technical Excellence Award – 2009 from DAE, Bronze medal from Chemical Research Society of India (CRSI) – 2014. He is a fellow of the Maharashtra Academy of Sciences & the National Academy of Sciences, India. |
 Jyotirmayee Mohanty | Jyotirmayee Mohanty obtained her MSc in Chemistry from Utkal University, Odisha, in 1992 and joined Bhabha Atomic Research Centre, Mumbai, India, in 1994 after one year advanced orientation course conducted by the institute. After her PhD from the University of Mumbai in 2002, she carried out her postdoctoral research at MPIBPC, Göttingen, and JUB, Bremen, Germany, 2002–2004. Her current research interests focus on the dynamics of noncovalent supra-biomolecular assemblies, tuning their molecular properties and exploring their photofunctional activities towards various applications. She is a recipient of the Distinguished Lectureship Award – 2009 from the Chemical Society of Japan, APA-Prize for Young Scientist – 2010, Samanta Chandra Sekhar Award – 2011, Scientific & Technical Excellence Award – 2011 from DAE, and a fellow of the Maharashtra Academy of Sciences and the National Academy of sciences, India. She has also received the prestigious Humboldt Fellowship for Experienced Researchers from Alexander von Humboldt Foundation. |
1. Introduction
Nucleic acids or the DNAs/RNAs contain the genetic information essential for life. In particular, their sequence encodes vital instructions for the cell, storage and replication of hereditary information.1 The DNAs usually assume a standard double helix conformation and have been the subject of extensive studies by chemists, biologists, physicists and theoreticians.2,3 However, in the last two decades, it was recognized that certain sequence specific DNAs under changed ambient conditions, adopt several alternative conformations having specific roles during cell life.4 DNA is a very dynamic molecule, capable of forming a number of spatial arrangements, which include single-stranded hairpins, homo-duplexes, triplexes, and quadruplexes, under certain physiological conditions.3,5 On these attributes, several research groups have envisaged the involvement of such noncanonical structures in the recombination, regulation of gene expression and proliferation of tumor cells etc. as potential therapeutic targets.3,6On the other hand, guanine (G), one of the nucleobases present in DNAs/RNAs, has long been known to self associate by π–π stacking or by forming G-tetrads (or G-quartets), formed by four guanines hydrogen bonded to each other and are further stabilized by Hoogsteen hydrogen-bonds (vide infra).7–10 In other words, the presence of short Guanine rich stretches in the oligonucleotide/DNA (or RNA) strands can also form stacks of such hydrogen-bonded G-tetrads, either an inter-strand or intra-strand folding pattern, apparently directed by the nucleotide sequence and solution conditions.7,11,12 That is to say, the DNA structures having two or more such stacked G-tetrads are now broadly termed as G-quadruplexes.10,13 Guanine-rich oligonucleotides or DNAs that can form G-quadruplex structure is now believed to be an important DNA structural motif that is prevalent in the human genome and is in the center stage of nucleic acid research.10,14,15
G-quadruplex structure is considered to be functionally important in the mammalian genome for transcriptional regulation, DNA replication and genome stability and their presence can be explored in assessing cellular functions, or can be utilized for chemical intervention of biological activities.16,17 In the recent years, G-quadruplex structures have drawn the immense attention of researchers in other interdisciplinary areas of nucleic acid research, genomics, supramolecular chemistry, and spectroscopy.7,18 It is known that G-quadruplex forming sequences are found throughout the genome with more than 40% of all human genes having a potential G-quadruplex forming sequence located within 1 kb of the gene start site.19 This folded higher order structures cannot bind to proteins required to form the transcriptionally active complex thereby down regulating the gene having G-rich promoter sequences.9,19 For this reason, the G-quadruplex structures, which are believed to be potential drug targets, are examined for their interaction with small molecules20 or metal ions, that can selectively affect its formation, recognize specific structures, and incorporate stimuli responsive fluorescence features. All these contribute immensely towards the development of biosensors and anti-cancer therapeutics.9,10,16,20
Even though G-quadruplexes have been extensively studied for more than 20 years,3,4,7,10,21–25 the exact nature of their biological significance, apart from the telomerase activity of the single strand telomere overhang, is still poorly understood.7 At present, other than their increasing biological significance, the G-quadruplex structures are gaining interest in therapeutics for anticancer treatment, as sensors for bio-analytical applications, as nano-materials/nano-wires for light harvestng and few number of reviews have appeared segregating such applications.4,6–8,10,12,24–27 G-quadruplex structure is known to be sensitive to the stabilizing cations (e.g. Na+vs. K+).3 These properties raise the possibility that sensors or switches could be fabricated using G-quadruplexes. The changes in the fluorescence behaviour of the dyes/ligands in the presence of a G-qudruplex have been utilized to visualize G-quadruplex formation in a human cell11,28 and to detect metal ions29–32 and small biomolecules such as amino acids, histidine (His), cysteine (Cys), biothiol,33–35 in nanomolar range. In this feature article we have overviewed some of the recent reports in this context and focus on the formation, stabilization, ligand binding of certain G-quadruplexes in the presence of biologically/technologically relevant small molecules and their prospective utilization as sensors for various applications.
2. G-quadruplexes
G-quadruplexes are non-canonical secondary DNA/RNA structures formed by the stacking of two or more planar and hydrogen bonded G-tetrads with either intra- or intermolecular association and are usually found in guanine-rich oligonucleotide sequences.3,14 In these π–π stacked G-tetrads, the four guanines are held in a planar arrangement through Hoogsteen hydrogen bonding through N2 and N7, and N1 and O6 (Chart 1).14 Since a G-tetrad has four O6 oxygen atoms clustered in its center, a cationic charge is essential for its stabilization (Chart 1).14 Hence, G-quadruplexes act as strong ligands for metal ions such as Na+, K+ and various cationic molecules. G-quadruplex structures are polymorphic, having a number of topologies with distinct structural identity under various physiological conditions.3 The preferential topology of the stable quadruplex structures adopted by a G-rich sequence depends on the nature of cations.3 Quadruplexes can be formed from one, two or four separate strands of G-rich DNA (or RNA) and can display a wide variety of topologies, which are in part, a consequence of various possible combinations of strand direction, as well as variations in loop size and sequence.3,14 Depending on the direction of the strands (5′ to 3′) or parts of a strand that form the tetrads, the quadruplex structures may be described as parallel, anti-parallel or hybrid (Chart 1).14 If the 5′ to 3′ direction in all the four pillar strands is the same, the quadruplex is termed parallel quadruplex. If the opposite (or alternate) strands run in opposite directions (one 5′ to 3′ and the other 3′ to 5′), the quadruplex is said to have adopted an anti-parallel folding (Chart 1). A mixed combination of both the strand direction is also seen in some specific DNA sequences.25 The complexity of G-quadruplex structures with different topologies depends on various parameters: the oligonucleotide sequence; the number of oligonucleotide strands involved (e.g., unimolecular, bimolecular, tetramolecular); the directionality of strands (e.g., parallel, antiparallel, mixed); the size and type of intervening loops (e.g., diagonal loops, lateral loops and double chain reversal loops) and the environmental factors, such as the interacting alkali metals, the molecular crowding and the presence of binding ligands.3,6,14 The combination of all these factors brings out significant structural distinction of the quadruplex structures, which reflects explicitly on their behavior towards ligand binding, stability and functionality.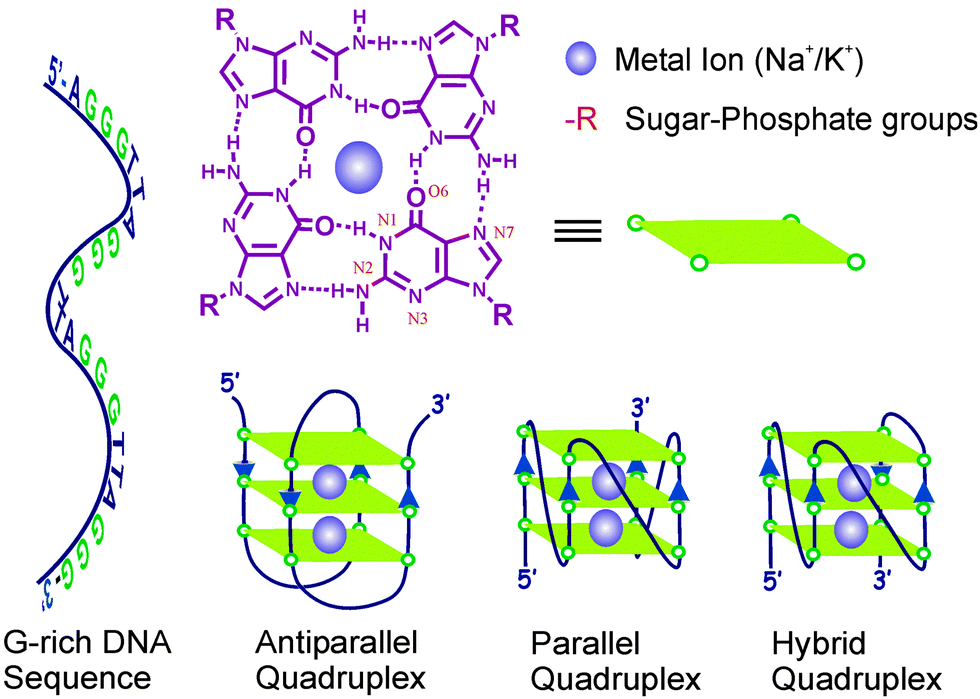 | ||
| Chart 1 Structures of the G-quartet/tetrad and the various common folding topologies found in G-quadruplexes. | ||
G-quadruplex structures have received widespread attention because of their promising utilities in not only in vitro, but also in vivo applications.19 Their presence has been manifested in diverse biological functions such as gene regulation, chromosomal stability by blocking cellular polymerases and helicases, serving as intermediates in recombination, and telomerase activity.19,36 Recent bioinformatics analyses showed that ∼400![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 putative quadruplex forming sequences exist throughout the human genome.6,36,37 Other than in human telomeric DNA, these sequences are frequently enriched within the promoter regions of oncogenes including C-MYC, C-KIT, H-RAS, and K-RAS, untranslated regions of mRNAs and telomeres, suggesting that quadruplex structures may play a pivotal role in the control of a variety of cellular processes, including telomere maintenance, replication, transcription and translation.9 In fact, telomeres have received much attention in this regard since they can fold into several distinct intramolecular G-quadruplexes, leading to the rational design and development of G-quadruplex-stabilizing molecules for their control.6,25,27
000 putative quadruplex forming sequences exist throughout the human genome.6,36,37 Other than in human telomeric DNA, these sequences are frequently enriched within the promoter regions of oncogenes including C-MYC, C-KIT, H-RAS, and K-RAS, untranslated regions of mRNAs and telomeres, suggesting that quadruplex structures may play a pivotal role in the control of a variety of cellular processes, including telomere maintenance, replication, transcription and translation.9 In fact, telomeres have received much attention in this regard since they can fold into several distinct intramolecular G-quadruplexes, leading to the rational design and development of G-quadruplex-stabilizing molecules for their control.6,25,27
Human telomeric DNA, which attracted large number of studies related to quadruplex formation, comprises tandem repeats of the sequence 5′-TTAGGG-3′, and is present as a 3′ overhang at the ends of chromosomes.25,38 Telomere length decreases with each cell division in normal somatic cells.23 Consequently, these progressive decreases limit the proliferative potential of cells.20,23,25 In contrast to normal cells, 80–85% of human tumor cells have functional telomerase that elongates telomere DNA.23,25,27,39 Quadruplex folding inhibits telomerase activity because G-quadruplex DNA cannot function as a substrate for telomerase; therefore, development of G-quadruplex-ligands that inhibit telomerase activity via induction or stabilization of a G-quadruplex has become an area of great interest.23,25,27 These bioinformatic studies clearly indicate that G-quadruplexes can influence carcinogenesis by modulating transcription of oncogenes. Notably, some of the G-quadruplex binding ligands regulate the expression of these oncogenes by binding to G-quadruplexes in the promoter regions. Thus, ligands that recognize and bind to G-quadruplexes in telomere DNA and/or promoter regions of oncogenes are promising anticancer drugs.9,25,27 In this perspective, small molecules, metal ions, metal–ligand complexes have garnered immense attention for developing quadruplex-specific ligands/drugs, which can chemically interfere with the quadruplex activity as therapeutic agents.9 In fact, a wide range of quadruplex-specific small molecules have been reported so far with some of them showing very promising biological activities.6
2.1. Mechanistic insight into ligand binding
As discussed above, the central core of the G-tetrad/quadruplex is negatively polarized due to the orientation of the carbonyl groups from each guanine base towards the centre of the G-tetrad.7,10,14 Hence, metal ions such as Na+, K+, etc. and small cationic organic molecules are essential for the stabilization of the G-quadruplex.3,7,10,14 Practically, in most of the studies the preformed G-quadruplex templates, prepared in the presence of Na+/K+ ions are used to study the uptake of potential chromophoric guests on to it. Additionally, small organic molecules having large π-planar aromatic surface can interact with the planar G-tetrads via strong π–π stacking interactions. For example, the structural features of 5,10,15,20-tetra(N-methyl-4-pyridyl)porphyrin (TMPyP) allow the molecule to bind to a human telomeric G-quadruplex with high affinity via π–π stacking and electrostatic interactions.24,25,40 Nevertheless, the loop sizes and topology, which vary from one form of quadruplex to the other, are also very decisive in binding and stabilizing the quadruplex motifs. Therefore, while designing, it is important to consider ligand interactions with different types of loops and grooves.40 Precisely, in addition to the structural features of the central core of the ligands, the side chains also play critical roles in achieving the selectivity among the quadruplex DNAs. In designing ligands/dyes specific to quadruplexes, it is particularly hard to discriminate among the quadruplxes and also to maintain only very low affinity for duplex DNA. Most of the ligands are usually polyaromatic molecules, such as N,N′-(9-(4-(dimethylamino) phenylamino) acridine-3,6-diyl)bis(3-(pyrrolidin-1-yl) propan amide) trihydrochloride (BRACO-19), 3,11-difluoro-6,8,13-trimethyl-8H-quino[4,3,2-kl]acridinium methosulfate (RHPS4), luoroquinolones (Quarfloxin) and telomestatin, that stack in plane at the end tetrad.23,27 In addition, ligands can bind to the grooves of the G-quadruplexes or even, in principle, intercalate between the tetrads. Pradeepkumar et al.41 report that the naphthyridine-based ligands with quinolinium and pyridinium side chains form a promising class of quadruplex DNA stabilizing agents having high selectivity for quadruplex DNA structures over duplex DNA structures. In addition to achieving the end stacking with the pyridine/naphthyridine/phenanthroline moieties, optimization of side chains is also demonstrated in discriminating promoter G-quadruplex DNAs over telomeric G-quadruplex DNAs.41,42The studies on the formation and interaction of the G-quadruplex with small organic molecules and its characteristics are mainly looked at by several spectroscopic measurements, such as UV-vis absorption, fluorescence measurements or fluorescence intercalator displacement assay, circular dichroism (CD), etc. Since the quadruplex structures are chiral, they can be directly monitored by CD measurements. The parallel, anti-parallel, hybrid type and various other possible quadruplex structures can be distinguished by the position and nature of the CD spectra. Particularly, a positive peak at 295 nm and a trough at 265 nm describe an anti-parallel structure whereas a positive peak at 260 nm and a trough at 240 nm is indicative of an all-parallel structure.14 On the other hand, probing the photophysical changes in the extrinsically added ligands/dyes do provide convenient alternative spectroscopic signatures to follow the quadruplex dynamics, its formation, and stability. In this article, we highlight the recent progress in the studies of certain extrinsic fluorogenic dyes in stabilizing and detecting G-quadruplexes and the use of their apparent fluorescence enhancement for metal ion detection and sensor applications.
3. Extrinsic fluorogenic dyes with G-quadruplexes
Noncovalent, extrinsic fluorescent probes find extensive usage as local reporters in many biological applications, especially in various fields of protein/DNA analysis, e.g., for the sensitive and selective detection of distinct assemblies from proteins and DNA.43 Here, the specific interaction with the DNA environment introduces considerable change in the photophysical characteristics of the dye, projecting the details of its local microenvironment. Fluorescent probes/markers provide a powerful and direct means for studying the folding and localization of biological macromolecules in living cells. Fluorescent probes capable of structure-specific reporting of G-quadruplex structures in vivo will provide a much needed tool for basic biological research and the exploration of G-quadruplex DNA as a potential drug target.15,24 Many of the G-quadruplex probes highlighted here exhibit promising activity in vitro, but give cellular staining patterns that cannot necessarily be interpreted as originating from G-quadruplex structures in vivo. This is an especially challenging goal. There are a limited number of dyes that display a strong modulation in fluorescence behavior when bound to DNAs; however, by and large, none of them exhibits a marked structural selectivity towards quadruplex DNAs, which is a challenge to engineer structure-specific G-quadruplex inducing/stabilizing agents for targeted therapeutic and diagnostic applications. On the other hand, fluorogenic dyes exhibiting viscosity/rigidity dependent radiative properties act as sensitive fluorescent probes for such structural transitions and to understand the factors governing molecular recognition, especially in dynamic biomolecular assemblies like quadruplexes.44 In this context, cationic fluorogenic dyes like thioflavin T (ThT: 1, Chart 2), triphenylmethane (TPM) dyes, thiazole orange (TO: 2, Chart 2), triphenylamine (TPA) dyes, which are very weakly fluorescent in aqueous solution become highly fluorescent upon binding to G-quadruplex templates. The fluorescence behavior of these dyes undergo dramatic enhancement in the intensity and shows obvious bathochromic or hypsochromic shift in the spectral position. The striking enhancement in the emission has been very useful for the visualization of the G-quadruplex structures through fluorescence imaging.11,28 In a recent study based on its fluorescence behavior, it has been demonstrated that thioflavin T induces G-quadruplex formation and also acts as a selective sensor with respect to other DNAs.45 Thiazole orange (2), another fluorogenic dye, is weakly fluorescent in aqueous solution,46 but becomes bright upon binding to G-quadruplex DNA and is usually used for the fluorescence intercalator displacement assay.41 Several porphyrin derivatives and metal complexes have also been shown to be responsive to metal ions on their binding with G-quadruplex moiety. Thus the advantages of inducing and stabilizing and visualizing the G-quadruplex structure in guanine rich sequences through small organic fluorescent molecule have found direct relevance as useful diagnostic sensor probes and potential therapeutic agents for anticancer treatment.9,16 Some of these reports, which are directly on the use of extrinsic dyes with G-quadruplex motifs, are discussed briefly in the following sections. | ||
| Chart 2 Chemical structures of benzothiazole-based dyes. | ||
3.1. Interaction of G-quadruplex with thioflavin T
It has been found that many of the quadruplex binding dyes do equally interact with other DNA forms like the single strand or duplex DNAs, which make them nonspecific for quantitative measurements.25,49–51 Therefore, there was a need to develop quadruplex specific dyes/ligands which can selectively induce and stabilize quadruplex structures and in situ function as a selective fluorescent probe for in vivo visualization of key cellular processes. With this aim, in a recent study we, in collaboration with Pradeepkumar’s group,45 investigated the interaction of 1 with human telomeric DNA (22AG: 5′-AGGGTTAGGGTTAGGGTTAGGG-3′) and compared it with other single stranded (ss-) or double stranded (ds-) DNAs as well as with the calf thymus DNA (ct-DNA).45 Gradual titration of the 22AG human telomeric DNA to the ThT solution, resulted in significant bathochromic shift in the ThT absorption profile with a concomitant enhancement in the fluorescence intensity (I/I0) to the order of ∼1500-fold as displayed in Fig. 1.
 | ||
| Fig. 1 (A) Absorption and (B) fluorescence spectra of 1 solution (∼3 μM) containing 5 mM Tris (pH 7.2) with [22AG]/μM: (1) 0, (2) 0.12, (3) 0.25, (4) 0.75, (5) 1.5, (6) 2.5, (7) 4.0, (8) 5.0. Reprinted with permission from ref. 45. Copyright (2013) American Chemical Society. | ||
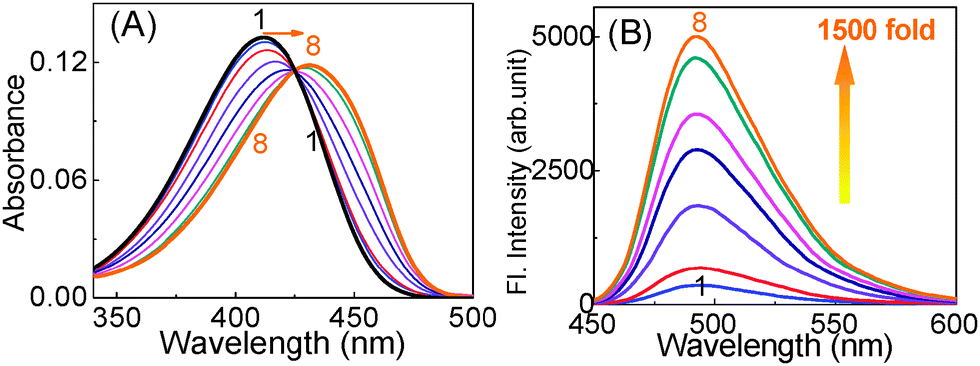
The interaction with a quadruplex structure induced by the cationic ThT itself was further confirmed from a relative study with a pre-folded G-quadruplex in the presence of metal ions (K+), which provided unusually large shift in the absorption maxima (Fig. 2A) and huge enhancement in the emission intensity to ∼2100 fold (Fig. 2B).45 However, similar measurement in the presence of Na+ provided only very low emission enhancement only to an extent of ∼300 fold, projecting a viable method for distinguishing between Na+ and K+. The apt and strong noncovalent interactions experienced by the ThT on the G-quadruplex DNA, either by end stacking, groove binding or intercalation, upholds the dye in rigid and more planar form, thus severely restricting its otherwise highly feasible nonradiative torsional relaxation channel.48,52,53
 | ||
| Fig. 2 Absorption (A) and fluorescence (B) spectra of 1 (∼3 μM) upon titration with prefolded 22AG quadruplex DNA in solution containing 50 mM Tris (pH 7.2) and 50 mM KCl and [22AG]/μM; (1) 0, (2) 0.5, (3) 1.0, (4) 1.5, (5) 2.5, (6) 3.5, (7) 4.5, (8) 6.0, (9) 8.0, (10) 12.0. λex for B is at 425 nm. Reprinted with permission from ref. 45. Copyright (2013) American Chemical Society. | ||
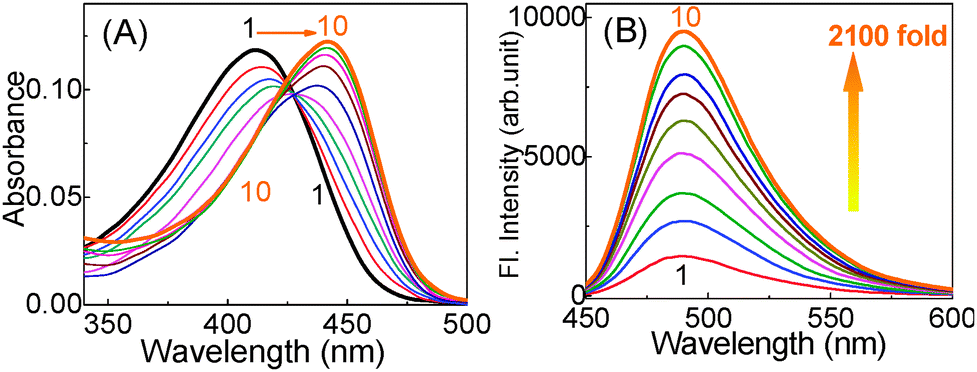
In the CD measurements the 22AG DNA in the presence of 1 (without any buffer) displayed a characteristic CD band i.e. a strong positive band centered at 294 nm and a negative band at 265 nm (Fig. 3A), which are very much the characteristics of anti-parallel G-quadruplex DNA. However, in the presence of 50 mM tris buffer, a neat switch over of the anti-parallel structure to the parallel structure, having CD bands at 265 nm and the 240 nm has been documented (Fig. 3B). It has been also noted that the parallel/antiparallel quadruplexes formed in the presence of K+ quantitatively transformed into only anti-parallel topology after the addition of 1. These changes are schematically presented in Scheme 1.
 | ||
| Fig. 3 CD spectra recorded for 22AG DNA (12.5 μM) in various Tris buffer concentrations with 1. (A) Solution in unbuffered; (B) buffered (5 mM Tris pH 7.2) with 1: (1) 0 eq.; (2) 2 eq.; (3) 4 eq.; (4) 8.eq. Adapted with permission from ref. 45. Copyright (2013) American Chemical Society. | ||
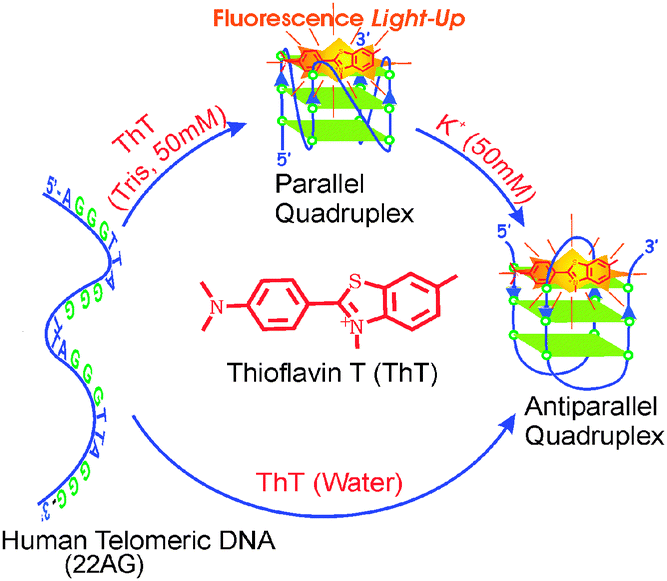 | ||
| Scheme 1 Schematic representation of the topological transformation in the 22AG human telomeric DNA to parallel and antiparallel quadruplex in presence of 1/Tris/K+. Reprinted with permission from ref. 45. Copyright (2013) American Chemical Society. | ||
The fluorescence enhancement revealed in 1 upon binding to an anti-parallel quadruplex (>2100 fold) is found to be by far above from the emission enhancement observed with DNAs of nonspecific sequences, as ss-/ds-DNAs and ct-DNAs. The Fig. 4A shows a comparison of the overall emission intensity enhancement monitored at 490 nm for different DNA forms with that of 22AG in the absence and presence of salt. The striking fluorescence light-up in ThT dye upon binding to the human telomeric G-quadruplex, in the absence and presence of salt (especially K+), is shown to be highly specific/selective compared to the less than 250 fold enhancement observed with other DNA strands.
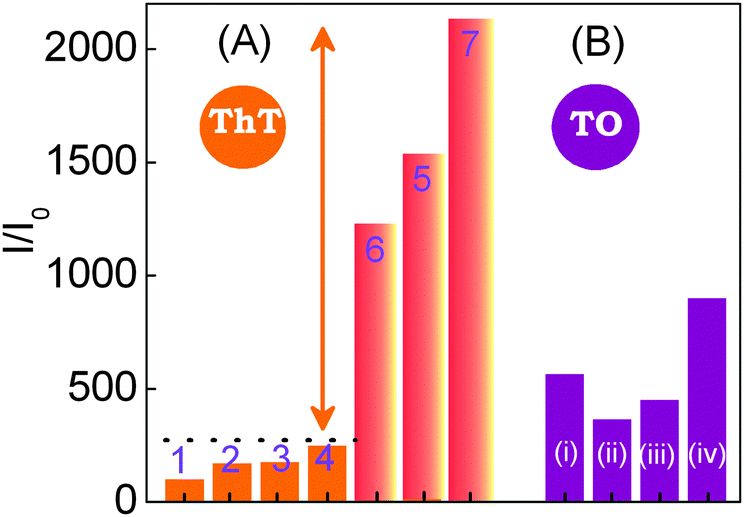 | ||
| Fig. 4 Comparison of emission intensity enhancement in 1 and 2. (A) Emission intensity of 1 in various DNAs. (1) in the absence of nucleic acids (1 alone x100); (2) ss-DNA2 (20 μM, 10 mM Tris, pH 7.2); (3) ds-DNA (20 μM, 10 mM KCl, 10 mM Tris, pH 7.2); (4) ct-DNA (300 μM, 10 mM Tris, pH 7.2); (5) 22AG DNA (8 μM) in the absence of salt and buffer; (6) 22AG DNA (8 μM, 50 mM Tris, pH 7.2); (7) prefolded 22AG DNA (10 μM) with 50 mM Tris (pH 7.2) and 50 mM KCl. (B) Emission intensity of 2 in various DNAs: (i) ds-DNA (8 μM, 10 mM KCl, 10 mM Tris, pH 7.2); (ii) 22AG DNA (8 μM, no buffer, no salt); (iii) ct-DNA (150 μM, 10 mM Tris, pH 7.2); (iv) pre-folded 22AG DNA (8 μM) with 50 mM Tris (pH 7.2) and 50 mM KCl. Reprinted with permission from ref. 45. Copyright (2013) American Chemical Society. | ||
In a comparative study with TO (2), a widely used quadruplex binder dye, the emission enhancement with various DNA strands displayed fluorescence enhancement in the range of 300–500 fold, which are by and large at par Fig. 4B. This first hand report on ThT–G-quadruplex binding clearly ascertains the dual role of ThT as an efficient inducer and selective fluorescent sensor of quadruplex DNA over other DNA forms.45
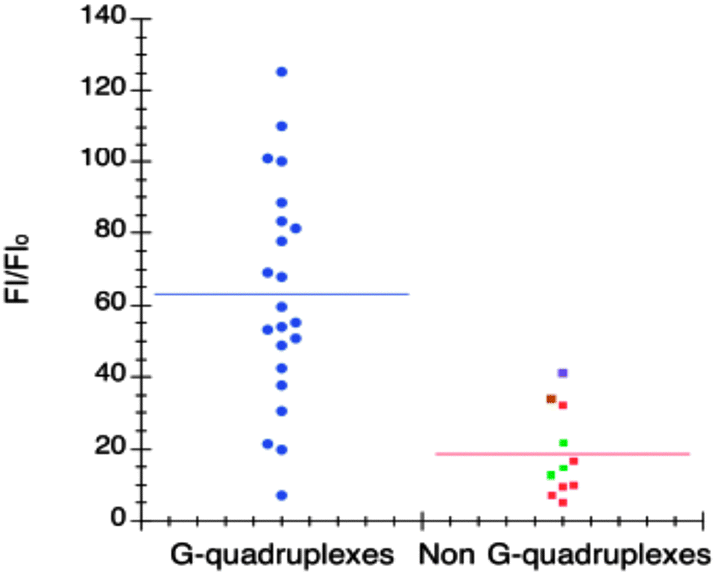 | ||
| Fig. 5 Each point corresponds to fluorescence enhancement of 1 in the presence of a different oligonucleotide. The change in fluorescence emission is plotted for DNA and RNA quadruplexes on the left (in blue) and non-G-quadruplex structures on the right. Green dots correspond to trinucleotides, purple dot to parallel-duplex, brown dot to the triplex and red to other nonquadruplex-forming sequences. Reproduced with license from ref. 54, copyright (2014) Oxford University Press. | ||
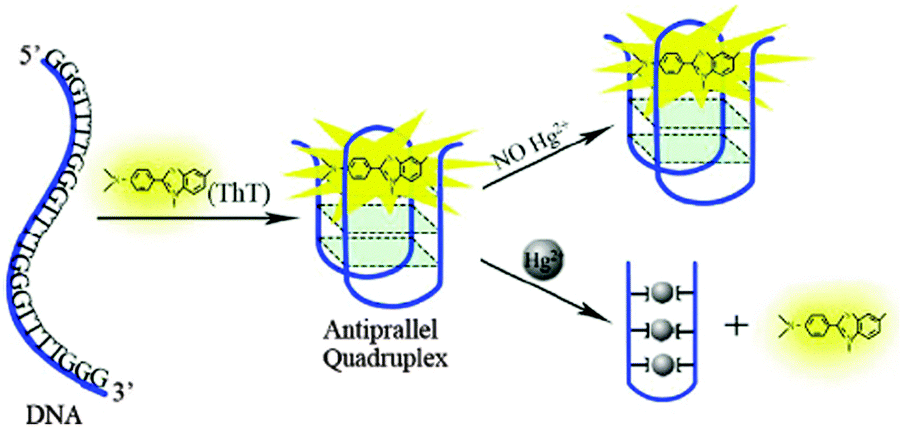 | ||
| Scheme 2 Scheme for the Hg2+ inhibits ThT folding G-rich DNA with many T-loop residues into the G-quadruplex. Reproduced with permission from ref. 29, copyright (2014) Elsevier B.V. | ||
With the addition of nano-mole concentration of Hg2+, the fluorescence sharply decreased as the G-quadruplex is changed to Hg2+ mediated T–Hg–T double-stranded DNA. Following this conformational change, the ThT gets released from the G-quadruplexes, which brings back the ThT fluorescence to off state. This led to the demonstration of a highly sensitive label-free sensor for Hg2+ and a detection limit of 5 nM was achieved against other metal ions (Fig. 6).29 This label-free fluorescence spectrometric Hg2+ detection is simple, quantitative, sensitive, and highly selective and much better than the other optical sensors used for Hg2+ detection (Table 1) and is a apt sensing platform for Hg2+ bioactivity analysis.29
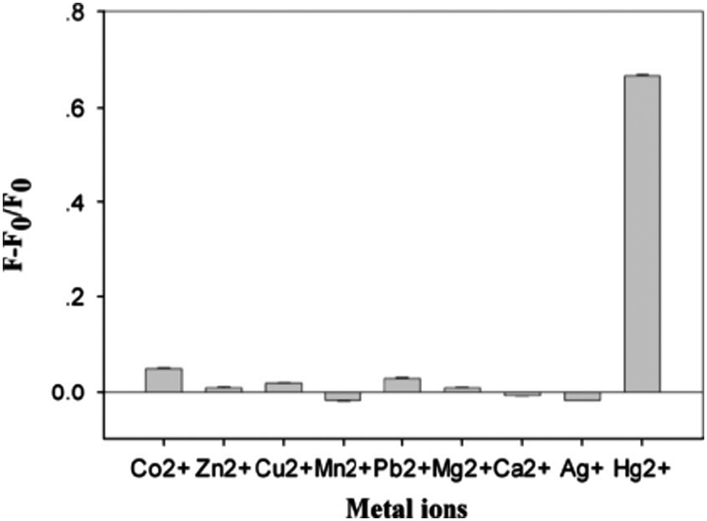 | ||
| Fig. 6 Selectivity of ThT induced probe1 form a G-quadruplex structure for the detection of Hg2+. The concentrations of Hg2+ is 5 μM and other metal ions are (Mg2+ and Ca2+ at 100 μM, Zn2+, Co2+, Ag+ and Mn2+ ions at 20 μM, Pb2+ and Cu2+ at 5 μM) respectively. F0 and F are the fluorescence intensity in the absence and presence of Hg2+, respectively. The error bars represented for standard deviation across three repetitive experiments. Reproduced with permission from ref. 29, copyright (2014) Elsevier B.V. | ||
| Hg(II) ion sensor | Detection limit | Method |
|---|---|---|
| Au nanoparticles: gold nanoparticles; MSO: mercury specific oligonucleotide; PMNT: poly(3-(3′-N,N,N-triethylamino-1′-propyloxy)-4-methyl-2,5-thiophene hydrochloride). | ||
| Au nanoparticles | 100 nM | Colorimetric |
| Au nanoparticles | 3 μM | Naked eye |
| Au nanoparticles | 6 μM | Colorimetric |
| MSO-PMNT | 42 nM | Fluorometric |
| MSO oligonucleotide | 40 nM | Fluorometric |
| MSO-ThT | 5 nM | Fluorometric |
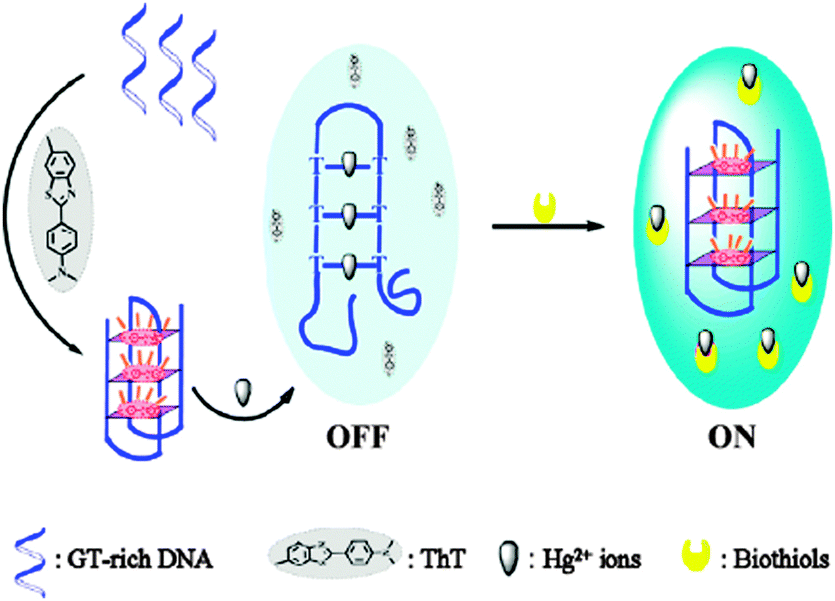 | ||
| Scheme 3 Schematic illustration of the fluorescence changes of the ThT under different conditions. Reproduced with permission from ref. 34, crown copyright (2013) Elsevier B.V. | ||
The present assay allows for the selective determination of Cys and GSH in the range of 20 nM–2.5 μM and 30 nM–2.0 μM with a detection limit of 8.4 nM and 13.9 nM, respectively.34
The system is simple in design and economically viable in operation. Moreover, it eliminates the need for dye-modified oligonucleotide or the sophisticated synthetic and separation process. The protocol exhibits excellent selectivity for the determination of Cys and GSH over other amino acids as documented in Fig. 7.34 The diagnostic capability and potential in practical applications of this method have been demonstrated by detecting biothiols in human blood serum,34 which proposed its practicability for diagnostic purposes.
 | ||
| Fig. 7 Fluorescence intensity change (F − F0) of ThT/G-quadruplex at λex = 485 nm in the presence of various amino acids, BSA, where F and F0 are fluorescence intensities of the ThT/G-quadruplex in the presence and absence of a different amino acid, BSA. The error bar represents the standard deviation of three measurements. The final concentrations of ThT dye, AGRO100, Hg2+ ions, Cys/GSH and the other 19 kinds of amino acids are 0.20 μM, 0.50 μM, 6.0 μM, 2.0 μM and 20.0 μM respectively. The concentration of BSA was 20 mg L−1. Reproduced with permission from ref. 34, crown copyright (2013) Elsevier B.V. | ||
The fluorescent intensities of the samples containing no T1 and Exo III are nearly close to that of 1, indicating no ThT responsive quadruplex formation.35 The fluorescence intensity does not enhance even in the presence of T1 or Exo III, respectively. However, upon simultaneous addition of T1 and Exo III, a great fluorescence enhancement is achieved, which indicated that T1 acts as a trigger of the Exo III digestion reaction and liberate Gs to fold into a quadruplex.35 Due to the binding sites in the quadruplexes being more specific and rigid for 1, the fluorescence intensity increases dramatically (Scheme 4).35
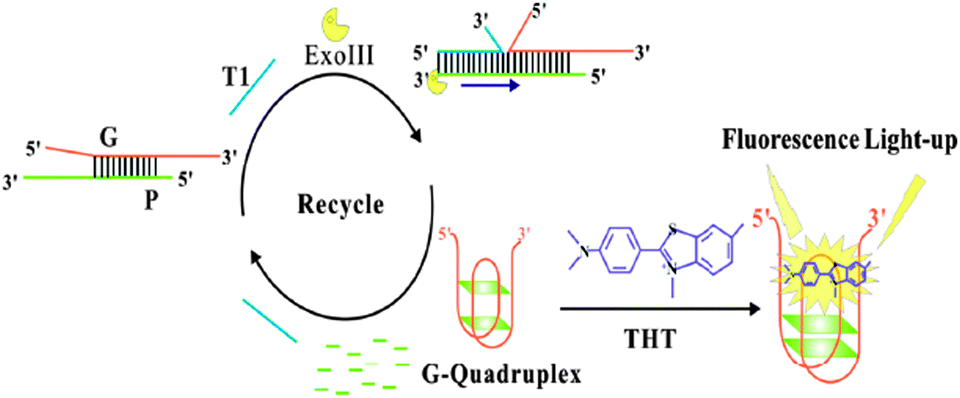 | ||
| Scheme 4 Schematic illustration of the sensor for amplified DNA detection. Reproduced with permission from ref. 35, copyright (2014) Elsevier B.V. | ||
The sensitivity of the proposed fluorescent biosensor for accurate quantification of T1 is investigated by varying the concentration of T1 under the optimized assay conditions. As shown in Fig. 8, a dramatic increase of fluorescence intensity is observed as the concentration of T1 increase from 100 fM to 50 pM. The intensity is linear to the concentration of T1 in the range from 100 fM to 1 pM (Fig. 8, inset) with a detection limit of 20 fM.35 The repeatability of the biosensor was assessed by analyzing 500 fM target DNA and the relative standard deviation of eight replicate determinations was 1.46%.
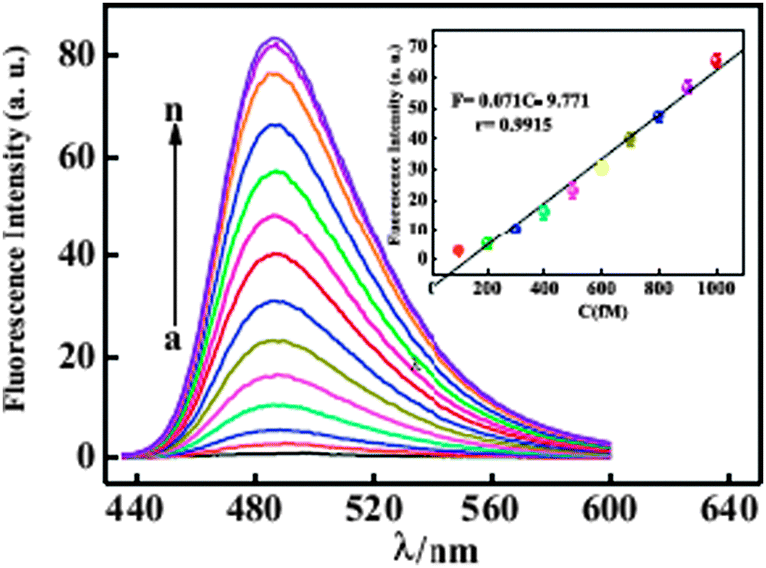 | ||
| Fig. 8 Fluorescence emission spectra of ThT + G/P in the presence of Exo III and various concentrations of T1 (from a to n): 0.00 fM, 100 fM, 200 fM, 300 fM, 400 fM,500 fM, 600 fM, 700 fM, 800 fM, 900 fM, 1 pM, 5 pM, 10 pM, 50 pM. Inset: a calibration curve demonstrating peak fluorescence intensity versus T1 concentration. Reproduced with permission from ref. 35, copyright (2014) Elsevier B.V. | ||
The sensor can detect as low as 20 fM target DNA and exhibits ultrahigh discrimination ability even for the detection of single-base mismatch. The biosensor has a strong resistance to the complex matrix of saliva, and can be used to detect ultra-trace amounts of T1 in real saliva samples.35 These features, as well as its other advantages, such as label-free, easy-to-use, fast and inexpensive technique, make it a promising candidate for use as a non-invasive early diagnosis method for breast cancer detection in saliva, especially in developing parts of the world where they lack adequate medical facilities. It is envisioned that this strategy would offer a universal approach for non-invasive and early diagnosis of many kinds of cancer just by designing rational DNA probe according to the target sequence.
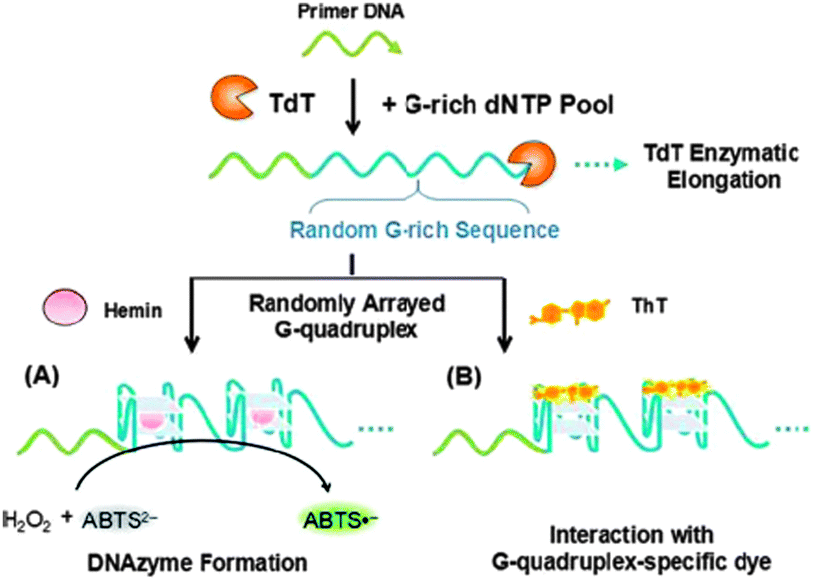 | ||
| Scheme 5 Schematic presentation of the preparation of TdT-generated G-quadruplexes and its derivative colorimetric (A) and fluorescent (B) assays of TdT activity. Reproduced from Ref. 62 with permission from The Royal Society of Chemistry. | ||
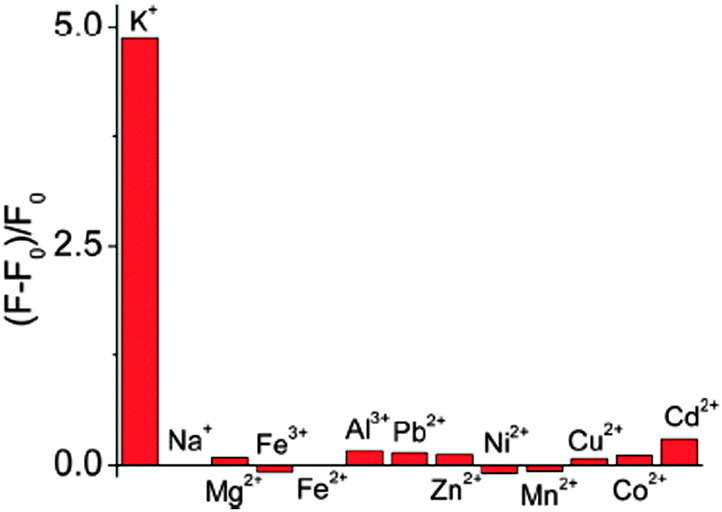 | ||
| Fig. 9 Selective responses of 1/Tel22 (5′-AG3(TTAG3)3-3′) to K+. The presence of an individual metal ion (2 mM, F0 and F stand for the fluorescent intensities in the absence and presence of the metal ions, respectively). Reprinted with permission from ref. 30. Copyright (2014) American Chemical Society. | ||
Three dyes, 1, 2, and pyronin Y (PY), were used as the donor, intermediate, and acceptor, respectively. The addition of Sr2+ ions to a mixture of GMP and the dyes results in the formation of G-tetrad structures, which gradually grow into long nanofibers.18 The three dyes are confined simultaneously in the nanofibers and brought into proximity, thus allowing for a two-step FRET from 1 to PY mediated by 2. However, similar FRET measurements between the three dyes in the presence of a G-quadruplex DNA structure was not as efficient as the G-tetrad, probably due to the loop structure and topological differences. These G-tetrad nanofibers showed light-harvesting properties both in solution and in the solid state, thus offering an advantage from the viewpoint of device processing.18 The photocurrent response as a function of time of GMP/Sr nanofibers containing different dyes in solution upon irradiation with visible light has been demonstrated. The generation of photocurrent in the visible light, which provides the material with the potential for application as a nanoscale photoelectric device.18
3.2. Interaction of the G-quadruplex with triphenyl-methane and triphenylamine dyes
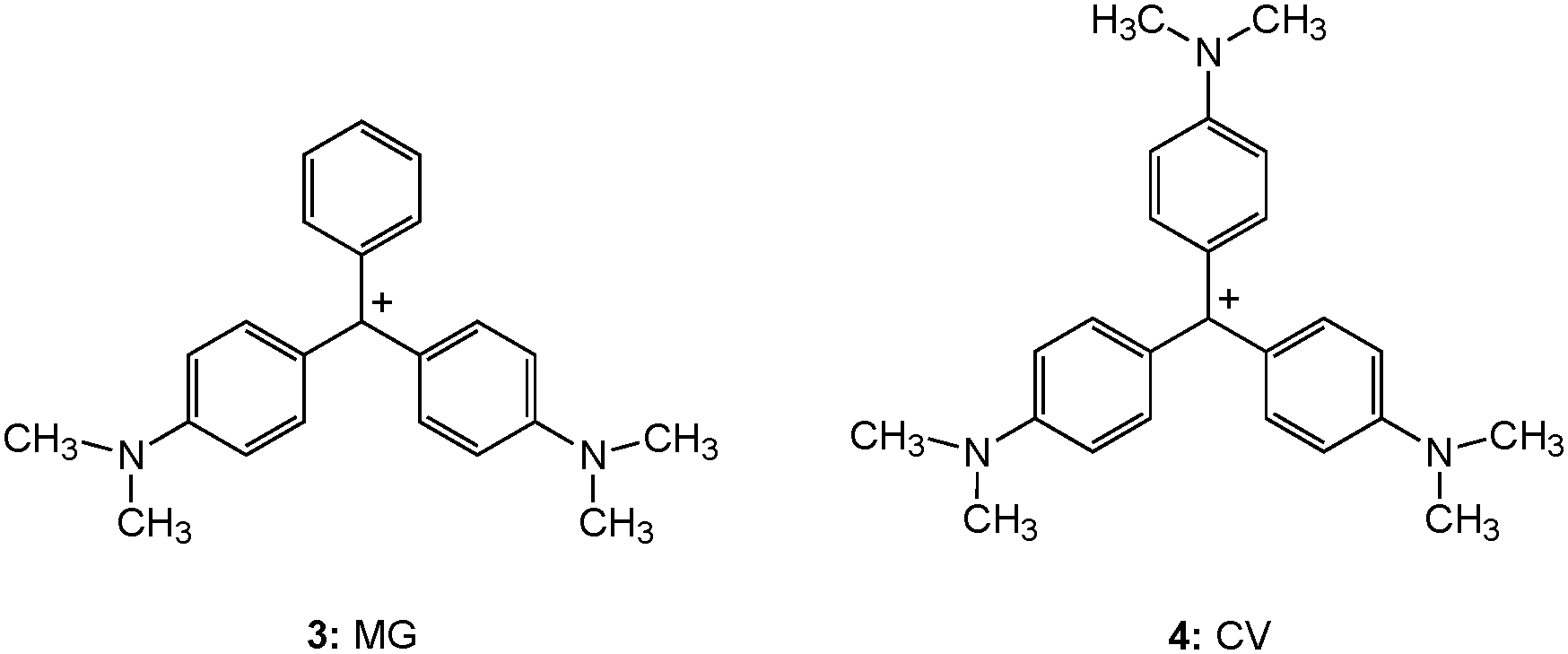 | ||
| Chart 3 Chemical structures of triphenylmethane (TPM) dyes. | ||
The interaction of 3 with single strand DNA homopolymers, (dA)40, (dT)40, (dC)40 and double-stranded DNA (ds-DNA40) along with guanine-rich single-strand oligomer d(G2T)13G of a similar length have been investigated.67 Distinctly, in the presence of a d(G2T)13G oligomer, 3 shows bathochromic shift of ∼18 nm in its absorption and the fluorescence bands (Fig. 10A).67 The specific binding interaction leads to an approximately 100-fold enhancement in the radiative yield of the dye. In the binding curve (changes in the fluorescence intensity of 3 at varying concentrations of d(G2T)13G at a particular wavelength), the initial enhancement in the fluorescence intensity tends to a plateau at a d(G2T)13G concentration of approximately 30 μm (Fig. 10B, region I). A further increase in the oligomer concentration results in a remarkable second stage enhancement of the fluorescence of 3, which tends towards saturation at a concentration of approximately 120 μm (region II) in d(G2T4)13G. However, similar measurements in the presence of other ss-DNA polymers and ds-DNA, a less than 14-fold fluorescence enhancement was observed in the dye (Fig. 10A).
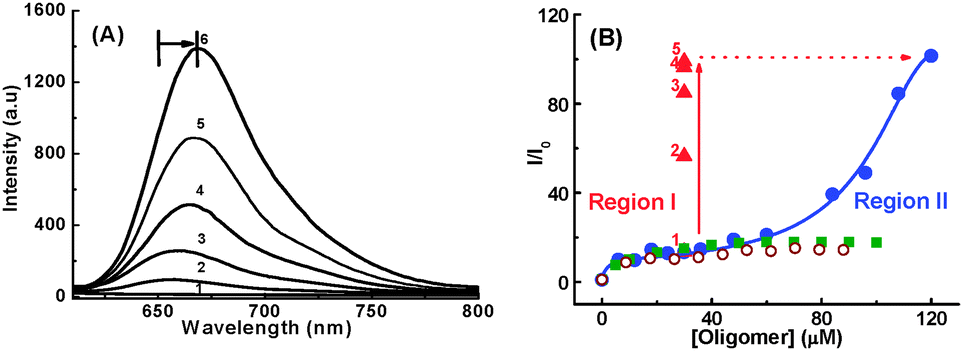 | ||
| Fig. 10 (A) Fluorescence intensity enhancement in an aqueous solution of 1 μM of malachite green containing 5 mM phosphate buffer at pH 7, with increasing concentration of d(G2T)13G/μM; (1) 0, (2) 6, (3) 60, (4) 96, (5) 108, (6) 120. (B) Binding curves for 1 μM of 3 with varying concentrations of oligomers; ● d(G2T)13G, ■ (dG)40, ○ ds-DNA40. ▲ denotes the increase in emission intensity at [d(G2T)13G] ∼30 μM with the addition of NaCl/M: (1) 0, (2) 0.1, (3) 0.2, (4) 0.3 and (5) 0.5. Adapted with permission from ref. 67, copyright (2007) Wiley-VCH Verlag GmbH & Co. KGaA. | ||
Increasing the concentrations of NaCl up to 0.5 M in the dye solution at two different concentrations of d(G2T)13G (Fig. 10B, region I and region II) results in a 100 fold fluorescence enhancement at region I and no changes in the fluorescence intensity at region II. Moreover, the fluorescence decay traces for 3 in the presence of d(G2T)13G followed a tri-exponential decay kinetics with an average lifetime of 560 ps in region I and 1.1 ns in region II, and the decay trace obtained in region I in the presence of 0.5 M NaCl clearly matches the one in the region II (Fig. 11). The above results suggested that in the presence of NaCl, at higher oligomer concentration, G-quadruplexes are formed via an inter-strand interaction.67 The electron rich phenyl rings of 3 can effectively π stack on the face of the G-quadruplex, rigidizing the dye in its planar structure, leaving the positive charge to lie in the centre of the quadruplex. The anisotropy traces recorded in the solution at region I with 0.5 M NaCl and region II without NaCl, displayed quite good match (inset of Fig. 11), confirming that the dye experiences a similar local environment in both the cases.67
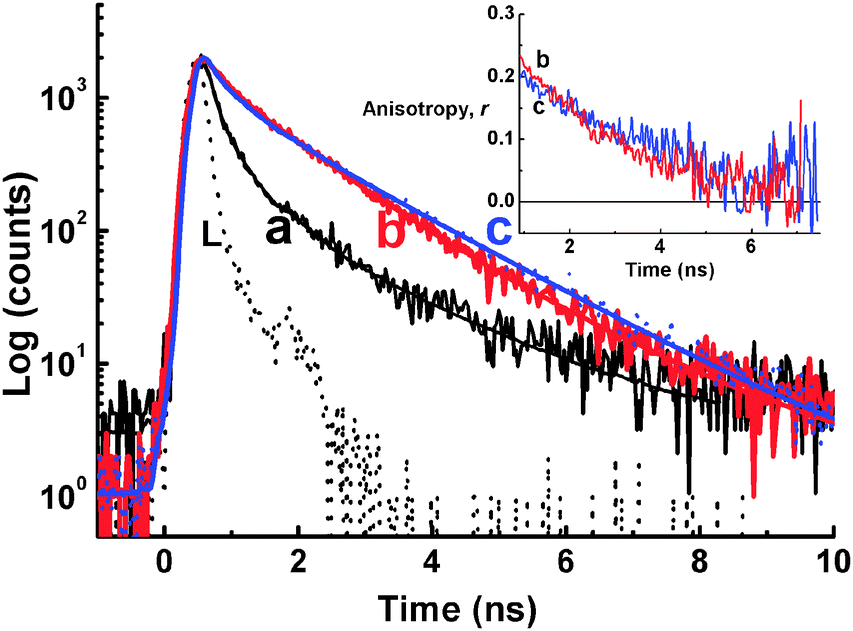 | ||
| Fig. 11 Fluorescence decay traces recorded at 690 nm upon excitation by a 636 nm diode laser in 1 μM of 3 solution containing 5 mM phosphate buffer at pH 7 with d(G2T)13G/μM (a) 30, (b) 120 and (c) 30 with 0.5 M NaCl. L represents the excitation pulse profile. Inset: Fluorescence anisotropy traces measured under the conditions for traces b and c. Adapted with permission from ref. 67, copyright (2007) Wiley-VCH Verlag GmbH & Co. KGaA. | ||
Further, the UV difference spectrum of d(G2T)13G in the presence of NaCl at 80 °C and 1 °C exhibits spectral features in agreement with that of a G-quadruplex. Similar measurements with a known quadruplex forming strand (G4T4)4 showed ∼70 fold increase in fluorescence suggests that the observed enhancement may be more general with G-quadruplexes.67 This specific interaction, as compared to other ss-/ds-DNAs, recommends a convenient method to detect the G-quadruplex formation.
3.2.1.1. Discrimination of G-quadruplexes from duplex and single-stranded DNAs. In a subsequent study, Kong et al. have investigated the fluorescence and energy-transfer fluorescence of 3 and 4 in the presence of G-quadruplex, ds-, and ss-DNAs.68 The fluorescence of both free 3 and 4 was weak, and a significant increase in the emission intensity was observed at fixed 3 or 4 concentrations in the presence of G-quadruplex DNAs, ds- or ss-DNAs, but the extent of fluorescence enhancement was different between 3 and 4. The fluorescence intensities of 4 in the presence of human telomeric DNAs (Hum12: 5′-(T2AG3)2; Hum21: 5′-G3(T2AG3)3), Oxy12 (5′-G4T4G4) and Oxy28 (5′-G4(T4G4)3) DNAs were always higher than those in the presence of ds- or ss-DNAs. This feature has been aptly projected in dye 4 as a biosensor for discriminating G-quadruplexes from ds- or ss-DNAs.68 On the G-quadruplex, it is likely that the three phenyl rings of the TPM dyes stack onto the two external G-tetrads of the quadruplexes, and the positively charged dialkylamino substituents interact with the negatively charged phosphate backbone and loops. The presence of dimethylamino substituents on all the phenyl rings hinders the intercalative binding of the dyes into stacked base pairs in duplex DNAs and in G-quadruplex DNAs, as well as the groove binding of the dyes to ds-DNAs. However, the absence of the third dimethylamino substituent in 3 allows the dye to partially intercalate between stacked base pairs in ds-DNAs or to bind to the grooves of ds-DNAs through the side of the unsubstituted phenyl ring. This could bring out more fluorescence in 3 to a larger extent than that of 4 in the presence of ds-DNAs. The fluorescence intensities observed from anti-parallel G-quadruplexes were more intense than those from parallel G-quadruplex complexes.68
It is known that energy transfer between DNA bases and a bound fluorescent ligand is only possible if the ligand is in close contact with the DNA bases. The fluorescence spectra recorded when the two dyes are excited at 280 nm, which is in the DNA absorption region of 240–320 nm, were clearly different from their normal fluorescence spectra obtained upon direct excitation of the dyes. The excitation through the G-quadruplexes increased the fluorescence from the dyes greatly (Fig. 12).68 This indicated that the binding modes between the two dyes and the G-quadruplex DNAs were intercalation or end stacking, but not outside binding, and that effective energy transfer occurred between the G-quadruplex DNAs and the dyes and is more appropriate than the normal fluorescence spectra for discriminating the G-quadruplexes from ds- and ss-DNAs, especially in the case of 3.68
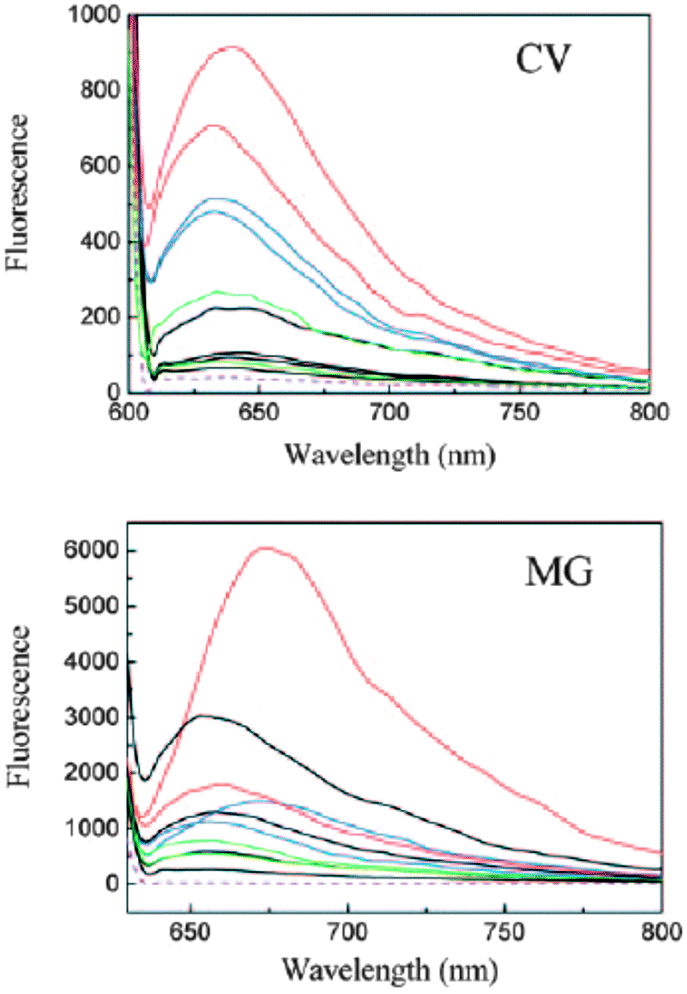 | ||
| Fig. 12 Fluorescence spectra of 4 (top) and 3 (bottom) in the presence of different DNAs: Free dye (purple, dashed lines), intramolecular G-quadruplexes (red), intermolecular G-quadruplexes (blue), ds-DNAs (black), and ss-DNAs (green). The orders (top to bottom) of the DNAs are intramolecular G-quadruplex DNAs: 7.5 μM Hum21 and Oxy28; intermolecular G-quadruplex DNAs: 15 μM Hum12 and Oxy12; ds-DNAs: 360 μM ct-DNA (base concentration), 15 μM AT, LD, and GC; ss-DNAs: 15 μM ss-DNA1 and ss-DNA2. [CV] = [MG] = 15 μM. Reproduced with permission from ref. 68, copyright (2009) Wiley-VCH Verlag GmbH & Co. KGaA. | ||
3.2.1.2. G-quadruplex bound crystal violet as K+ sensor. In a separate study, Kong et al. further demonstrated a novel K+ detection method by using a label-free G-quadruplex-forming oligonucleotide and dye 4.31 This method is based on the fluorescence difference of some 4–G-quadruplex complexes in the presence of K+ or Na+, and the fluorescence change with the variation of K+ concentration. In Hum21 the fluorescence of 4 increased with an increase in concentration of K+ (Fig. 13) and 4–Hum21 complex displayed much higher selectivity for K+ over Ca2+/Mg2+ and can provide a fast response to the variation of K+ concentration.31
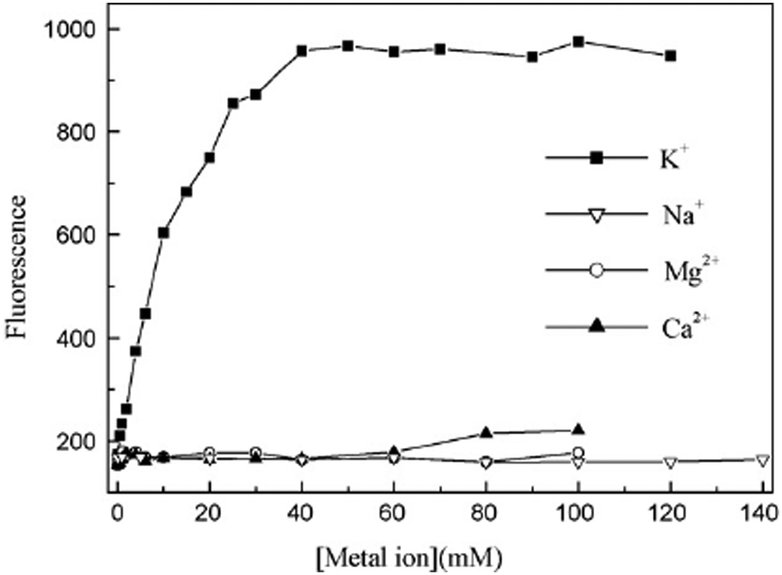 | ||
| Fig. 13 Fluorescence of the 4–Hum21 complex as a function of K+, Na+, Mg2+ or Ca2+ concentration in the presence of 300 mM Na+ ion. Reproduced with permission from ref. 31, copyright (2009) Elsevier B.V. | ||
6 showed ∼190 fold fluorescence enhancement at 620 nm along with a 20 nm bathochromic shift in the absorption band upon binding to the G-quadruplex of 22AG DNA (Fig. 14 and 15). The CD studies showed that the 6 molecule could induce the 22AG DNA sequence to adapt an antiparallel structure. However, CPT1 did not show any changes in the fluorescence behavior except the 10 nm red shift in the absorption band (Chart 4).70
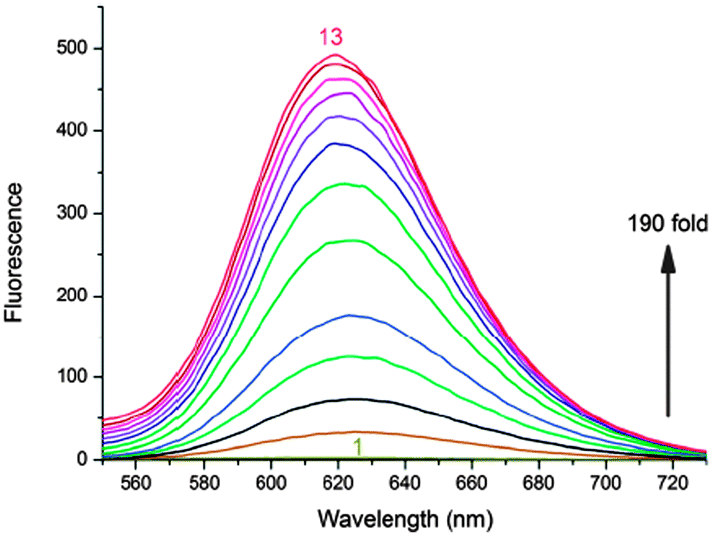 | ||
| Fig. 14 Fluorescence spectra of 6 (10 μM) upon titration with prefolded 22AG quadruplex DNA in 10 mM Tris (pH 7.4) and 50 mM KCl. [22AG]/μM: (1) 0, (2) 0.5, (3) 1.0, (4) 1.5, (5) 2.0, (6) 3.0, (7) 4.0, (8) 5.0, (9) 6.0, (10) 7.0, (11) 8.0, (12) 9.0, and (13) 10.0. λex is 521 nm. Reproduced from ref. 70 with permission from The Royal Society of Chemistry. | ||
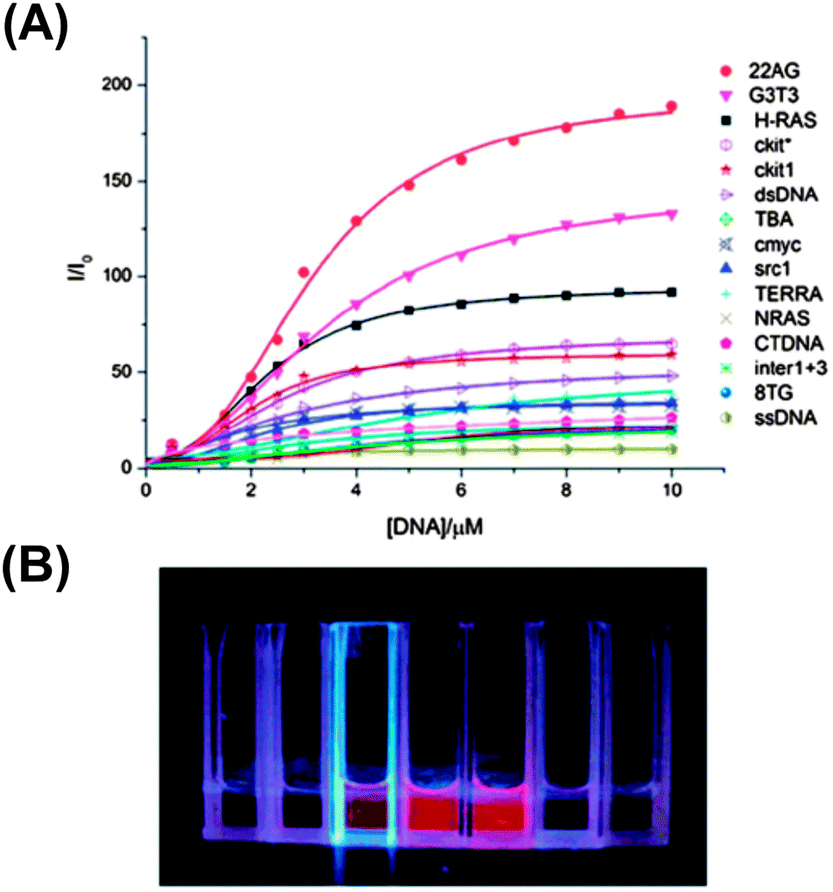 | ||
| Fig. 15 (A) Fluorescence titration of 6 with various G-quadruplexes and ct-DNA/ss-DNA/ds-DNA. Solution contains 10 μM 6, 10 mM Tris (pH 7.4) and 50 mM KCl. λex is 521 nm and the fluorescence intensity is recorded at 620 nm. (B) Fluorescence distinction of 6 with different DNAs under irradiation of UV light, the solution is buffered with 10 μM 6, 10 mM Tris (pH 7.4) and 50 mM KCl. From left to right, DNAs are as follows: ss-DNA, ct-DNA, H-RAS, 22AG, G3T3, c-kit and c-myc. Concentrations of different DNAs are all 10 μM. Reproduced from ref. 70 with permission from The Royal Society of Chemistry. | ||
 | ||
| Chart 4 Chemical structures of triphenylamine based dyes. | ||
Furthermore, 6 showed a high specificity and selectivity towards antiparallel G-quadruplexes against other DNA conformations, which was observed by the naked eye under UV light (Fig. 15). These characteristics of 6 make it a promising fluorescent switch-on probe for sensing antiparallel G-quadruplexes and for DNA imaging.70
3.3. Interaction of G-quadruplex with porphyrins and phthalocyanines
Porphyrins and phthalocyanines, another class of commercially available planar molecule, are the most extensively studied G-quadruplex-ligands which bind and stabilize different types of G-quadruplex structures. The porphyrin derivatives are composed of four modified pyrrole units and four cationic functional groups, which shows high affinity to bind a G-quadruplex tetrad via π–π stacking and electrostatic interactions.N-Methyl mesoporphyrin (NMM, 7; Chart 5) is a well studied porphyrin dye commercially available as a mixture of four N-methyl pyrrole regioisomers. 7 generally exhibits low-to-modest G-quadruplex affinity and is selective for the parallel form of the human telomeric G-quadruplex.717 also exhibits relatively weak fluorescence emission, which is enhanced two- to tenfold upon G-quadruplex binding. Employing this, Li et al. have demonstrated a novel label-free, G-quadruplex-based approach for simultaneous detection of His and Cys.33 The present assay is based on the highly specific interaction among aminoacids (His or Cys), Cu2+ and 7 bound G-quadruplex.33 The fluorescence intensity of 7 was dramatically enhanced in the presence of G-quadruplex formed from 24GT, which can be effectively quenched by cupric ion (Cu2+) due to the chelation of Cu2+ by 7 as well as the unfolding of G-quadruplex by Cu2+. The presence of His or Cys will disturb the interaction between Cu2+ and the NMM/G-quadruplex because of the strong binding affinity of Cu2+ to the imidazole group of His or with the thiol group in Cys, leading to distinct fluorescence emission intensity. These processes are schematically shown in Scheme 6 and the fluorescence intensity changes in the presence of various amino acids are shown in Fig. 16.33 A detection limit as low as 3 nM for His and 5 nM for Cys was obtained by practical measurement, confirming the high sensitivity of the present approach. This sensing protocol can determine His or Cys in diluted biological samples such as urine, exhibiting great potential to meet the need of practical application.33
 | ||
| Chart 5 Chemical structures of NMM and PPIX. | ||
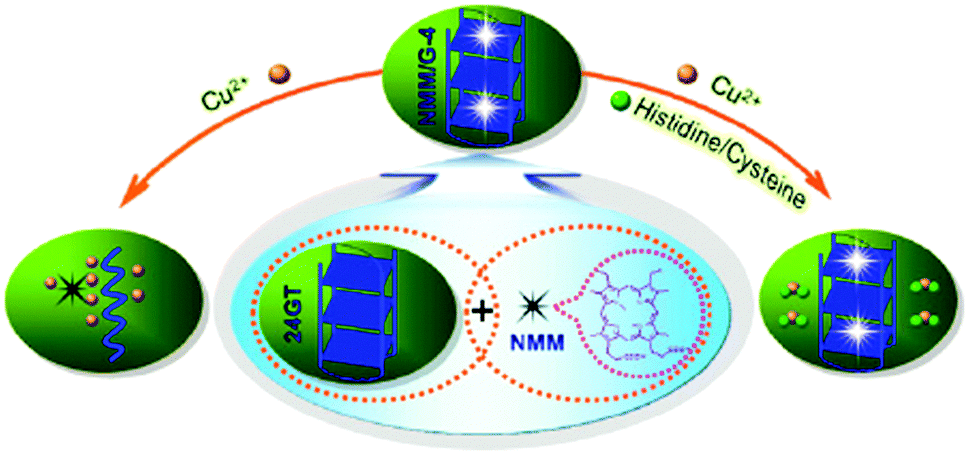 | ||
| Scheme 6 Schematic illustration of the fluorescence change of the NMM/G-quadruplex ensemble under different conditions. The combination of 7 and an intramolecular G-quadruplex generated from 24GT oligonucleotide functions as a signal indicator NMM/G-quadruplex with strong fluorescent intensity. The cupric ion can quench the fluorescence of the NMM/G-quadruplex through its coordination with 7 as well as the unfolding of the G-quadruplex by Cu2+ (as shown in the left side). However, the presence of His or Cys can disturb the interaction between Cu2+ and the NMM/G-quadruplex complex due to their interaction with Cu2+, generating a distinct fluorescence response from that of the cupric ion alone (as shown in the right side). Reproduced with permission from ref. 33, copyright (2012) Elsevier B.V. | ||
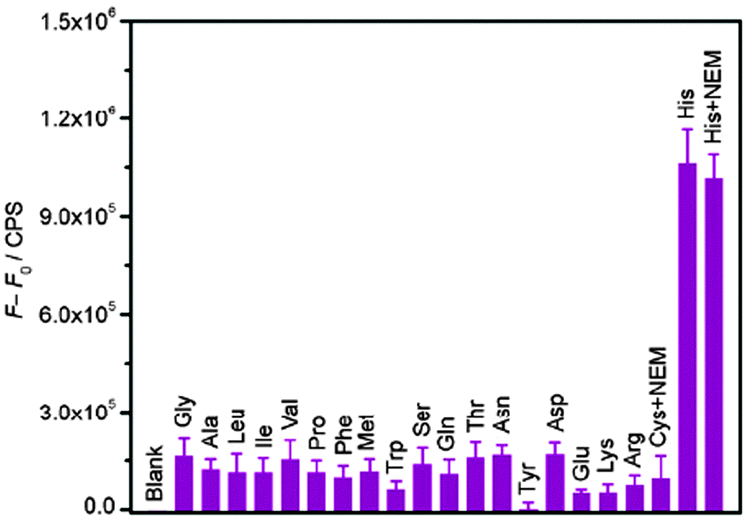 | ||
| Fig. 16 Fluorescence intensity change (F − F0) histograms of the NMM/G-quadruplex at 608 nm in the presence of Cu2+ and various amino acids with error bars, where F and F0 are fluorescence intensities of the NMM/G-quadruplex/Cu2+ in the presence and absence of different amino acid, respectively. Reproduced with permission from ref. 33, copyright (2012) Elsevier B.V. | ||
Quantification of various metal ions in biological systems is of tremendous interest in monitoring ecosystem and the human health.72 Copper is one of the heavy metals, which is essential in various physiological processes, however, Cu2+ ions are toxic when its level exceeds cellular needs and is believed to be responsible for serious neurodegenerative diseases, such as Menkes disease, Wilson disease, and Alzheimer’s disease.73,74 This demands a method for rapid and sensitive detection of such metal ions. Protoporphyrin IX (PPIX, 8; Chart 5) is a ubiquitous heme precursor that has been reported to be a G-quadruplex-selective fluorescent probe in vitro.75 It selectively binds parallel G-quadruplex DNA with good affinity (Kd ∼30–120 nM), while exhibiting much lower affinity for duplex and anti-parallel G-quadruplex DNA (Kd >50 μM).75 Taking advantage of this, Zhang et al.76 developed a sensitive G-quadruplex based probe for Cu2+ detection. The probe takes advantage of the high stability of the G-quadruplex and the high sensitivity of 8 toward Cu2+ to provide an effective platform for rapid and reliable detection of Cu2+. The probe shows a high selectivity for Cu2+ over other divalent metal ions (Fig. 17). Through this detection strategy, a limit of detection (3 nM) for Cu2+ analysis is achieved.76
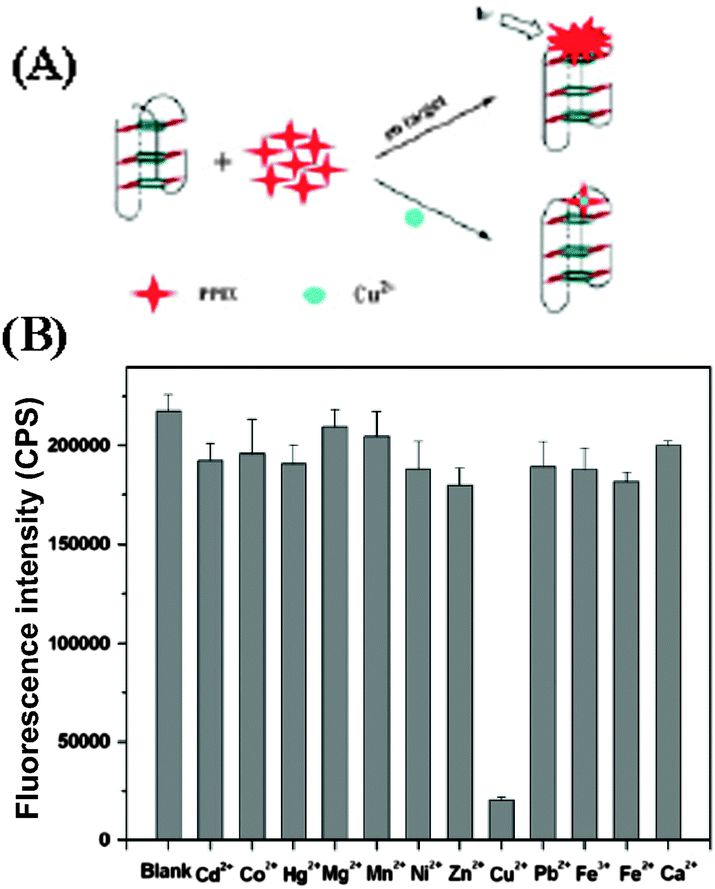 | ||
| Fig. 17 (A) Schematic representation of the label-free G-quadruplex-specific fluorescent probe for the detection of Cu2+. (B) Selectivity of Cu2+ using the G-quadruplex-based probe. Cu2+ was used at 2.0 μM, and other metal ions were used at 5.0 μM. λex = 410 nm. Reproduced with permission from ref. 76, copyright (2012) Elsevier B.V. | ||
Phthalocyanines are large π-planar compounds composed of four iso-indole groups that are linked by nitrogen atoms. The large π-conjugated systems confer rubrical photophysical properties to phthalocyanines. They have a large π-planar core which is big enough to engage in π–π stacking interactions with G-tetrad. They have the potential to function as G-quadruplex-ligands with high specificity and high affinity. Two phthalocyanine derivatives, tetramethylpyridinium-porphyrazines (3,4-TMPyPz) and the derivative with zinc as the coordination metal, (Zn(II)3,4-TMPyPz), could stabilize and/or induce a human telomere G-quadruplex.25,77 The detailed measurements reveal that both the phthalocyanine derivatives bound to ds-DNA with at least 30-fold lower affinity than the human telomere G-quadruplex. The higher affinity and selectivity of the phthalocyanine derivatives for the human telomere G-quadruplex, relative to those for TMPyP, were attributed to their expanded π-planar structures.25,77
3.4. Luminescent metal complexes
Transition metal complexes incorporating several custom made ligands feature another class of well studied for G-quadruplex binding structures, which have shown a variety of applications, such as light (on–off) switches, metal ion sensors etc.78 Here the properties are much dependent on both the metal ion as well as the ligand profiles and can be tuned in custom ways. Several such complexes of ruthenium and iridium metal ions (Chart 6) have been tested for their appropriateness towards various structures of G-quadruplex DNA, in search of therapeutic and sensor applications.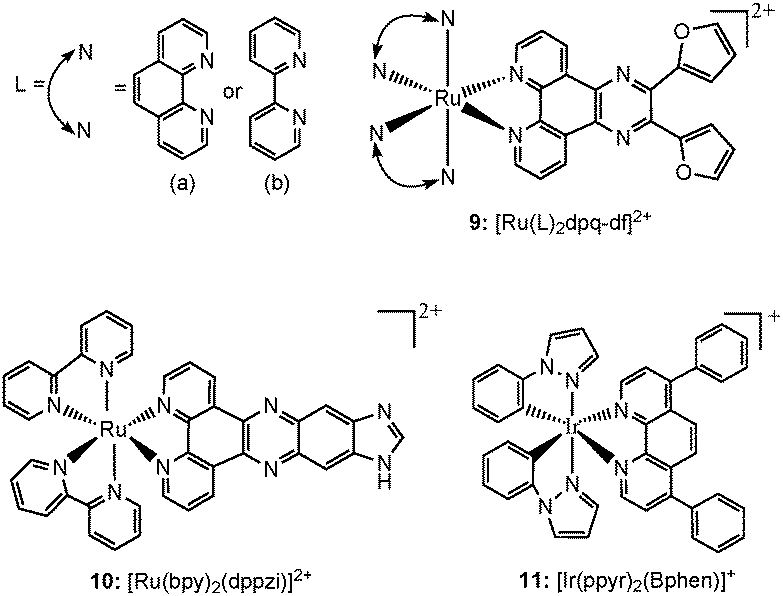 | ||
| Chart 6 Chemical structures of luminescent metal complexes. | ||
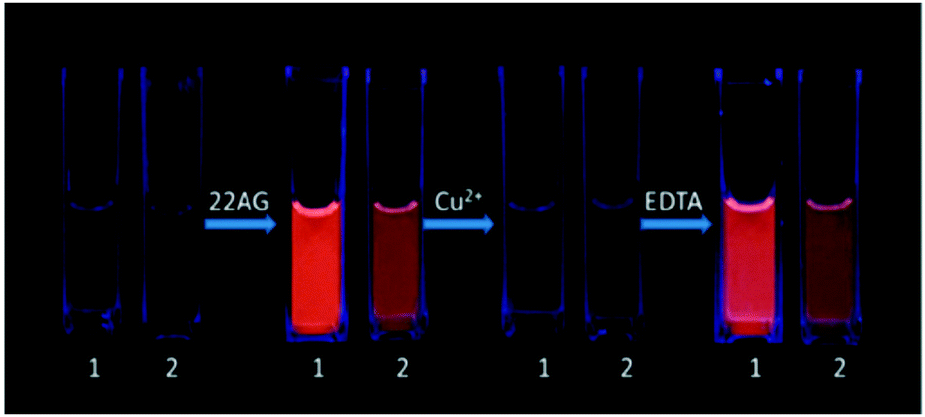 | ||
| Fig. 18 Images of the reversible “light switch” behavior of 2 μM 1 and 2 for DNA (1 μM) under UV light at 365 nm in 10 mM Tris-HCl, 100 mM KCl, pH 7.4. Reproduced with permission from ref. 32, copyright (2014) Elsevier Inc. | ||
The “light switch” effect of 9a is superior to that of 9b, whereas, 9b has a better selectivity toward the hybrid G-quadruplex. Both the luminescence and absorption spectra titrations prove that complex 9a exhibits a better DNA-binding affinity than 9b, probably because the auxiliary ligand of 9a (phen) has a stronger hydrophobicity and larger square π-aromatic surface than 9b (bpy). All the observations suggest that the ancillary ligands could cause interesting differences in the spectral properties and G-quadruplex DNA-binding behavior of the corresponding Ru(II) complexes.32 The work can be valuable for designing new reversible molecular probes and metal ion sensors.
In another work, Shi et al. have shown that cycling of the G-quadruplex DNA “light switch” off and on has been accomplished for [Ru(bpy)2(dppzi)]2+ (10) (dppzi = dipyrido[3,2-a:2′,3′-c] phenazine) through the successive introduction of [Fe(CN)6]4− ions and G-quadruplex DNA, respectively (Fig. 19).79 The switch can be cycled through the competition of [Fe(CN)6]4− ions and G-quadruplex DNA. This binding features of 10 with G-quadruplex DNA may show promise for probing G-quadruplex DNA and provide a more comprehensive understanding of the molecular recognition of G-quadruplex DNA.
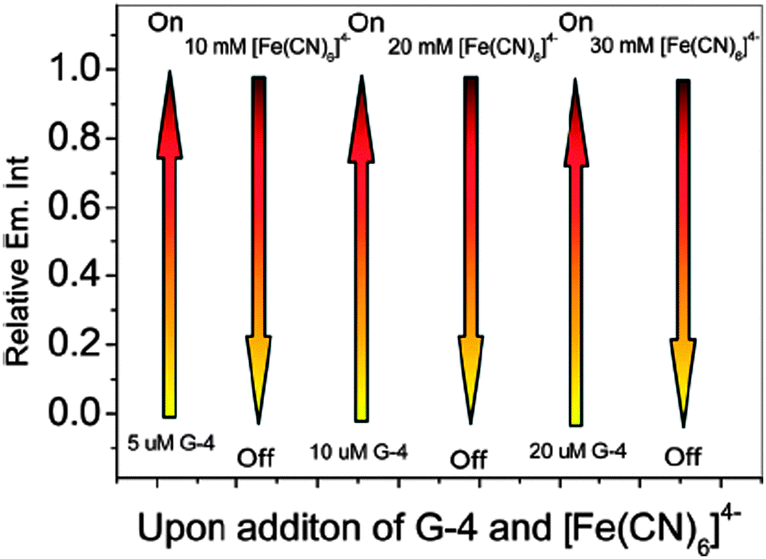 | ||
| Fig. 19 The relative emission intensity of 10 upon successive additions of G-quadruplex DNA (G-4) and [Fe(CN)6]4− in 100 mM K+ buffer. Reproduced from ref. 79 with permission from The Royal Society of Chemistry. | ||
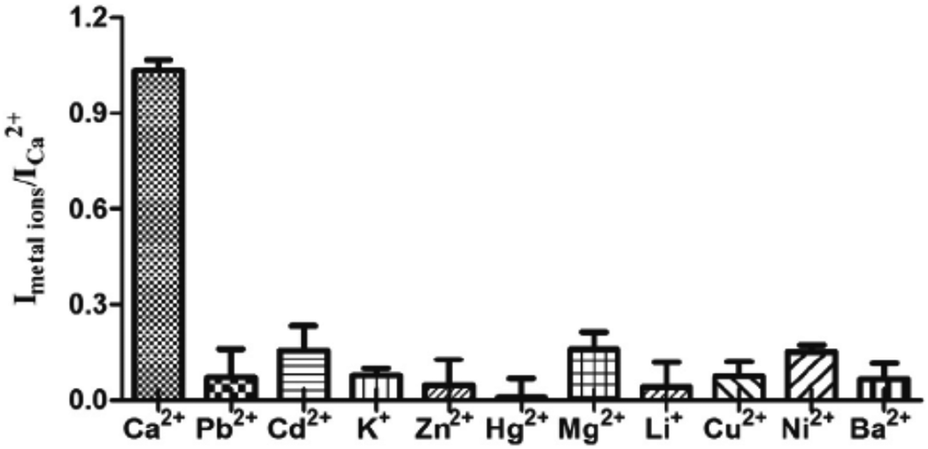 | ||
| Fig. 20 Selectivity of the G-quadruplex-based Ca2+ ion assay. The concentration of Ca2+ ions was 1.0 μM and the concentrations of the other metal ions were 1.0 μM. Experimental conditions: pre-annealed G4 antiparallel G-quadruplex (5 μM) and complex 1 (1 μM) in 50 mM MES buffer (20 mM NaCl, pH 6.1). Reproduced with permission from ref. 81, copyright (2014) Elsevier Inc. | ||
4. Visualization of a G-quadruplex in a human cell
Despite a wealth of current knowledge about the human genome, there remain many challenges associated with the non-canonical nucleic acid structures, such as the role of G-quadruplexes in vivo for transcriptional regulation, DNA replication and genome stability. In a recent study, Balasubramanian et al. have illustrated that G-quadruplex structures exist within double-stranded genomic DNA and its presence in the telomeric region can be explicitly identified using a G-quadruplex-specific antibody.16 In a subsequent study, they further demonstrated the visualization of RNA G-quadruplex structures within the cytoplasm of human cells using a G-quadruplex structure-specific antibody (Chart 7).11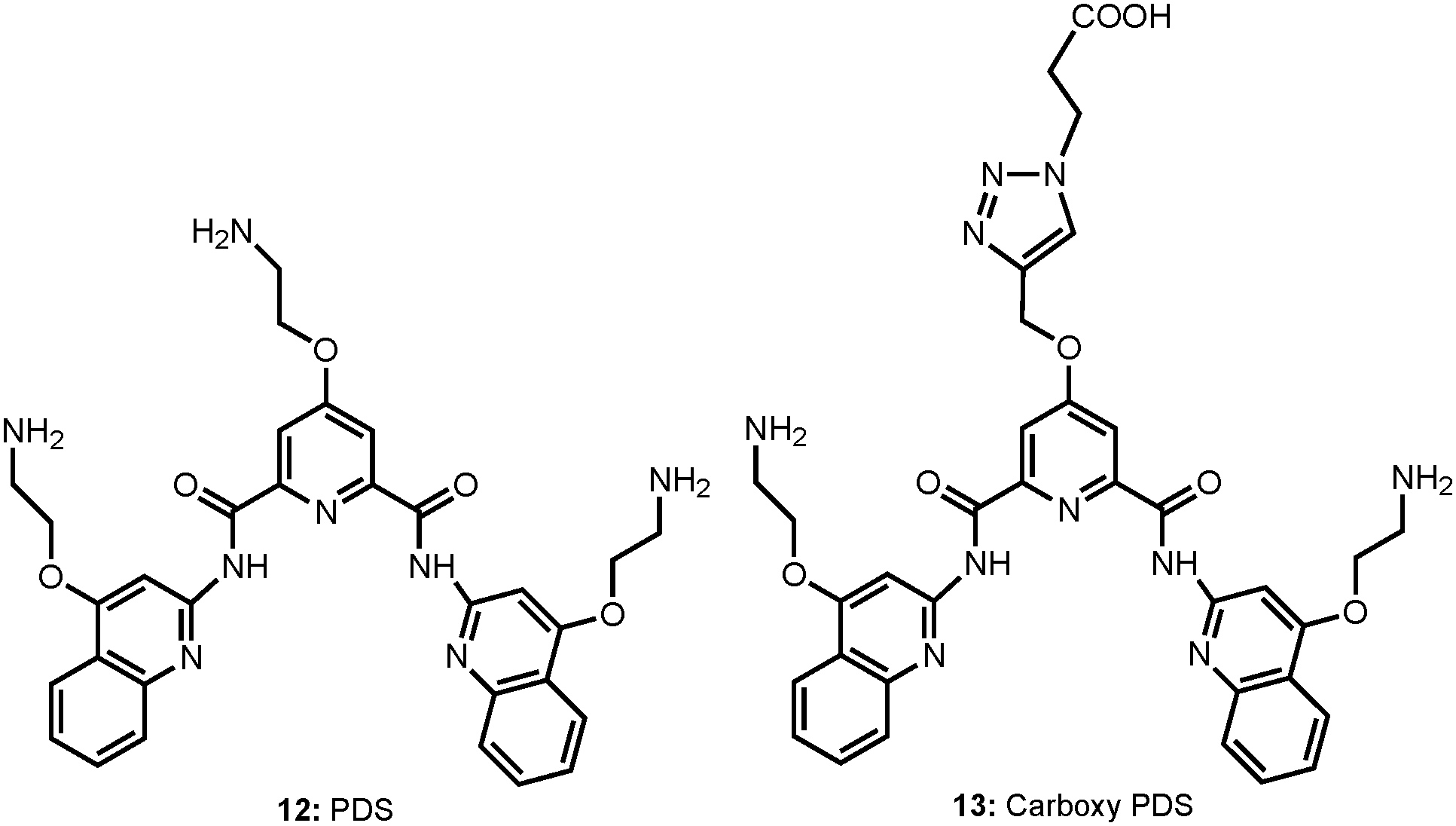 | ||
| Chart 7 Chemical structure of pyridostatin (PDS) and carboxypyridostatin (carboxyPDS). | ||
For this, a small molecule pyridostatin (PDS: 12), which binds both DNA and RNA G-quadruplexes in biophysical assays, was used. After the PDS treatment of cells, there was substantial increase in the number of foci detected in the cytoplasm, which are sensitive to RNase but not to DNase treatment, confirming the presence of RNA G-quadruplexes in the cytoplasm. Their studies further identified an improved derivative, the carboxypyridostatin (carboxyPDS: 13), that showed a preference for RNA G-quadruplexes compared to DNA G-quadruplexes.
As provided in Fig. 21,11 upon treatment of human cells with carboxyPDS, the researchers observed a marked increase in the number of cytoplasmic foci (Fig. 21A and B), which is comparable to that seen following PDS treatment. Noticeably, carboxyPDS did not cause an increase in the number of nuclear foci, and this is in contrast to PDS, which traps endogenous DNA G-quadruplex structures and leads to more staining in the nuclei (Fig. 21C–E).11 Separately, the study also confirmed that the BG4 antibody targets in the nucleus are the DNA structures, as the signals are found to be sensitive to DNase but not to RNase treatment, in both the cases of PDS- or carboxyPDS-treated cells.
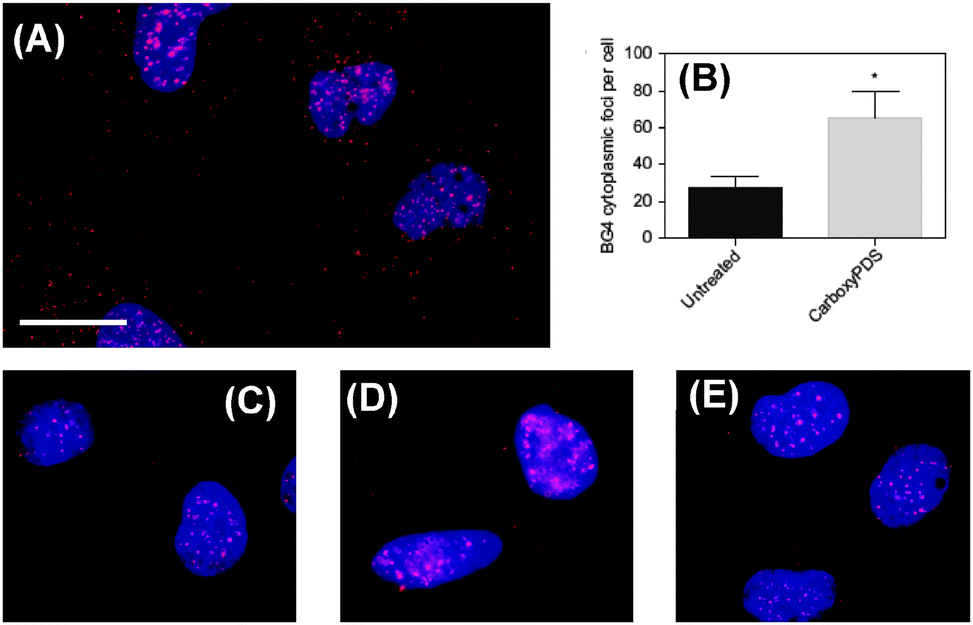 | ||
| Fig. 21 Indirect immunofluorescence microscopy shows selective stabilization of RNA over DNA G-quadruplexes in SV40-transformed MRC5 fibroblasts by the small molecule carboxyPDS (13), as revealed by staining with the BG4 antibody. (A) After 24 hours of treatment with 13 (2 mM), an increase in the number of BG4 cytoplasmic foci (red) was observed. Nuclei are blue through staining with DAPI, cytoplasm is dark and BG4 foci are red (Alexafluor 594). Scale bar, 20 mm. (B), Quantification of BG4 cytoplasmic foci number per cell for A and for untreated cells. For each condition 100–200 cells were counted and the standard error of the mean was calculated from three replicates. *P < 0.01. (C), DNA G-quadruplexes (red) detected by BG4 in the nuclei of untreated human cells. (D), After treatment with 12 the number of BG4 nuclear foci increased. (E), After treatment with 13 no increase in nuclear BG4 foci was observed. Reproduced with license from ref. 11. Copyright © 2013, Rights Managed by Nature Publishing Group. | ||
These results demonstrate that a small-molecule ligand such as carboxyPDS can target and trap RNA G-quadruplex structures selectively in the cytoplasm of human cells. It is worth noting that the ligand that exhibits a preference for RNA G-quadruplexes rather than DNA G-quadruplexes in biophysical experiments also shows the same selectivity within a cellular context. This is a proof-of-concept that strategies based on small molecules have the potential to decouple the recognition of RNA G-quadruplexes from DNA G-quadruplexes in cells. Together these findings provide substantive evidence for the formation of G-quadruplex structures in the genome of mammalian cells and corroborate the prospective strategy of chemical intervention in biology and medicine through the targeting of G-quadruplex structures and their function.
5. Summary and outlook
In this feature article, we have overviewed some of the very recent reports on targeting G-quadruplexes with fluorogenic/fluorescent dyes. Soon after the revelation that the fluorogenic dye, thioflavin T, selectively binds to human telomeric G-quadruplex DNA bringing out amazing enhancement in the ThT fluorescence,45 a flurry of reports followed demonstrating its implication in generalized high-throughput, label-free, fluorescence-based detection of various metal ions and small biomolecules, down to the pico molar range. The reversible disruption of the hydrogen bonded quadruplex structure in the presence of Hg2+ ensues an emission quenching and acts as a Hg2+ fluorescence off sensor,29 however, the emission is once again turned on as a sensor in the presence of biothiols.34 ThT, as a selective and sensitive fluorescent biosensor for short DNA species of c-erbB-2 can detect the DNA as low as 20 fM and can detect it in ultra-trace amounts in real saliva samples.35 Apart from discriminating G-quadruplexes from duplex or single-stranded DNAs,68 crystal violet, has supported a K+ detection method, even in the presence of Na+, Ca2+ or Mg2+.31 Allowing the chelation of Cu2+ by N-methyl mesoporphyrin, Li et al. have developed a novel label-free, highly specific G-quadruplex-based approach for simultaneous detection of histidine and cysteine. Cu2+ itself can be detected by taking advantage of the high stability of the G-quadruplex and the high sensitivity of the PPIX toward Cu2+.76 While the ruthenium complex exhibited light switch off and on through the successive addition of Cu2+ ions and EDTA,32 the iridium complex distinctly recognized a parallel G-quadruplex with a selective and sensitive switch-on luminescence response in the presence of Ca2+ ions.81 These detection methods are very simple, sensitive, and highly selective, quantitative, cost effective, and much better than the other optical and colorimetric sensors for bio-analytical applications. Of late, few more exciting studies have appeared based on the ThT/G-quadruplex template revealing selective recognition of parallel/anti-parallel thrombin-binding aptamer G-quadruplexes, enzyme-free/label-free fluorescence sensor for the detection of liver cancer related short gene and proposed improvements on thioflavin T to enhance visual discrimination of different quadruplex topologies.82–84In spite of extensive research on human telomeric G-quadruplexes in vitro, untill recently, it has remained an important challenge to visualize G-quadruplex structures in the DNA of mammalian cells in vivo. Balasubramanian’s group provided this breakthrough and reported an engineered, structure-specific antibody probe for quantitative visualization of DNA G-quadruplex structures in human cells.28 Using this antibody along with a RNA G-quadruplex specific ligand, visualization and selective chemical targeting of RNA G-quadruplex structures in the cytoplasm of human cells has been corroborated, highlighting its relevance in chemical intervention of qudruplex dynamics in the cellular process.11 Since the formation of G-quadruplexes is dynamically sensitive to the cell cycle, and recent studies signify the role of G-rich sequences at the telomeric and promoter regions in inhibiting/promoting cancer growth, it would be rewarding to generate, by sequencing, a map of the precise locations of G-quadruplex structures at a genome-wide level. In pursuing this goal, it is essential that we understand the factors that affect chemical, biological and genetic roles of G-quadruplex structures, especially a clinically viable method for cancer treatment.
Acknowledgements
We acknowledge all the co-authors and collaborators of our published articles cited in the references. We thank Dr H. Pal, Head, Molecular Photochemistry Section, Dr D. K. Palit, Head, Radiation & Photochemistry Division and Dr B. N. Jagatap, Director, Chemistry group, BARC, India, for their encouragement and support.Notes and references
- B. Ruttkay-Nedecky, J. Kudr, L. Nejdl, D. Maskova, R. Kizek and V. Adam, Molecules, 2013, 18, 14760–14779 CrossRef CAS PubMed.
- F. S. D. Leva, E. Novellino, A. Cavalli, M. Parrinello and V. Limongelli, Nucleic Acids Res., 2014, 42, 5447–5455 CrossRef PubMed.
- O. Doluca, J. M. Withers and V. V. Filichev, Chem. Rev., 2013, 113, 3044–3083 CrossRef CAS PubMed.
- J. L. Huppert, FEBS J., 2010, 277, 3452–3458 CrossRef CAS PubMed.
- C. E. Pearson and R. R. Sinden, Curr. Opin. Struct. Biol., 1998, 8, 321–330 CrossRef CAS.
- J. Bidzinska, G. Cimino-Reale, N. Zaffaroni and M. Folini, Molecules, 2013, 18, 12368–12395 CrossRef CAS PubMed.
- J. T. Davis, Angew. Chem., Int. Ed., 2004, 43, 668–698 CrossRef CAS PubMed.
- S. Neidle, FEBS J., 2010, 277, 1118–1125 CrossRef CAS PubMed.
- S. Balasubramanian, L. H. Hurley and S. Neidle, Nat. Rev. Drug Discovery, 2011, 10, 261–275 CrossRef CAS PubMed.
- Y. Xu, Chem. Soc. Rev., 2011, 40, 2719–2740 RSC.
- G. Biffi, M. D. Antonio, D. Tannahill and S. Balasubramanian, Nat. Chem., 2014, 6, 75–80 CrossRef CAS PubMed.
- A. T. Phan, FEBS J., 2010, 277, 1107–1117 CrossRef CAS PubMed.
- S. Burge, G. N. Parkinson, P. Hazel, A. K. Todd and S. Neidle, Nucleic Acids Res., 2006, 34, 5402–5415 CrossRef CAS PubMed.
- G. N. Parkinson, in Quadruplex Nucleic Acids, ed. S. Neidle and S. Balasubramanian, The Royal Society of Chemistry, Cambridge, UK, 2006, pp. 1–30 Search PubMed.
- E. Largy, A. Granzhan, F. Hamon, D. Verga and M.-P. Teulade-Fichou, Visualizing the Quadruplex: From Fluorescent Ligands to Light-Up Probes. In Quadruplex Nucleic Acids, Springer, Berlin/Heidelberg, Germany, 2013 Search PubMed.
- E. Y. N. Lam, D. Beraldi, D. Tannahill and S. Balasubramanian, Nat. Commun., 2013, 4, 1796 CrossRef PubMed.
- R. Nanjunda, E. A. Owens, L. Mickelson, T. L. Dost, E. M. Stroeva, H. T. Huynh, M. W. Germann, M. M. Henary and W. D. Wilson, Molecules, 2013, 18, 13588–13607 CrossRef CAS PubMed.
- F. Pu, L. Wu, X. Ran, J. Ren and X. Qu, Angew. Chem., Int. Ed., 2015, 54, 892–896 CrossRef CAS PubMed.
- J. L. Huppert and S. Balasubramanian, Nucleic Acids Res., 2007, 35, 406–413 CrossRef CAS PubMed.
- D. Sun, B. Thompson, B. E. Cathers, M. Salazar, S. M. Kerwin, J. O. Trent, T. C. Jenkins, S. Neidle and L. H. Hurley, J. Med. Chem., 1997, 40, 2113–2116 CrossRef CAS PubMed.
- S. Balasubramanian and S. Neidle, Curr. Opin. Chem. Biol., 2009, 13, 345–353 CrossRef CAS PubMed.
- J. L. Huppert, A. Bugaut, S. Kumari and S. Balasubramanian, Nucleic Acids Res., 2008, 36, 6260–6268 CrossRef CAS PubMed.
- P. Phatak, J. C. Cookson, F. Dai, V. Smith, R. B. Gartenhaus, M. F. G. Stevens and A. M. Burger, Br. J. Cancer, 2007, 96, 1223–1233 CrossRef CAS PubMed.
- B. R. Vummidi, J. Alzeer and N. W. Luedtke, ChemBioChem, 2013, 14, 540–558 CrossRef CAS PubMed.
- H. Yaku, T. Fujimoto, T. Murashima, D. Miyoshi and N. Sugimoto, Chem. Commun., 2012, 48, 6203–6216 RSC.
- S. Neidle and G. N. Parkinson, Nat. Rev. Drug Discovery, 2002, 1, 383–393 CrossRef CAS PubMed.
- A. Artese, G. Costa, F. Ortuso, L. Parrotta and S. Alcaro, Molecules, 2013, 18, 12051–12070 CrossRef CAS PubMed.
- G. Biffi, D. Tannahill, J. McCafferty and S. Balasubramanian, Nat. Chem., 2013, 5, 182–186 CrossRef CAS PubMed.
- J. Ge, X.-P. Li, J.-H. Jiang and R.-Q. Yu, Talanta, 2014, 122, 85–90 CrossRef CAS PubMed.
- L. Liu, Y. Shao, J. Peng, C. Huang, H. Liu and L. Zhang, Anal. Chem., 2014, 86, 1622–1631 CrossRef CAS PubMed.
- D.-M. Kong, J.-H. Guo, W. Yang, Y.-E. Ma and H.-X. Shen, Biosens. Bioelectron., 2009, 25, 88–93 CrossRef PubMed.
- X.-H. Lu, S. Shi, J.-L. Yao, X. Gao, H.-L. Huang and T.-M. Yao, J. Inorg. Biochem., 2014, 140, 64–71 CrossRef CAS PubMed.
- H. Li, J. Liu, Y. Fang, Y. Qin, S. Xu, Y. Liu and E. Wang, Biosens. Bioelectron., 2013, 41, 563–568 CrossRef CAS PubMed.
- L. Tong, L. Li, Z. Chen, Q. Wang and B. Tang, Biosens. Bioelectron., 2013, 49, 420–425 CrossRef CAS PubMed.
- J. Chen, J. Lin, X. Zhang, S. Cai, D. Wu, C. Li, S. Yang and J. Zhang, Anal. Chim. Acta, 2014, 817, 42–47 CrossRef CAS PubMed.
- M. L. Bochman, K. Paeschke and V. A. Zakian, Nat. Rev. Genet., 2012, 13, 770–780 CrossRef CAS PubMed.
- V. T. Mukundan and A. T. Phan, J. Am. Chem. Soc., 2013, 135, 5017–5028 CrossRef CAS PubMed.
- R. K. Moyzis, J. M. Buckingham, L. S. Cram, M. Dani, L. L. Deaven, M. D. Jones, J. Meyne, R. L. Ratliff and J. R. Wu, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 6622–6626 CrossRef CAS.
- K. E. Gordon and E. K. Parkinson, Methods Mol. Biol., 2004, 281, 333–348 CAS.
- G. N. Parkinson, R. Ghosh and S. Neidle, Biochemistry, 2007, 46, 2390–2397 CrossRef CAS PubMed.
- V. Dhamodharan, S. Harikrishna, C. Jagadeeswaran, K. Halder and P. I. Pradeepkumar, J. Org. Chem., 2012, 77, 229–242 CrossRef CAS PubMed.
- V. Dhamodharan, S. Harikrishna, A. C. Bhasikuttan and P. I. Pradeepkumar, ACS Chem. Biol., 2015 DOI:10.1021/cb5008597.
- A. Hawe, M. Sutter and W. Jiskoot, Pharm. Res., 2008, 25, 1487–1499 CrossRef CAS PubMed.
- A. C. Bhasikuttan, J. Mohanty, W. M. Nau and H. Pal, Angew. Chem., Int. Ed., 2007, 46, 4120–4122 CrossRef CAS PubMed.
- J. Mohanty, N. Barooah, V. Dhamodharan, S. Harikrishna, P. I. Pradeepkumar and A. C. Bhasikuttan, J. Am. Chem. Soc., 2013, 135, 367–376 CrossRef CAS PubMed.
- J. Mohanty, N. Thakur, S. Dutta Choudhury, N. Barooah, H. Pal and A. C. Bhasikuttan, J. Phys. Chem. B, 2012, 116, 130–135 CrossRef CAS PubMed.
- T. Ban, D. Hamada, K. Hasegawa, H. Naiki and Y. Goto, J. Biol. Chem., 2003, 278, 16462–16465 CrossRef CAS PubMed.
- J. Mohanty, S. Dutta Choudhury, H. Pal and A. C. Bhasikuttan, Chem. Commun., 2012, 48, 2403–2405 RSC.
- P. Yang, D. A. Cian, M.-P. Teulade-Fichou, J.-L. Mergny and D. Monchaud, Angew. Chem., Int. Ed., 2009, 48, 2188–2191 CrossRef CAS PubMed.
- F. Koeppel, J.-F. Riou, A. Laoui, P. Mailliet, P. B. Arimondo, D. Labit, O. Petitgenet, C. Hélène and J.-L. Mergny, Nucleic Acids Res., 2001, 29, 1087–1096 CrossRef CAS PubMed.
- R. T. Wheelhouse, D. Sun, H. Han, F. X. Han and L. H. Hurley, J. Am. Chem. Soc., 1998, 120, 3261–3262 CrossRef CAS.
- V. I. Stsiapura, A. A. Maskevich, V. A. Kuzmitsky, V. N. Uversky, I. M. Kuznetsova and K. K. Turoverov, J. Phys. Chem. B, 2008, 112, 15893–15902 CrossRef CAS PubMed.
- S. Dutta Choudhury, J. Mohanty, H. Pal and A. C. Bhasikuttan, J. Am. Chem. Soc., 2010, 132, 1395–1401 CrossRef PubMed.
- A. R. de la Faverie, A. Guedin, A. Bedrat, L. A. Yatsunyk and J.-L. Mergny, Nucleic Acids Res., 2014, 42, e65 CrossRef PubMed.
- X. F. Wang and M. S. Cynader, J. Neurosci., 2001, 21, 3322–3331 CAS.
- M. T. Goodman, K. McDuffie, B. Hernandez, L. R. Wilkens and J. Selhub, Cancer, 2000, 89, 376–382 CrossRef CAS.
- W. Droge and E. Holm, FASEB J., 1997, 11, 1077–1089 CAS.
- A. Meister, Science, 1983, 220, 472–477 CAS.
- X. Jia, J. Li and E. Wang, Chem. – Eur. J., 2012, 18, 13494–13500 CrossRef CAS PubMed.
- Y. Tanaka, S. Oda, H. Yamaguchi, Y. Kondo, C. Kojima and A. Ono, J. Am. Chem. Soc., 2007, 129, 244–245 CrossRef CAS PubMed.
- H. Torigoe, A. Ono and T. Kozasa, Chem. – Eur. J., 2010, 16, 13218–13225 CrossRef CAS PubMed.
- Z. Liu, W. Li, Z. Nie, F. Peng, Y. Huang and S. Yao, Chem. Commun., 2014, 50, 6875–6878 RSC.
- J. R. Babendure, S. R. Adams and R. Y. Tsien, J. Am. Chem. Soc., 2003, 125, 14716–14717 CrossRef CAS PubMed.
- I. K. Kandela, J. A. Bartlett and G. L. Indig, Photochem. Photobiol. Sci., 2002, 1, 309–314 CAS.
- D. Grate and C. Wilson, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 6131–6136 CrossRef CAS.
- D. M. Kolpashchikov, J. Am. Chem. Soc., 2005, 127, 12442–12443 CrossRef CAS PubMed.
- A. C. Bhasikuttan, J. Mohanty and H. Pal, Angew. Chem., Int. Ed., 2007, 46, 9305–9307 CrossRef CAS PubMed.
- D.-M. Kong, Y.-E. Ma, J. Wu and H.-X. Shen, Chem. – Eur. J., 2009, 15, 901–909 CrossRef CAS PubMed.
- J. Wang, C. He, P. Y. Wu, J. Wang and C. Y. Duan, J. Am. Chem. Soc., 2011, 133, 12402–12405 CrossRef CAS PubMed.
- H. Lai, Y. Xiao, S. Yan, F. Tian, C. Zhong, Y. Liu, X. Weng and X. Zhou, Analyst, 2014, 139, 1834–1838 RSC.
- J. M. Nicoludis, S. P. Barrett, J.-L. Mergny and L. A. Yatsunyk, Nucleic Acids Res., 2012, 40, 5432–5447 CrossRef CAS PubMed.
- Y. Xiang, A. Tong and Y. Lu, J. Am. Chem. Soc., 2009, 131, 15352–15357 CrossRef CAS PubMed.
- Y.-R. Kim, H. J. Kim, J. S. Kim and H. Kim, Adv. Mater., 2008, 20, 4428–4432 CrossRef CAS.
- T. Mayr, I. Klimant, O. S. Wolfbeis and T. Werner, Anal. Chim. Acta, 2002, 462, 1–10 CrossRef CAS.
- T. Li, E. Wang and S. Dong, Anal. Chem., 2010, 82, 7576–7580 CrossRef CAS PubMed.
- L. Zhang, J. Zhu, J. Ai, Z. Zhou, X. Jia and E. Wang, Biosens. Bioelectron., 2013, 39, 268–273 CrossRef CAS PubMed.
- D. P. N. Goncalves, R. Rodriguez, S. Balasubramanian and J. K. M. Sanders, Chem. Commun., 2006, 4685–4687 RSC.
- S. N. Georgiades, N. H. A. Karim, K. Suntharalingam and R. Vilar, Angew. Chem., Int. Ed., 2010, 49, 4020–4034 CrossRef CAS PubMed.
- S. Shi, J. Zhao, X. Gao, C. Lv, L. Yang, J. Hao, H. Huang, J. Yao, W. Sun, T. Yao and L. Ji, Dalton Trans., 2012, 41, 5789–5793 RSC.
- S. Orrenius, B. Zhivotovsky and P. Nicotera, Nat. Rev. Mol. Cell Biol., 2003, 4, 552–565 CrossRef CAS PubMed.
- H.-Z. He, M. Wang, D. S.-H. Chan, C.-H. Leung, X. Lin, J.-M. Lin and D.-L. Ma, Methods, 2013, 64, 212–217 CrossRef CAS PubMed.
- D. Zhao, X. Dong, N. Jiang, D. Zhang and C. Liu, Nucleic Acids Res., 2014, 42, 11612–11621 CrossRef PubMed.
- X. Li, L. Gan, Q. Qishui Ou, X. Zhang, S. Cai, D. Wu, M. Chen, Y. Xia, J. Chen and B. Yang, Biosens. Bioelectron., 2015, 66, 399–404 CrossRef CAS PubMed.
- Y. Kataoka, H. Fujita, Y. Kasahara, T. Yoshihara, S. Tobita and M. Kuwahara, Anal. Chem., 2014, 86, 12078–12084 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |