
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImmobilization of polyoxometalates in the Zr-based metal organic framework UiO-67†
William
Salomon
a,
Catherine
Roch-Marchal
*a,
Pierre
Mialane
a,
Paul
Rouschmeyer
a,
Christian
Serre
a,
Mohamed
Haouas
a,
Francis
Taulelle
a,
Shu
Yang
b,
Laurent
Ruhlmann
b and
Anne
Dolbecq
*a
aInstitut Lavoisier de Versailles, UMR 8180, Université de Versailles Saint-Quentin en Yvelines, 45 Avenue des Etats-Unis, 78035 Versailles cedex, France. E-mail: catherine.roch@uvsq.fr; anne.dolbecq@uvsq.fr
bUniversité de Strasbourg, Institut de Chimie, UMR CNRS 7177, Laboratoire d'Electrochimie et de Chimie Physique du Corps Solide, 4 rue Blaise Pascal, CS 90032, 67081 Strasbourg cedex, France
First published on 9th January 2015
Abstract
The encapsulation of polyoxometalates within the large pores of the Zr(IV) biphenyldicarboxylate UiO-67 metal–organic framework has been achieved, for the first time, by direct solvothermal synthesis. The resulting POM@UiO-67 composite materials were fully characterized by XRPD, IR, MAS NMR, N2 porosimetry measurements and cyclic voltammetry.
Polyoxometalates (POMs) are soluble early-transition-metal clusters with a large diversity of structures and compositions.1 They possess redox and acid–base properties which can be exploited for catalytic applications.2 However their low specific surface area, low stability under catalytic conditions and high solubility in aqueous solution constitute some of their drawbacks. The search for stable heterogeneous catalysts with a high surface area, which could combine the activity of the POMs with the advantages of heterogeneous catalysts, such as easier recovery and recycling, thus attracts a lot of interest. Encapsulation of POMs within the cavities of metal organic frameworks (MOFs)3 constitutes a strategy to access to POMs-based heterogeneous catalysts.4 The mesoporous M(III)-trimesate MIL-100 and -terephthalate MIL-101 families (M = Fe, Cr, Al; MIL stands for Material of Institut Lavoisier) have been the most investigated host matrices so far because of their very large pore sizes and surface areas and good chemical stability. Keggin-type as well as sandwich-type POMs have been incorporated either by impregnation or in situ during the MOF synthesis.5 Besides the MIL families, Cu–BTC frameworks (BTC = 1,3,5-benzene-tricarboxylate), known as HKUST-1 (HKUST stands for Hong-Kong University of Science and Technology), have been the subject of intense research.5 Due to the small size of the pores they can only accommodate Keggin anions by direct synthesis. However the poor aqueous stability of these copper carboxylate based MOFs might prevent their practical use. Among the few examples of a series of thermally, and chemically stable MOFs with high surface area, the UiO-66 to 68 family (UiO stands for University of Oslo), built up from {ZrIV6O4(OH)4} oxocluster nodes and linear dicarboxylate linkers (with the general formulae [ZrIV6O4(OH)4(linker)6]), is one of the most studied MOFs.6 These materials exhibit by themselves very promising catalytic properties.7 In UiO-67 the inorganic octahedral Zr6 units are bound to 12 other inorganic subunits through biphenyl dicarboxylate ligands forming a face-centered cubic (fcc) structure (a = 27.1 Å). This MOF exhibits two types of cages: supertetrahedral (∅ ∼ 11.5 Å) and superoctahedral (∅ ∼ 18 Å) accessible through microporous triangular windows (∅ ∼8 Å) (Fig. 1a), corresponding to the voids of the fcc packing. However, the incorporation of guest molecules within the pores of UiO MOFs has only been rarely studied8 and, to our knowledge, there have been so far no reports of POM@UiO composites. Here we propose for the first time a synthetic method for the encapsulation within the pores of UiO-67 of three representative examples of POMs: [PW12O40]3−, [PW11O39]7− and [P2W18O62]6− whose size (Fig. 1b–d) can fit in the microporous octahedral cavities of this MOF.
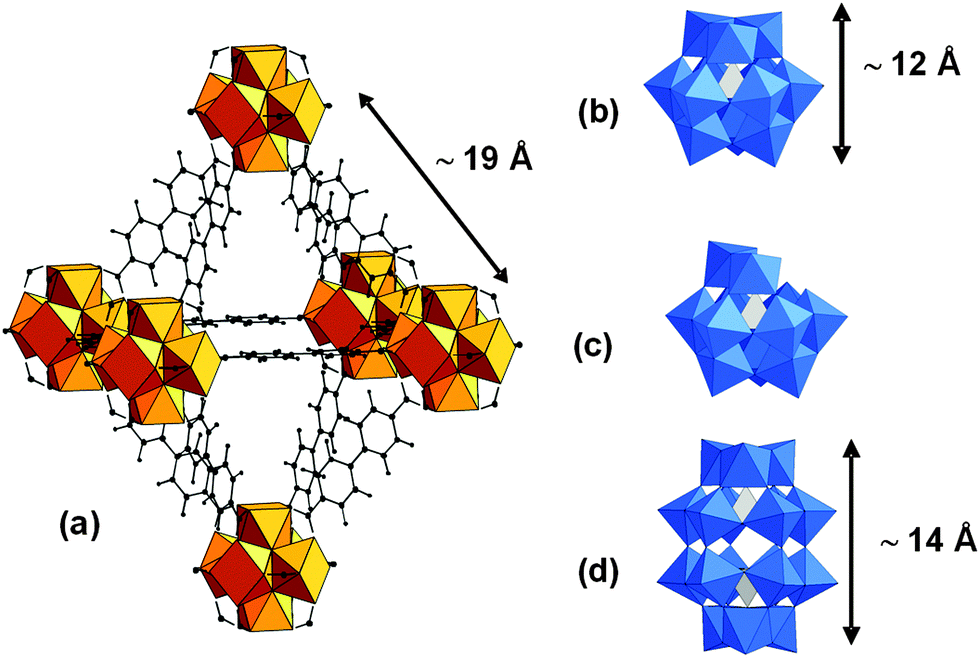 | ||
| Fig. 1 Polyhedral representations of (a) the octahedral cages of UiO-67, (b) [PW12O40]3− (PW12), (c) [PW11O39]7− (PW11) and (d) [P2W18O62]6− (P2W18); blue octahedra: WO6, grey tetrahedra: PO4, orange polyhedra: ZrO8, black spheres: C, small black spheres: H. | ||
Attempts to incorporate POMs in the UiO-67 by an impregnation method, like for the MIL-101 materials,5 have failed. This can be related to the narrow size of the windows. Thus a direct synthesis approach has been considered. A mixture of the pre-formed POM species (H3[PW12O40], TBA4H3[PW11O39] or TBA6[P2W18O62]), zirconium tetrachloride, benzoic acid as a modulator,9 and biphenyldicarboxylic acid was heated at 120 °C in dimethylformamide (DMF) (ESI†). Concentrated HCl was also added following previous reports.6b,d One would also expect that under the synthetic conditions used for the synthesis of UiO-67, the monolacunary PW11 POM would react with Zr(IV) ions to lead to the monomeric 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex [PW11O39Zr(H2O)n]3− (n = 2, 3), postulated by Kholdeeva et al.10 Such a monomeric monosubstituted complex has also been evidenced in the Dawson11 and Lindquist family.12 The resulting insoluble microcrystalline materials, PW12@UiO-67, PW11Zr@UiO-67 and P2W18@UiO-67, respectively, were filtered and washed several times with DMF and acetone. Infrared (IR) spectra (Fig. 2) indicate the presence of both the POM and the MOF, with P–O and W–O vibration bands characteristic of the POM observed between 850 and 1100 cm−1 together with the carboxylate vibration bands of the linker between 1300 and 1600 cm−1. The positions of the Bragg peaks are similar in the X-ray powder diffraction pattern of bare UiO-67 and of the POM@UiO-67 materials, confirming that the introduction of POMs in the synthetic medium does not perturb the formation of the MOF (Fig. S1, ESI†).
:1 complex [PW11O39Zr(H2O)n]3− (n = 2, 3), postulated by Kholdeeva et al.10 Such a monomeric monosubstituted complex has also been evidenced in the Dawson11 and Lindquist family.12 The resulting insoluble microcrystalline materials, PW12@UiO-67, PW11Zr@UiO-67 and P2W18@UiO-67, respectively, were filtered and washed several times with DMF and acetone. Infrared (IR) spectra (Fig. 2) indicate the presence of both the POM and the MOF, with P–O and W–O vibration bands characteristic of the POM observed between 850 and 1100 cm−1 together with the carboxylate vibration bands of the linker between 1300 and 1600 cm−1. The positions of the Bragg peaks are similar in the X-ray powder diffraction pattern of bare UiO-67 and of the POM@UiO-67 materials, confirming that the introduction of POMs in the synthetic medium does not perturb the formation of the MOF (Fig. S1, ESI†).
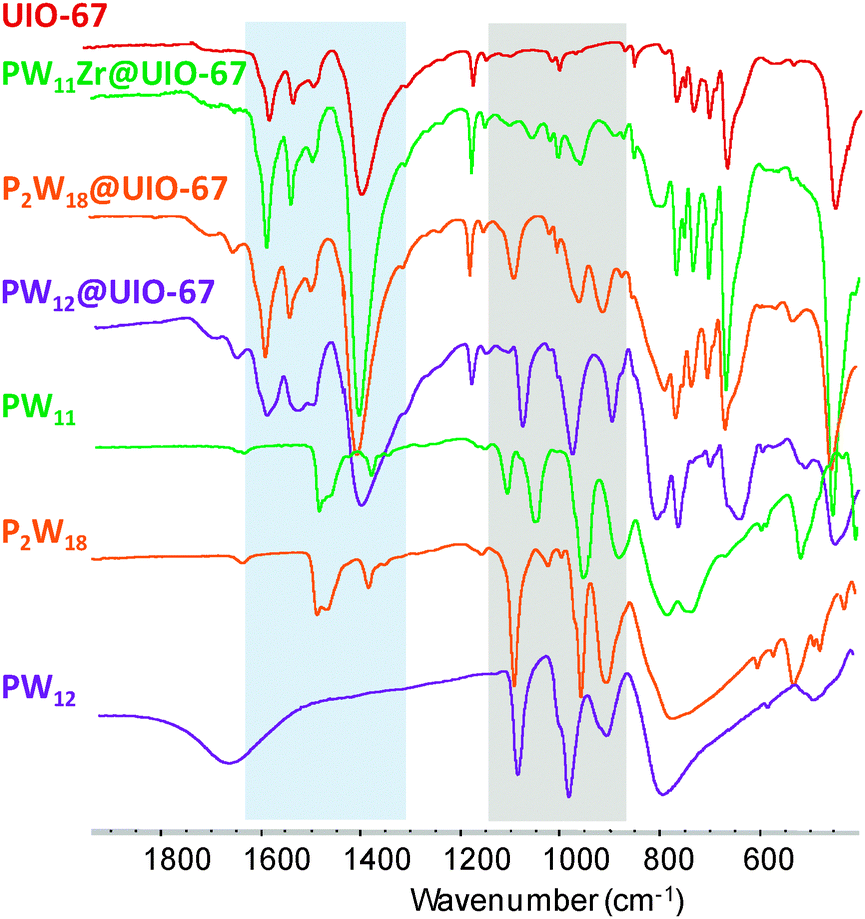 | ||
| Fig. 2 IR spectra of POM@UiO-67 composites compared to that of the POM precursors and UiO-67. The regions with characteristic peaks of the POMs and of the MOF are highlighted in grey and light blue respectively. | ||
Elemental analysis combined with thermogravimetric analysis (TGA) measurements allows proposing the [Zr6O4(OH)5.4][C14H8O4]5.3 formula for UiO-67, in agreement with the existence of linker defects occupied by hydroxide anions.6b One expects that for the POM@UiO-67 composites, the negative charges introduced by the POMs compensate, almost totally, the linker deficiencies. Note that the presence of tetrabutylammonium cations can be ruled out both by IR, elemental analysis and nuclear magnetic resonance (NMR). The TGA curves for UiO-67 and its composites (Fig. S2 and Table S1, ESI†) reveal steps that are attributed to water removal, linker decomposition and formation of inorganic oxides. Combined with elemental analyses, these results lead to the estimation of the following formulae [Zr6O4(OH)4][C14H8O4]5.37[PW12O40]0.42 (PW12@UiO-67), [Zr6O4(OH)4][C14H8O4]5.73[PW11O39Zr]0.18 (PW11Zr@UiO-67), and [Zr6O4(OH)4.30][C14H8O4]5.10[P2W18O62]0.25 (P2W18@UiO-67) for the POM@UiO-67 composites. As there is one octahedral cavity per Zr6 unit, these formulae indicate that approximately 1/2, 1/5 and 1/4 of the cavities are occupied by POMs in PW12@UiO-67, PW11Zr@UiO-67 and P2W18@UiO-67, respectively.
N2 sorption isotherms have been recorded for all the reported compounds (Fig. S3 and Table S2, ESI†). As expected, the surface area as well as the total pore volumes decrease from UiO-67 to POM@UiO-67 composites, as a consequence of the encapsulation of the POMs within the octahedral cavities, leaving however a significant porosity accessible to nitrogen. It should be noted that the values of the normalized specific surface area, taking into account the mass of UiO-67 in the composites samples, are significantly different from the value of UiO-67 (Fig. S3 and Table S2, ESI†), suggesting that the POMs are located inside the cavities and not at the surface of the material.
The 1H MAS and 13C{1H} CPMAS NMR spectra of the three POM@UiO-67 (Fig. S4 and S5, ESI†) exhibit the characteristic resonances from the crystalline UiO-67. The Zr–OH hydroxyl protons from the hexameric unit lie in the 0–3.6 ppm range while the resonances of the linker, i.e. aromatic protons, are observed at 7.1 and 7.9 ppm. The aromatic carbon atoms (125, 130, 134, and 143 ppm) and the carbon atoms of the carboxylic function (172 ppm) are in the expected range.8c Noteworthily, broad components of all these resonances are observed and the general tendency showed that higher the amount of encapsulated POM larger the fraction of these broad resonances (23% in PW11Zr@UiO-67, 54% in P2W18@UiO-67, and 68% in PW12@UiO-67). This is assigned to structural disorder caused by the POM filling pores.
The 31P MAS NMR spectra of PW12@UiO-67 (Fig. S6a, ESI†) and P2W18@UiO-67 (Fig. 3a) show characteristic features of the POM precursors with signals at −15.2 and −12.9 ppm respectively, comparable to the literature values.13 In contrast, the spectrum of PW11Zr@UiO-67 (Fig. S7a, ESI†) exhibits a resonance at −13.2 ppm instead of −12.4 ppm expected for PW11.14 This shift is in perfect agreement with that determined for a mixture of PW11 and ZrCl4 in DMF (Fig. S8, ESI†).
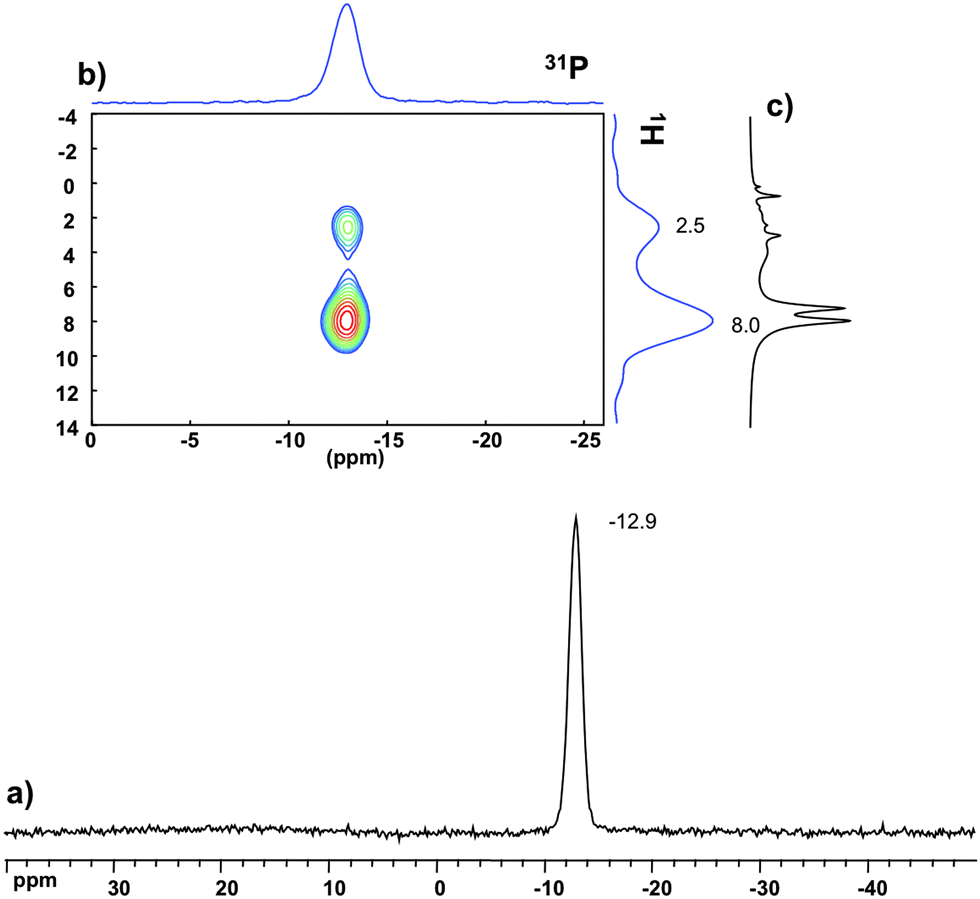 | ||
| Fig. 3 Solid-state NMR spectra of P2W18@UiO-67: (a) 31P{1H} CPMAS; (b) 1H–31P HETCOR; (c) 1H MAS. | ||
31P NMR provides not only information about the structure of the POM but also some insights on location and host–guest interactions between the POM and the MOF. Indeed, the 1H–31P HETCOR (heteronuclear correlation) experiment enables us to probe the dipolar contact between the 31P and 1H nuclei. Correlations between the phosphorus atom of the POM and protons of the aromatic linkers as well as Zr–OH framework hydroxyl groups are observed in the three POM@UiO-67 composite materials. This unambiguously indicates the close proximity of the POM to the surface of the MOF inside the pores homogeneously as otherwise no such efficient dipolar transfer would be seen. Furthermore, the strength of the correlation with these protons appears varied from one POM to another. Indeed in P2W18@UiO-67 (Fig. 3b) and PW12@UiO-67 (Fig. S6b, ESI†) the 31P site correlates mainly with the aromatic resonance (8 ppm) while in PW11Zr@UiO-67 (Fig. S7b, ESI†) the correlation mainly occurs with the hydroxyl group peak (2.5 ppm). This clearly indicates the preferential interaction of PW12 and P2W18 with the organic linker, whereas PW11Zr should be closer to the inorganic node, suggesting interactions between the Zr(IV) ions inserted in the POM lacuna and the {ZrIV6O4(OH)4} clusters. Finally, these correlations involve only the broad components of the 1H spectra (as it could be compared with 1D MAS in Fig. 3c). This strongly suggests as stated above that the POMs are located in the distorted/disordered domain.
The electrochemical properties of the POM@UiO composite materials were also studied and compared to those of the POMs alone. The solids were first immobilized on the surface of the basal plane of the pyrolytic graphite disk (PG) and their electrochemical responses studied in pH 2.5 H2SO4–Na2SO4 0.5 M buffer solutions. PW12 and P2W18 and their related POM@UiO-67 composites exhibit similar redox behaviors related to the WVI/V redox couples (Table S3, Fig. S9–S11, ESI† and Fig. 4), showing that when saturated POMs are encapsulated inside the cages they retain their electrochemical properties. It must be noted however that the first reduction process for P2W18 is 175 mV higher for P2W18@UiO-67 than for P2W18.
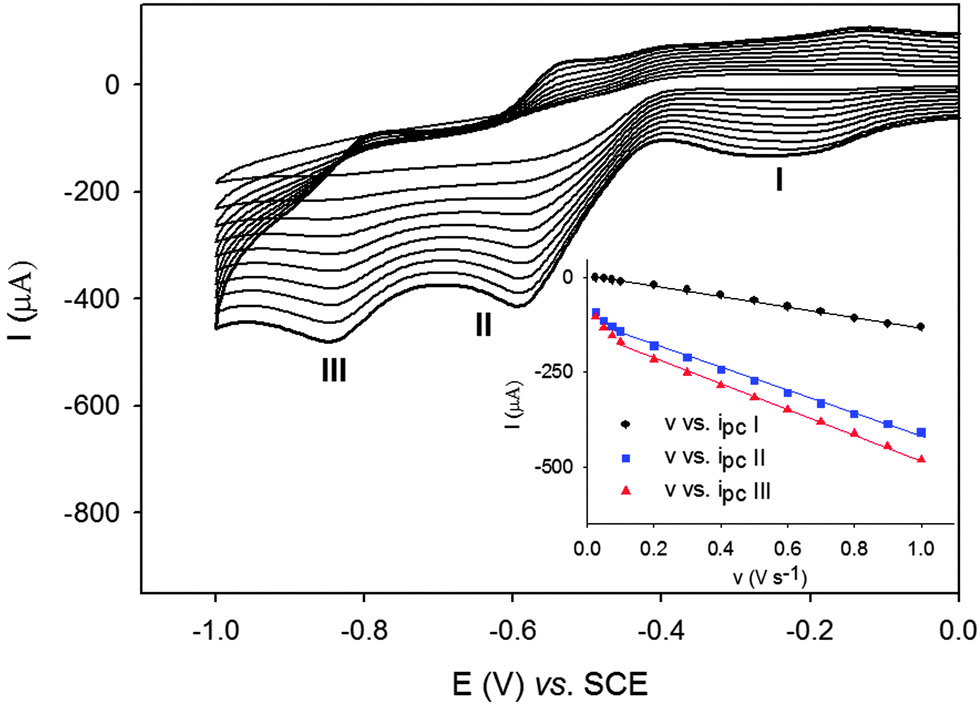 | ||
| Fig. 4 Cyclic voltammograms of P2W18@UiO-67 immobilized at a PG electrode in a pH 2.5 H2SO4–Na2SO4 0.5 M buffer solution at different scan rates from 0.025 to 1.000 V s−1. Inset: plots of Ipcvs. v for peaks I, II and III. | ||
In the case of PW11Zr@UiO-67 the cyclovoltammogram clearly shows the complexation of the monolacunary PW11 with Zr(IV) and the formation of PW11Zr encapsulated in UiO-67 (Fig. S12 and S13, ESI†). Indeed, PW11 immobilized on the PG exhibits four successive waves at −0.450 V, −0.560 V, −0.694 V and −0.830 V vs. SCE respectively while only two waves are measured for PW11Zr@UiO-67 at −0.713 V and −0.885 V. It should be noted that the electrochemical behaviors of the POM@UiO composites and of composites obtained by mechanical mixing of POMs and UiO (Fig. S14–S16) are different which confirm the encapsulation of the POMs inside the cavities and the transformation of PW11 into PW11Zr. Finally, all peak current intensities vary linearly with the scan rate in the range 0.1–1.0 V s−1, as expected for surface confined redox processes. This behavior is in agreement with the immobilization of the POMs inside the cavities of the MOF and contrasts with the observation of diffusion-controlled processes for PW11@MIL-101(Cr) which were attributed to the mobility of the POMs.15
In conclusion, the immobilization of three polyoxotungstates in the largest pores of UiO-67(Zr) has been carried out for the first time, by a direct synthetic method. POM encapsulation compensates almost totally the linker deficiencies. The elaborated POM@UiO-67 composites have been thoroughly characterized. Cyclic voltammetry studies have confirmed the direct formation of PW11Zr during the synthetic process and the integrity of the saturated inserted POMs PW12 and P2W18. MAS NMR experiments provided essential information on the structure of the composites. First, they have shown interactions between the POMs and the framework, thus confirming the encapsulation within the structure. Moreover they highlighted the existence of disordered domains related to the presence of the POMs. Finally they evidenced specific interactions between the asymmetric PW11Zr POMs and the UiO framework. Noticeably, because of the small size of the cage windows, POM leaching cannot occur in these materials. The introduction of POMs might pave the way for future applications in catalysis. It may also greatly influence the hydrophilicity of the materials and thus their gas sorption properties. Future work will focus on these two applications.
This work was supported by CNRS, UVSQ, University of Strasbourg and the French National Research Agency (ANR) as part of the “Investissements d'Avenir” program no. ANR-11-IDEX-0003-02 and CHARMMMAT ANR-11-LABX-0039. Patricia Horcajada and Thomas Devic are gratefully acknowledged for fruitful discussions.
Notes and references
- (a) H. N. Miras, L. Vilà-Nadal and L. Cronin, Chem. Soc. Rev., 2014, 43, 5679 RSC; (b) O. Oms, A. Dolbecq and P. Mialane, Chem. Soc. Rev., 2012, 41, 7497 RSC; (c) A. Proust, B. Matt, R. Villanneau, G. Guillemot, P. Gouzerh and G. Izzet, Chem. Soc. Rev., 2012, 41, 7605 RSC.
- (a) H. Lv, Y. V. Geletii, C. Zhao, J. W. Vickers, G. Zhu, Z. Luo, J. Song, T. Lian, D. G. Musaev and C. L. Hill, Chem. Soc. Rev., 2012, 41, 7572 RSC; (b) A. Sartorel, M. Bonchio, S. Campagna and F. Scandola, Chem. Soc. Rev., 2013, 42, 2262 RSC; (c) N. Mizuno and K. Kamata, Coord. Chem. Rev., 2011, 255, 2358 CrossRef CAS PubMed; (d) X.-B. Han, Z.-M. Zhang, T. Zhang, Y.-G. Li, W. Lin, W. You, Z.-M. Su and E.-B. Wang, J. Am. Chem. Soc., 2014, 136, 5359 CrossRef CAS PubMed.
- Special issue on Metal Organic Frameworks Chem. Soc. Rev., 2014, 43, 5415.
- J. Juan-Alcañiz, J. Gascon and F. Kapteijn, J. Mater. Chem., 2012, 22, 10102 RSC.
- See for example: (a) N. V. Maksimchuk, M. N. Timofeeva, M. S. Melgunov, A. N. Shmakov, Y. A. Chesalov, D. N. Dybtsev, V. P. Fedin and O. A. Kholdeeva, J. Catal., 2008, 257, 315 CrossRef CAS PubMed; (b) L. Bromberg, Y. Diao, H. Wu, S. C. A. Speakman and T. A. Hatton, Chem. Mater., 2012, 24, 1664 CrossRef CAS; (c) C. M. Granadeiro, A. D. S. Barbosa, P. Silva, F. A. Almeida Paz, V. K. Saini, J. Pires, B. de Castro, S. S. Balula and L. Cunha-Silva, Appl. Catal., A, 2013, 453, 316 CrossRef CAS PubMed; (d) J. Juan-Alcañiz, E. V. Ramos-Fernandez, U. Lafont, J. Gascon and F. Kapteijn, J. Catal., 2010, 269, 229 CrossRef PubMed; (e) A.-X. Yan, S. Yao, Y.-G. Li, Z.-M. Zhang, Y. Lu, W.-L. Chen and E.-B. Wang, Chem. – Eur. J., 2014, 20, 6927 CrossRef CAS PubMed; (f) S. R. Bajpe, C. E. A. Kirschhock, A. Aerts, E. Breynaert, G. Absillis, T. N. Parac-Vogt, L. Giebeler and J. A. Martens, Chem. – Eur. J., 2010, 16, 3926 CrossRef CAS PubMed; (g) J. Song, Z. Luo, D. K. Britt, H. Furukawa, O. M. Yaghi, K. I. Hardcastle and C. L. Hill, J. Am. Chem. Soc., 2011, 133, 16839 CrossRef CAS PubMed; (h) J. J.-A. Alcañiz, M. Goesten, A. Martinez-Joaristi, E. Stavitski, A. V. Petukhov, J. Gascon and F. Kapteijn, Chem. Commun., 2011, 47, 8578 RSC . The complete list of references is given in the ESI† section.
- (a) J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850 CrossRef PubMed; (b) M. J. Katz, Z. J. Brown, Y. J. Colón, P. W. Siu, K. A. Scheidt, R. Q. Snurr, J. T. Hupp and O. K. Farha, Chem. Commun., 2013, 49, 9449 RSC; (c) J. E. Mondloch, M. J. Katz, N. Planas, D. Semrouni, L. Gagliardi, J. T. Hupp and O. K. Fahra, Chem. Commun., 2014, 50, 8944 RSC; (d) P. S. Barciá, D. Guimarães, P. A. P. Mendes, J. A. C. Silva, V. Guillerm, H. Chevreau, C. Serre and A. E. Rodrigues, Microporous Mesoporous Mater., 2011, 139, 67 CrossRef PubMed; (e) G. C. Schearer, S. Chavan, J. Ethiraj, J. G. Vitillo, S. Svelle, U. Olsbye, C. Lamberti, S. Bordiga and K. P. Lillerud, Chem. Mater., 2014, 26, 4068 CrossRef.
- (a) F. Vermoortele, R. Ameloot, A. Vimont, C. Serre and D. De Vos, Chem. Commun., 2010, 47, 1521 RSC; (b) M. J. Katz, J. E. Mondloch, R. K. Totten, J. K. Park, S. T. Nguyen, O. K. Farha and J. T. Hupp, Angew. Chem., Int. Ed., 2014, 53, 497 CrossRef CAS PubMed.
- (a) S. Devautour-Vinot, C. Martineau, S. Diaby, M. Ben-Yahia, S. Miller, C. Serre, P. Horcajada, D. Cunha, F. Taulelle and G. Maurin, J. Phys. Chem. C, 2013, 117, 11694 CrossRef CAS; (b) J. He, J. Wang, Y. Chen, J. Zhang, D. Duan, Y. Wang and Z. Yan, Chem. Commun., 2014, 50, 7063 RSC; (c) C. Larabi and E. A. Quadrelli, Eur. J. Inorg. Chem., 2012, 3014 CrossRef CAS.
- A. Schaate, P. Roy, A. Godt, J. Lippke, F. Waltz, M. Wiebcke and P. Behrens, Chem. – Eur. J., 2011, 17, 6643 CrossRef CAS PubMed.
- O. A. Kholdeeva, G. M. Maksmimov, R. I. Maksimovskaya, M. P. Vanina, T. A. Trubitsina, D. Y. Naumov, B. A. Kolesov, N. S. Antonova, J. J. Carbó and J. M. Poblet, Inorg. Chem., 2006, 45, 7224 CrossRef CAS PubMed.
- (a) M. N. Sokolov, N. V. Izarova, E. V. Peresypkina, D. A. Mainichev and V. P. Fedin, Inorg. Chim. Acta, 2009, 362, 3756 CrossRef CAS PubMed; (b) S. Vanhaecht, G. Absillis and T. N. Parac-Vogt, Dalton Trans., 2013, 42, 15437 RSC.
- R. J. Errington, P. S. S. Petkar, P. S. Middleton and W. McFarlane, J. Am. Chem. Soc., 2007, 129, 12181 CrossRef CAS PubMed.
- (a) J. Chunjie, S. Shengnan, W. Xuyang, W. Xiangsheng, G. Hongchen, G. Xinwen and C. Lidong, Acta Chim. Sin., 2013, 71, 810 CrossRef; (b) S. Ribeiro, A. D. S. Barbosa, A. C. Gomes, M. Pillinger, I. S. Gonçalves, L. Cunha-Silva and S. S. Balula, Fuel Process. Technol., 2013, 116, 350 CrossRef CAS PubMed.
- G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé and I. Margiolaki, Science, 2005, 309, 2040 CrossRef PubMed.
- P. M. Paes de Sousa, R. Grazina, A. D. S. Barbosa, B. de Castro, J. J. G. Moura, L. Cunha-Silva and S. S. Balula, Electrochim. Acta, 2013, 87, 853 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed syntheses and characterizations. See DOI: 10.1039/c4cc09986a |
| This journal is © The Royal Society of Chemistry 2015 |