
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring hydrogen peroxide responsive thiazolidinone-based prodrugs†
Christian
Perez
,
Jean-Philippe
Monserrat
,
Yao
Chen
and
Seth M.
Cohen
*
Department of Chemistry & Biochemistry, University of California, San Diego, 9500 Gilman Drive, La Jolla, CA 92093, USA. E-mail: scohen@ucsd.edu
First published on 20th March 2015
Abstract
A novel approach for developing prodrugs based on masked carboxylic acids is described. Rather than using conventional esterase-based activation, thiazolidinone protecting groups have been identified that can reveal carboxylic acid groups upon activation by hydrogen peroxide. This may prove valuable in the continuing development of prodrug strategies that rely on reactive oxygen species (ROS) as a trigger.
Prodrugs are inactive derivatives of bioactive molecules that are designed to become activated in vivo by a variety of stimuli. Prodrug strategies often serve to improve the drug-like properties of bioactive molecules including bioavailability, cell permeability, and pharmacokinetics.1–4 The success of the prodrug approach is illustrated by the fact that ∼10% of clinical therapeutics are classified as prodrugs.5
Prodrugs generally utilize a chemical modification strategy that renders the molecule biologically inactive or substantially less active. This chemical modification is commonly referred to as a ‘promoiety’. Carboxylic acids are a common functional group amenable to the prodrug method. The high polarity and ionizability of carboxylic acids can decrease the lipophilicity of the bioactive compounds, which inhibits membrane permeability and hence absorption.2 A common approach to overcome this drawback is through esterification of the acid moiety, which can be hydrolyzed in vivo by ubiquitous esterases.6 This strategy is the most popular method used to overcome issues associated with carboxylic acid containing drugs such as Pivampicillin, oseltamivir, and ximelagatran.2 However, esterases are ubiquitous and constitutively active, so ester-based prodrugs do not target disease-specific environments. Ideally, a prodrug method could be exploited to achieve targeted release of the active drug by disease-specific stimuli.
Reactive oxygen species (ROS) are naturally occurring species that result from cellular metabolism of molecular oxygen.7–9 The reduction of molecular oxygen results in the production of intermediates such as superoxide anion, hydrogen peroxide, hydroxyl radical, and organic peroxides.10 Cells maintain tightly controlled and rather complex systems of enzymatic and non-enzymatic antioxidants that balance and minimize levels of ROS.11 However, overproduction of these ROS can result in impaired cellular functions and formation of toxic metabolites. Several pathologies are associated with increased levels of ROS including inflammation,12 cancer,13–16 cardiovascular,17 and neurodegenerative diseases.18 Cancer cells are an ideal target for ROS-prodrug activation, since they exhibit elevated levels of H2O2 (5 μM to 1.0 mM).19 Therefore, ROS-activated prodrugs may serve as a platform for targeted release of potent therapeutics to these specific microenvironments, while limiting off-target interactions. Such ROS-mediated activation could result in spatially and temporally controlled release of therapeutics. Successful efforts to release therapeutics through ROS activation have been achieved through incorporation of sulfonate esters and boronic acid/ester promoieties, for the protection of alcohols and amine groups in bioactive molecules.20–24 However, none of these strategies have been proven to be amendable for the release of carboxylic acids.
There are several classes of inhibitors that stand to benefit from a ROS prodrug strategy. Of particular interest are inhibitors targeting cyclooxygenases (COX), matrix metalloproteinases (MMP), and angiotensin converting enzyme (ACE). ROS prodrug strategies for the COX inhibitor ibuprofen have been thoroughly explored.25 Ibuprofen is a non-selective cyclooxygenase (COX) inhibitor, and its chronic use as an analgesic and antiinflammatory has been associated with formation of ulcers and gastrointestinal bleeding.26 A ROS-responsive prodrug strategy could aid in diminishing adverse side effects. MMP inhibitors (MMPi) also stand to benefit from a ROS-targeted prodrug strategy, as MMP hyperactivity and ROS have been associated with cancer and ischemic reperfusion injury. Lastly, ACE inhibitors are major class of therapeutics with several FDA approved compounds in the clinic. ACE inhibitors, such as the tripeptide dicarboxylate enalapril, utilize an esterase prodrug approach in which one of the carboxylic acid pharmacophores is esterified.27 We hypothesized that by appending a ROS-sensitive functionality to the carboxylic acid group of these inhibitors, we could attenuate the inhibition by these compounds until the active drug is selectively liberated in the presence of H2O2.
In this study, we explored a novel approach for H2O2-activated prodrugs based on a thiazolidinone promoiety. This strategy was inspired by the use of H2O2 to cleave oxazolidinone protecting groups that are used as auxiliaries for asymmetric reactions, and are commonly referred to as Evan's auxiliaries.28,29 Similar protecting groups have also been used as auxiliaries.30,31 By examining modified oxazolidinones, we report a prodrug strategy that can effectively mask the carboxylic acid groups of several pharmacologically potent agents. In the presence of elevated levels of H2O2, the promoiety is hydrolyzed to generate the bioactive compound with a free carboxylic acid (Fig. 1).
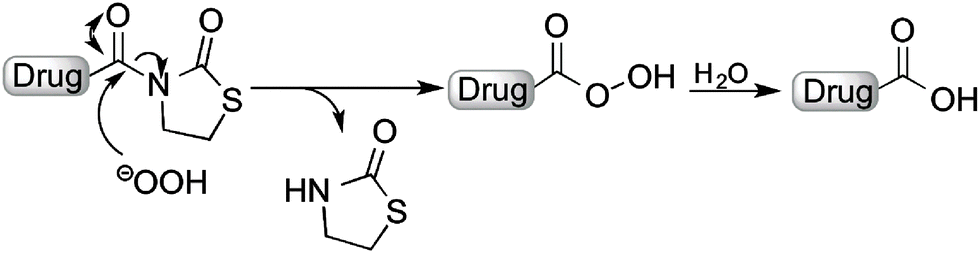 | ||
| Fig. 1 Proposed scheme for activation of a thiazolidinone prodrug. | ||
In order to develop carboxylic acid prodrugs, the responsiveness of several oxazolidinone derivatives to H2O2 and their stability in simulated physiological conditions were examined. Four distinct, but closely related, promoieties were studied: oxazolidinone (A), thiazolidinone (B), oxazolidinethione (C), and thiazolidinethione (D) (Fig. 2). Our approach was to evaluate each of these derivatives with respect to stability and rate of activation in order to identify a suitable promoiety. The promoiety showing the best stability balanced with the fastest rate of H2O2-activation was selected for further investigations (vide infra).
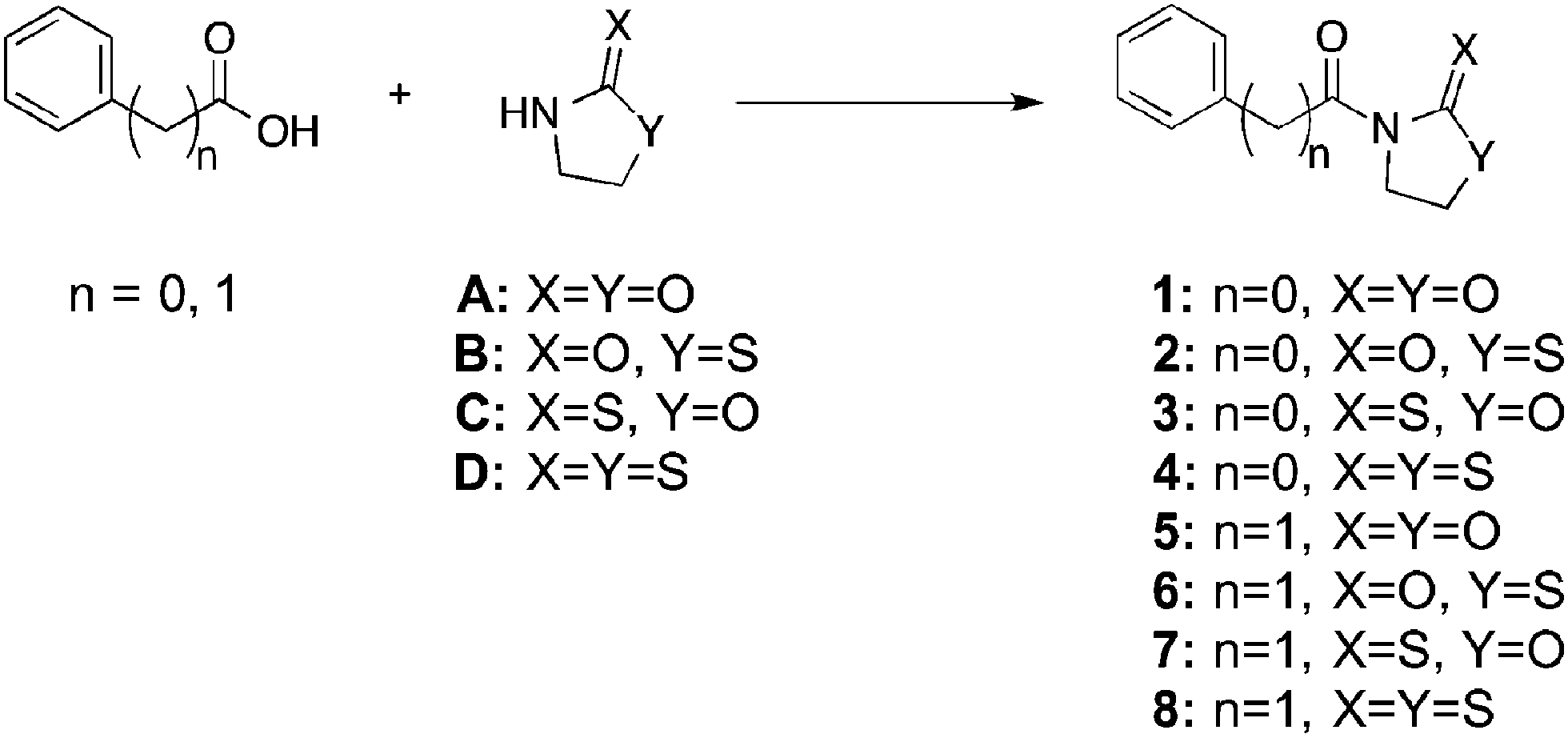 | ||
| Fig. 2 Model compounds of four related oxazolidinone derivatives in the context of both aryl and alkyl carboxylic acids. | ||
Each of these four groups was appended to two test compounds: benzoic acid and phenyl acetic acid. These model compounds were chosen for their structural simplicity and to highlight differences between the reactivity and stability of aryl and alkyl (benzylic) carboxylic acids. Compounds A and D are commercially available and the synthesis for B and C had been previously reported.32,33 Synthesis for several of the model compounds had also been described.34–37 Amide bond formation between the carboxylic acid compounds and the promoieties (A–D) was performed via one of two different methods: addition of DCC and DMAP in CH2Cl2 (method i) or through the Schotten–Baumann reaction (method ii), with the corresponding acid-chloride. All compounds were rigorously characterized (ESI†) by routine characterization methods. In addition, the single-crystal X-ray structure of compounds 2 and 3 were determined, unambiguously confirming their composition and connectivity (Fig. S24 and S25, Table S1, ESI†).
Compounds 1–8 were treated with H2O2 (20 equiv., 100 mM Tris-Cl, pH 7.4) in a 40% DMSO/60% buffer (100 mM Tris-Cl, pH 7.4) solution, and cleavage was monitored via analytical HPLC after 1 h and 4 h incubation. The high DMSO content was used in order to ensure that all of the compounds were completely soluble under the reaction conditions. After 4 h of incubation with H2O2, compounds 1, 2, 5, and 6 displayed a wide range of cleavage rates to the corresponding acids (i.e., benzoic acid or phenyl acetic acid, Table 1, Fig. S1, S3, S9 and S11, ESI†), with compounds 2 and 6 (both which utilize protecting group B) showing the fastest rates of conversion. In addition, the activation of compounds 2 and 6 was tested in a 5% DMSO/95% buffer (100 mM Tris-Cl, pH 7.4), and conversion to the corresponding product was also observed (Fig. S3 and S11, ESI†). The conversion of compounds 3, 4, 7, and 8, all of which contain a thione-based protecting group (e.g. exocyclic sulfur in C and D), were not determined, because these reactions were not clean, showing conversion to multiple products (Fig. S5, S7, S13 and S15, ESI†).
In addition to activation, prodrugs must have good aqueous stability, which was also tested by incubation in buffer in the absence of H2O2. The stability of each compound was examined in a 40% DMSO/60% buffer solution (again the high concentration of DMSO was used to ensure good solubility of all compounds; 100 mM Tris-Cl, pH 7.4) at physiological pH, as well as in the presence of glutathione (20 equiv., 100 mM Tris-Cl, pH 7.4), with incubation for 24 h at 37 °C. Compounds 1, 2, 5, and 6 displayed good stability in the presence and absence of glutathione, all showing <35% degradation (Table 2, Fig. S2, S4, S10 and S12, ESI†). In contrast, compounds 3, 4, 7, and 8, all showed poor stability, with rapid degradation under these incubation conditions (Table 2, Fig. S6, S8, S14 and S16, ESI†). This parallels the complex reactivity observed with these compounds (all of which contain protecting group C or D with an exocyclic sulfur) in the presence of H2O2 (vide supra). Overall, the model compounds that displayed the optimal combination of rate of cleavage and good stability were 2 and 6.
The success of compounds 2 and 6, both of which utilize promoiety B, prompted the investigation of the scope of this prodrug approach with an FDA approved drug, ibuprofen, and a well-studied matrix metalloproteinase inhibitor (Fig. 3).38 Both prodrugs (proIBU and proMMPi, Fig. 3) were readily synthesized† and evaluated for ROS activation. ProMMPi activation by two ROS species was evaluated. Two independent experiments were carried out in which a proMMPi solution (100 μM) was added to 20 equiv. of H2O2 or NaClO. Activation by H2O2 to the corresponding MMPi was observed (Fig. S17, ESI†); however, exposure of proMMPi to NaClO lead to degradation to unidentified side products (Fig. S18, ESI†). In order to determine the sensitivity of proMMPi in a quantitative fashion, pseudo-first-order kinetic measurements were performed using analytical HPLC (Fig. S17, ESI†). The observed rate constant for conversion to the desired MMPi upon exposure to H2O2 was kobs = 1.1 × 10−4 s−1. The hydrolytic stability under simulated physiological conditions was also determined. The compounds were incubated in DMSO and buffer (100 mM Tris-Cl, pH 7.4) for 24 h at 37 °C. Additionally, each prodrug was incubated under the same conditions with the addition of 20 equiv. of lysine, serine, or glutathione, in order to demonstrate that the promoiety is not readily cleaved in the presence of biologically relevant nucleophiles. Both proIBU and proMMPi displayed remarkable stability of >95% under all of the conditions tested (Fig. S19 and S21, ESI†).
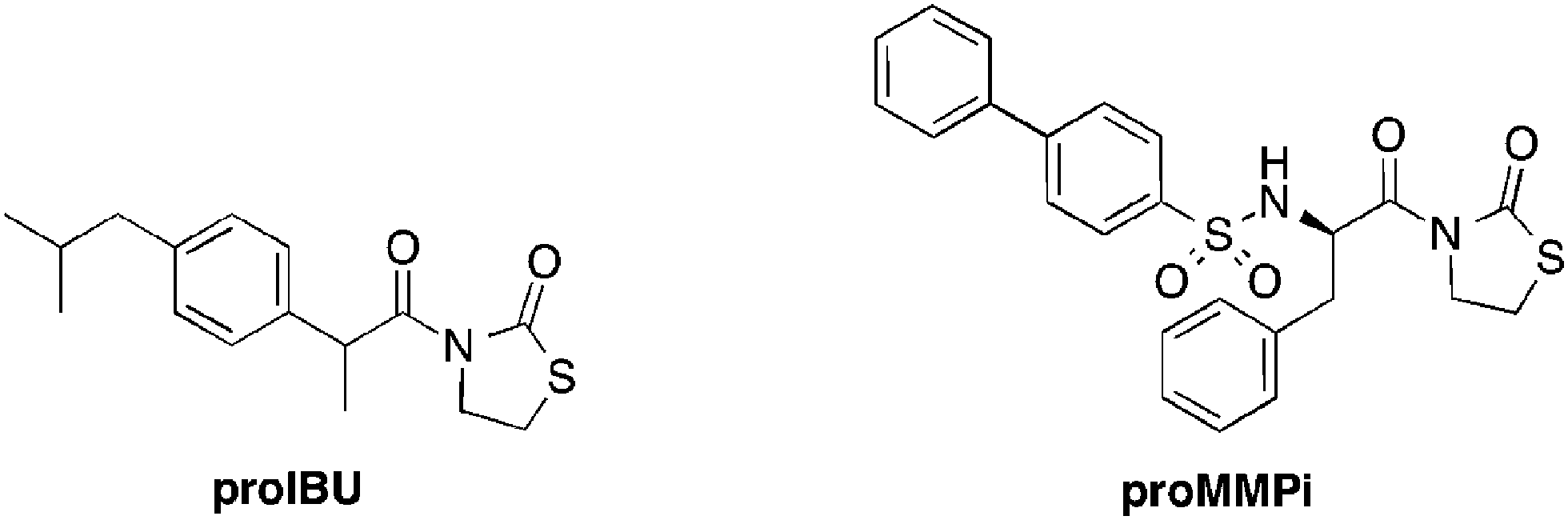 | ||
| Fig. 3 Prodrug version of ibuprofen and an MMP inhibitor. | ||
Inhibition by activated proIBU (proIBU + H2O2) using a commercially available fluorescent assay for COX-1 could not be determined because H2O2 interfered with the assay (Fig. S23, ESI†). However, no inhibition by proIBU (<5%) of COX-1 was observed, indicating the promoiety did suppress the activity of proIBU against the target. The efficacy of the proMMPi to inhibit its target was also evaluated. The MMP assay was carried out as previously described against MMP-2, by utilizing a cleavable Förster resonance energy transfer (FRET) substrate.39,40 The proMMPi demonstrated essentially no inhibition against MMP-2 (<5%, Fig. S22, ESI†). In contrast, inhibition of MMP-2 after treatment of proMMPi with 20 equiv. of H2O2 was increased to ∼37%, close to the expected value of 50% and demonstrating that the inhibitory prolife was readily restored. The data confirm that the proMMPi is inactive against its target (MMP-2), but regains near full activity after being treated with H2O2.
Based on our proposed scheme (Fig. 1), a thiazolidinone molecule is liberated as the prodrug becomes activated by H2O2. Therefore, we conducted a cell viability assay to determine if the thiazolidinone moiety was cytotoxic. An MTS assay utilizing NIH 3T3 cells was performed in which the cytotoxicity of promoiety B was determined. Compound B demonstrated no significant cytotoxicity up to a concentration of 100 μM (Fig. S26, ESI†).
In summary, our findings demonstrate the development of a prodrug strategy for the H2O2-dependent release of carboxylic acids. A study using model compounds revealed the thiazolidinone promoiety (B, Fig. 2) to possess high stability and was not prone to attack by common biological nucleophiles. Prodrugs based on the thiazolidinone B displayed little to no activity against their targets, but in the presence of H2O2 activity was restored for an MMP inhibitor. Overall, these findings indicate that a thiazolidinone (B) prodrug strategy is viable for derivatizing carboxylic acid-based therapeutics for H2O2-targeted release. Considering the widespread use of ester-based prodrugs, such as ACE inhibitors, it is expected that the strategy here will have applicability to produce H2O2-activated, disease-targeted analogs of various valuable bioactive compounds.
We thank the UCSD Small Molecule Mass Spectrometry and X-ray diffraction facilities for assistance with experiments. This work was supported by a grant from the National Institutes of Health, National Institute of General Medical Sciences (R01 GM098435).
Notes and references
- V. J. Stella, J. Pharm. Sci., 2010, 99, 4755–4765 CrossRef CAS PubMed.
- J. Rautio, H. Kumpulainen, T. Heimbach, R. Oliyai, D. Oh, T. Jarvinen and J. Savolainen, Nat. Rev. Drug Discovery, 2008, 7, 255–270 CrossRef CAS PubMed.
- J. B. Zawilska, J. Wojcieszak and A. B. Olejniczak, Pharmacol. Rep., 2013, 65, 1–14 CrossRef CAS.
- P. Ettmayer, G. L. Amidon, B. Clement and B. Testa, J. Med. Chem., 2004, 47, 2393–2404 CrossRef CAS PubMed.
- K. M. Huttunen, H. Raunio and J. Rautio, Pharmacol. Rev., 2011, 63, 750–771 CrossRef CAS PubMed.
- B. M. Liederer and R. T. Borchardt, J. Pharm. Sci., 2006, 95, 1177–1195 CrossRef CAS PubMed.
- J. R. Stone and S. P. Yang, Antioxid. Redox Signaling, 2006, 8, 243–270 CrossRef CAS PubMed.
- S. G. Rhee, Science, 2006, 312, 1882–1883 CrossRef PubMed.
- C. C. Winterbourn, Nat. Chem. Biol., 2008, 4, 278–286 CrossRef CAS PubMed.
- B. C. Dickinson and C. J. Chang, Nat. Chem. Biol., 2011, 7, 504–511 CrossRef CAS PubMed.
- M. Giorgio, M. Trinei, E. Migliaccio and P. G. Pelicci, Nat. Rev. Mol. Cell Biol., 2007, 8, 722A–728 CrossRef PubMed.
- M. Nagata, Curr. Drug Targets: Inflammation Allergy, 2005, 4, 503–504 CrossRef CAS.
- E. O. Hileman, J. Liu, M. Albitar, M. J. Keating and P. Huang, Cancer Chemother. Pharmacol., 2004, 53, 209–219 CrossRef CAS PubMed.
- S. Kawanishi, Y. Hiraku, S. Pinlaor and N. Ma, Biol. Chem., 2006, 387, 365–372 CrossRef CAS PubMed.
- T. P. Szatrowski and C. F. Nathan, Cancer Res., 1991, 51, 794–798 CAS.
- S. Toyokuni, K. Okamoto, J. Yodoi and H. Hiai, FEBS Lett., 1995, 358, 1–3 CrossRef CAS.
- K. Sugamura and J. F. Keaney, Jr., Free Radical Biol. Med., 2011, 51, 978–992 CrossRef CAS PubMed.
- K. J. Barnham, C. L. Masters and A. I. Bush, Nat. Rev. Drug Discovery, 2004, 3, 205–214 CrossRef CAS PubMed.
- R. Kumar, J. Han, H. J. Lim, W. X. Ren, J. Y. Lim, J. H. Kim and J. S. Kim, J. Am. Chem. Soc., 2014, 136, 17836–17843 CrossRef CAS PubMed.
- K. B. Daniel, J. L. M. Jourden, K. E. Negoescu and S. M. Cohen, J. Biol. Inorg. Chem., 2011, 16, 313–323 CrossRef CAS PubMed.
- J. L. M. Jourden and S. M. Cohen, Angew. Chem., Int. Ed., 2010, 49, 6795–6797 CrossRef PubMed.
- J. L. M. Jourden, K. B. Daniel and S. M. Cohen, Chem. Commun., 2011, 47, 7968–7970 RSC.
- Y. Y. Kuang, K. Baakrishnan, V. Gandhi and X. H. Peng, J. Am. Chem. Soc., 2011, 133, 19278–19281 CrossRef CAS PubMed.
- P. Marzenell, H. Hagen, L. Sellner, T. Zenz, R. Grinyte, V. Pavlov, S. Daum and A. Mokhir, J. Med. Chem., 2013, 56, 6935–6944 CrossRef CAS PubMed.
- M. A. Clond, B. S. Lee, J. J. Yu, M. B. Singer, T. Amano, A. W. Lamb, D. Drazin, B. Kateb, E. J. Ley and J. S. Yu, PLoS One, 2013, 8, e61819 CAS.
- P. K. Halen, P. R. Murumkar, R. Giridhar and M. R. Yadav, Mini-Rev. Med. Chem., 2009, 9, 124–139 CrossRef CAS.
- R. O. Davies, H. J. Gomez, J. D. Irvin and J. F. Walker, Br. J. Pharmacol., 1984, 18(Suppl 2), 215S–229S CrossRef PubMed.
- D. A. Evans, J. Bartroli and T. L. Shih, J. Am. Chem. Soc., 1981, 103, 2127–2129 CrossRef CAS.
- D. A. Evans, T. C. Britton and J. A. Ellman, Tetrahedron Lett., 1987, 28, 6141–6144 CrossRef CAS.
- M. T. Crimmins and K. Chaudhary, Org. Lett., 2000, 2, 775–777 CrossRef CAS.
- M. T. Crimmins, B. W. King and E. A. Tabet, J. Am. Chem. Soc., 1997, 119, 7883–7884 CrossRef CAS.
- X. B. Deng, N. Chen, Z. X. Wang, X. Y. Li, H. Y. Hu and J. X. Xu, Phosphorus, Sulfur Silicon Relat. Elem., 2011, 186, 1563–1571 CrossRef CAS.
- G. Jalce, X. Franck and B. Figadere, Eur. J. Org. Chem., 2009, 378–386 CrossRef CAS.
- B. P. Mundy and Y. S. Kim, J. Heterocycl. Chem., 1982, 19, 1221–1222 CrossRef CAS.
- M. Sakamoto, S. Watanabe, T. Fujita, H. Aoyama and Y. Omote, J. Chem. Soc., Perkin Trans. 1, 1991, 2541–2545 RSC.
- I. Shibata, A. Baba and H. Matsuda, J. Chem. Soc., Chem. Commun., 1986, 1703–1704 RSC.
- T. Suzuki, Y. Hamashima and M. Sodeoka, Angew. Chem., Int. Ed., 2007, 46, 5435–5439 CrossRef CAS PubMed.
- Y. Tamura, F. Watanabe, T. Nakatani, K. Yasui, M. Fuji, T. Komurasaki, H. Tsuzuki, R. Maekawa, T. Yoshioka, K. Kawada, K. Sugita and M. Ohtani, J. Med. Chem., 1998, 41, 640–649 CrossRef CAS PubMed.
- C. G. Knight, F. Willenbrock and G. Murphy, FEBS Lett., 1992, 296, 263–266 CrossRef CAS.
- C. Perez, K. B. Daniel and S. M. Cohen, ChemMedChem, 2013, 8, 1662–1667 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, NMR and MS characterization, analytical HPLC traces, enzyme inhibition assays, crystallographic data. CCDC 1038268 and 1038269. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc09921d |
| This journal is © The Royal Society of Chemistry 2015 |