
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA concise route to the highly-functionalized azetidine precursor: the enantioselective synthesis of penaresidin B†
Feiqing
Ding‡
,
Ronny
William‡
,
Si Min
Kock
,
Min Li
Leow
and
Xue-Wei
Liu
*
Division of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, Singapore 637371. E-mail: xuewei@ntu.edu.sg
First published on 18th February 2015
Abstract
An efficient and high-yielding synthesis of penaresidin B is disclosed herein. The concise 8-step synthesis of azetidine aldehyde was devised by incorporating our novel strategy for ready access to 3-amino-2,3-dideoxysugars via regio- and stereoselective tandem hydroamination/glycosylation of glycal as the key step.
Azetidines constitute an important class of azaheterocycles that have been widely used in drug design as well as in the synthesis of natural products and pharmacologically active compounds.1,2 In addition, they also serve as versatile building blocks for other types of nitrogen-containing compounds with potential biological properties.3 However, azetidine alkaloids structurally related to phytosphingosines are rare, and only three biologically active sphingosine-like compounds derived from marine organisms have been isolated to date (Fig. 1). Penaresidin A (1) and penaresidin B (2), which exhibited potent actomyosin ATPase-activating activity when tested as an inseparable mixture, were isolated from an Okinawan marine sponge Penares sp. by Kobayashi et al.4 The related compound penazetidine A (3), which showed specific rat brain protein kinase C inhibitory activity, was isolated from the Pacific sponge Penaressollasi by Crews and co-workers.5
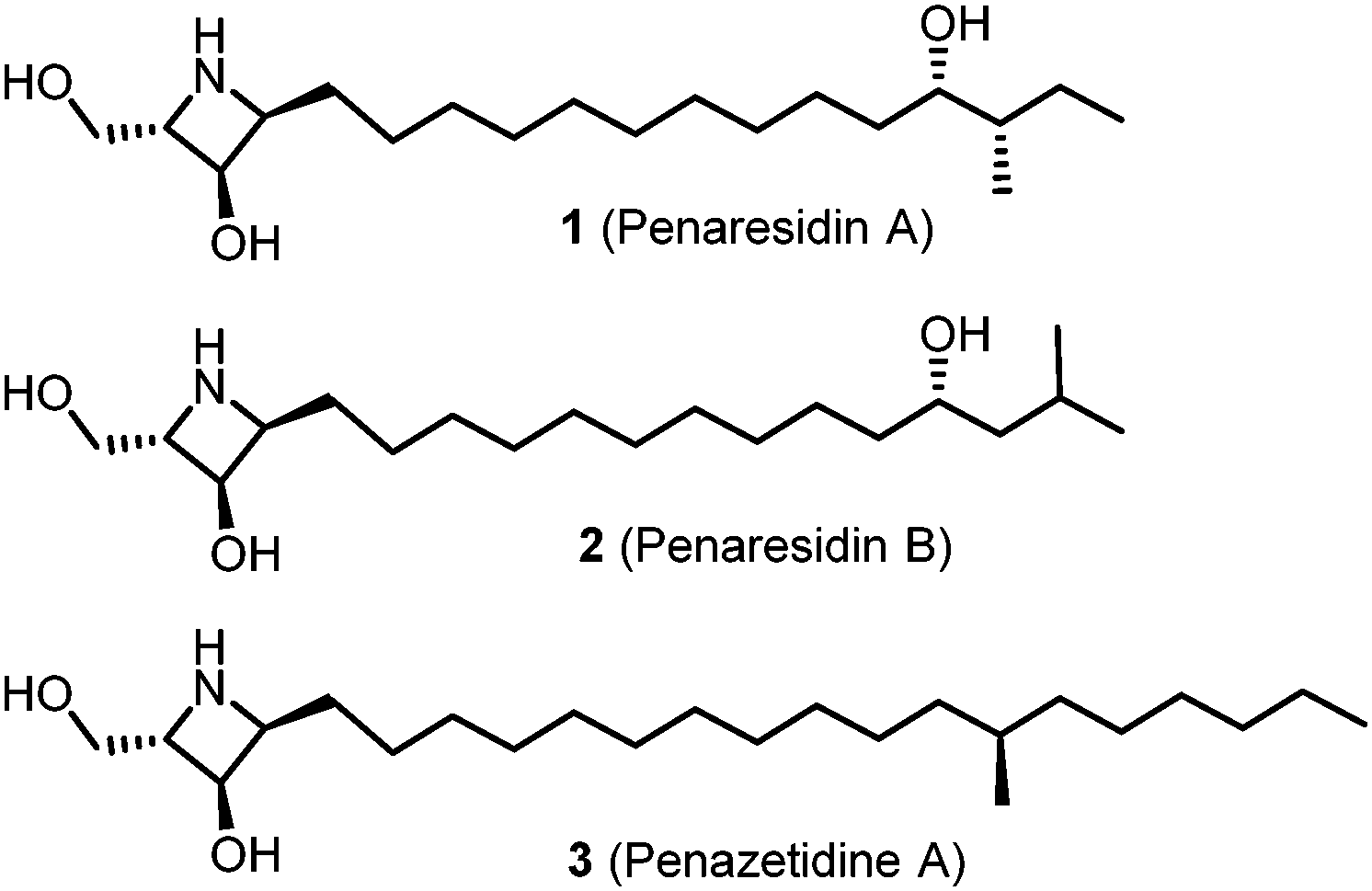 | ||
| Fig. 1 Structures of azetidine alkaloids. | ||
Interestingly, despite their rare occurrence in nature, azetidine alkaloids have captured tremendous interest from many synthetic chemists due to their significant biological activity and unique structure. To date, several syntheses of such alkaloid compounds have been reported.6 The current synthetic strategies rely on the construction of a highly functionalized azetidine skeleton with the requisite stereogenic centers. For example, Lin and co-workers reported the enantioselective synthesis of penaresidin A via a 14-step construction of a highly functionalized azetidine aldehyde 4 starting from divinylcarbinol, using Sharpless asymmetric epoxidation and Sharpless asymmetric hydroxylation reactions as the key steps (Scheme 1, eqn (1)).6i,j In addition, Raghavan and co-workers recently developed an alternative approach which comprises an 18-step construction of the azetidine subunit 5 of penaresidin A through stereoselective addition of the lithio anion of (R)-methyl p-tolylsulfoxide to an unsaturated sulfinylimine (Scheme 1, eqn (2)).6p Another multistep strategy that consists of a 17-step construction of the azetidine subunit 6 of penaresidin A, which started from D-galactal and involved Sharpless asymmetric epoxidation, regioselective ring-opening of epoxide and azetidine formation via the SN2 reaction, was most recently accomplished by Reddy's group (Scheme 1, eqn (3)).6q Although results from most reports are encouraging, poor stereoselectivities, low yields and lengthy synthetic steps present major impediments for the reported strategies to be widely applied. Hence, a short and straight forward synthetic route to azetidine with contiguous stereogenic centers in the enantiomerically pure form has remained a great and significant challenge for the chemical community.
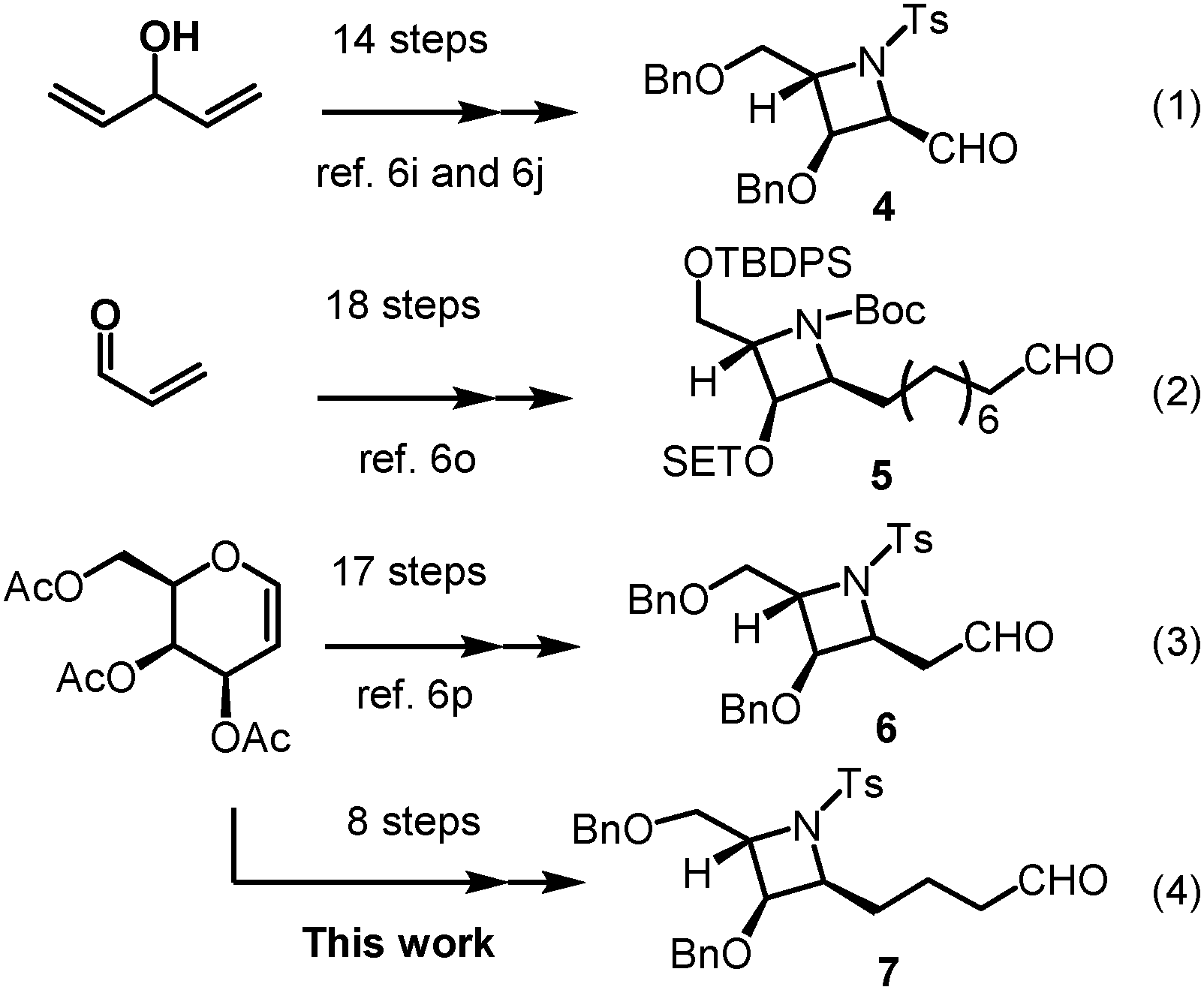 | ||
| Scheme 1 Strategies for construction of azetidine core in synthesis of azetidine alkaloids. | ||
With an aim to refine the synthetic route, we formulated a novel strategy which incorporates our reported method for ready access to 3-amino-2,3-dideoxysugars via the regio- and stereoselective tandem hydroamination/glycosylation of glycal as shown in Fig. 2.7 By extension of the synthetic utility of this protocol and its application in the total synthesis of natural products, we aim to demonstrate the efficient synthesis of azetidine aldehyde from 3,4,6-tri-O-acetyl-D-galactal via a linear sequence consisting of only 8 steps. Penaresidin B is envisioned as the ideal target molecule for this demonstration (Scheme 1, eqn (4)). Towards the end, the manner by which the above mentioned general strategy could be exploited to accomplish the shortest synthesis of azetidine alkaloids using commercially available and inexpensive compounds will be discussed.
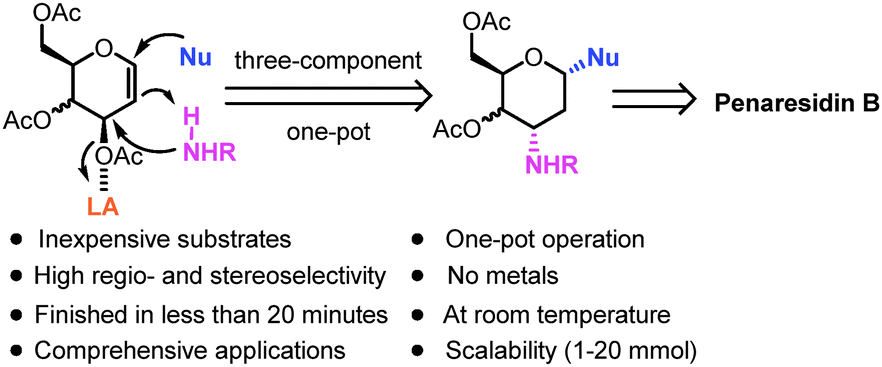 | ||
| Fig. 2 Our strategy for synthesis of penaresidin B via 3-amino-2,3-dideoxysugars. | ||
As azetidine alkaloids share a similar core structure and differ mainly along their long alkyl chains, we developed a strategy that focused on increasing structural and library diversity in a more efficient manner and hence paved the way for potential azetidine analogues to be synthesized for biological studies. Our retrosynthetic analysis of penaresidin B is depicted in Scheme 2. We proposed that our target molecule can be obtained from intermediate 8 which in turn could be formed via Julia–Kocienski olefination between azetidine aldehyde 7 and sulfone 9. The sulfone moiety 9 could be prepared through the Wittig reaction of 12 with aldehyde 13, while the azetidine core 7, which can also be regarded as an advanced intermediate for the synthesis of other azetidine alkaloids, can presumably be constructed from ester 10 using intramolecular Mitsunobu cyclization. Ester 10 could in turn be derived from 3-amino-2,3-dideoxygalactoside 11via the Wittig reaction following removal of the benzyl group. Finally, the 3-amino-2,3-dideoxygalactoside 11 could be accessed via regio- and stereoselective tandem hydroamination/glycosylation of D-galactal in a one-pot manner.
 | ||
| Scheme 2 Retrosynthetic analysis for a concise synthesis of penaresidin B. | ||
The proposed synthesis of the key fragment 7 starting from D-galactal is detailed in Scheme 3. In the initial step, a mixture of 3,4,6-tri-O-acetyl-D-galactal, benzyl alcohol and p-toluenesulfonamide in DCE was subjected to treatment with 2.2 equiv. of BF3·OEt2 at room temperature under a nitrogen atmosphere for 20 min. This led to the formation of benzyl 3-p-toluenesulfonamido-4,6-di-O-acetyl-2,3-dideoxy-α-D-galactopyranoside 11 in 78% yield with exclusive stereoselectivity. The exclusive formation of pure diastereomer allowed easy purification of the desired product using SiO2 flash column chromatography. The determination of the chemical structure and stereochemical characterization of 11 were achieved by extensive and detailed 1D and 2D NMR studies.8 The described protocol resulted in an efficient and stereoselective formation of 3-amino-2,3-dideoxygalactoside 11 with great reproducibility on a gram scale synthesis albeit in slightly lower yields. Sequentially, 11 was treated with Pd(OH)2/C under a hydrogen atmosphere to allow the selective deprotection of the benzyl group and this was followed by Wittig olefination of the resulting aldehyde 14 to form the unsaturated ester 15. Notably, 2-deoxy-β-C-glycoside was obtained by intramolecular cyclization when the reaction time was prolonged. Successive treatment of 15 with 10% Pd/C under a hydrogen atmosphere resulted in the reduction of the C–C double bond to form ester 10 with overall 43% yield in three steps. The subsequent intramolecular Mitsunobu reaction in the presence of PPh3/DIAD, which resulted in the conversion of ester 10 to the azetidine core 16, proceeded smoothly with a 75% yield.9 This step represents the key step in the present synthetic route as the core structure azetidine is assembled along with the requisite contiguous stereogenic centers. Removal of all the acetyl groups present in 16 followed by protection with benzyl groups resulted in the formation of compound 18, a benzyl protected azetidine ester, with a yield of 96%. The determination of the chemical structure and stereochemical characterization of 16 and 18 were established based on extensive and detailed 1D and 2D NMR studies, which showed a strong NOE correlation between the protons of C-3 and C-4; and between the protons of C-3 or C-4 and the protons of C-5; and between the proton of C-2 and the protons of C-6, and no correlation for H-2/H-4 (Fig. 3).8 Compound 18 was then subjected to chemoselective reduction with DIBAL-H to afford azetidine aldehyde 7 in a quantitative yield and was used directly in Julia–Kocienski olefination. Azetidine aldehyde 7 is of great significance in the natural product synthesis, as it comprises the core structure of several azetidine alkaloids.
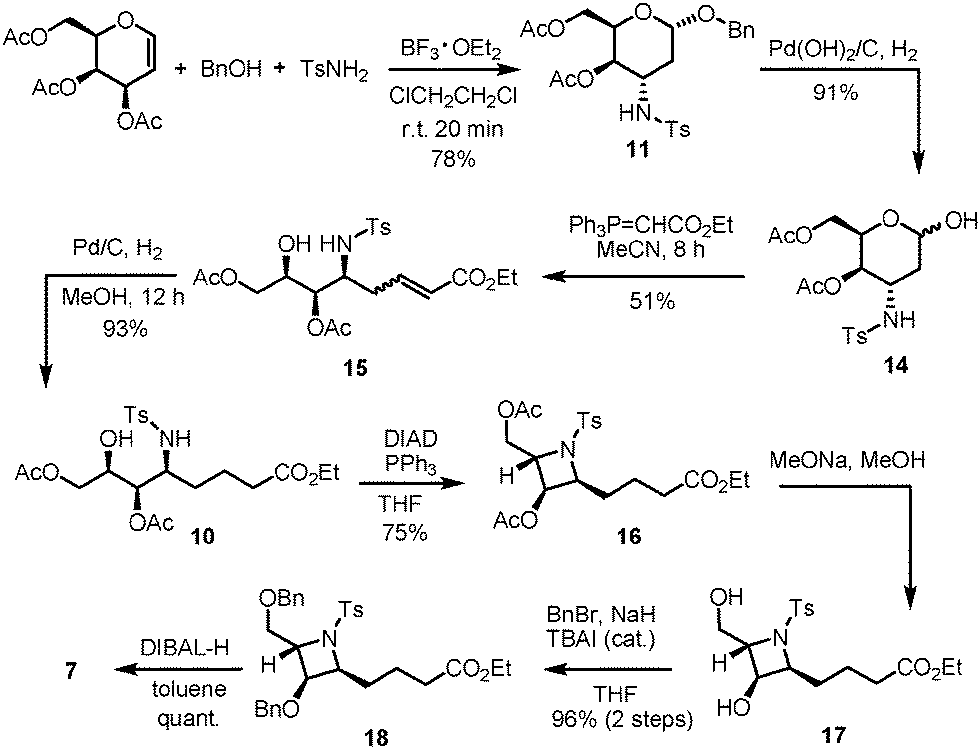 | ||
| Scheme 3 8-Step construction of the core azetidine aldehyde 7. | ||
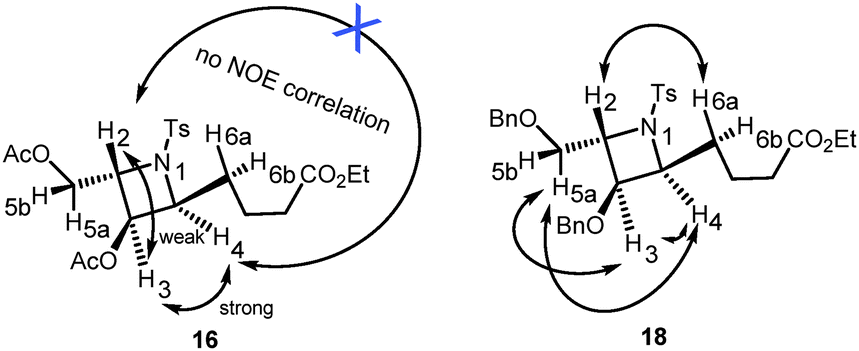 | ||
| Fig. 3 NOE correlations of compounds 16 and 18. | ||
Next, we shift our focus onto the preparation of the desired sulfone 9. Wittig olefination of two known fragments, stable ylide 12 (prepared from pentane-1,5-diol in 3 steps)10 and aldehyde 13 (prepared from L-leucine in 4 steps),11 in the presence of n-BuLi, was first carried out to afford the unsaturated ether 19 in 86% yield (Scheme 4). Treatment of 19 with Pd(OH)2/C under a hydrogen atmosphere resulted in the reduction of the C–C double bond and removal of the benzyl group, which produced a 90% yield of alcohol 20 in one pot. Subsequently, alcohol 20 was converted into thioether 22 using 1-phenyl-1H-tetrazole-5-thiol 21 in the presence of PPh3/DIAD. This was followed by oxidation of thioether 22 with a molybdenum salt and hydrogen peroxide to give sulfone 9 with a 73% yield.12 The above mentioned protocol produced an enantiopure sulfone with the required S-configuration efficiently and stereoselectively. Additionally, it allows great reproducibility on various scales.
 | ||
| Scheme 4 Preparation of sulfone 9 required for Julia–Kocienski olefination. | ||
With the azetidine aldehyde 7 and sulfone fragment 9 successfully prepared, we embarked on the construction of the C–C double bond via coupling of these two fragments. Assembly of the azetidine core structure with the sulfone chain constitutes the key step in this synthetic route. This was accomplished via Julia–Kocienski olefination, using KHMDS as a base at −78 °C to give olefin 8 in 81% yield.13 Sequential desilylation of 8 with p-TSA in methanol furnished alcohol 22 in 90% yield (Scheme 5) which was later converted to Ts-protected penaresidin B (23) in 49% yield upon treatment with 10% Pd/C under a hydrogen atmosphere. In fact, the removal of the two Bn protecting groups at this point proved rather challenging, as most methods attempted either resulted in no reaction or caused incomplete decomposition of the starting material. To our delight, after numerous attempts, facile deprotection of the two benzyl groups could be achieved with 10% Pd/C under a hydrogen atmosphere to afford the desired products, with moderate yield. Ts-protected penaresidin B (23) could be deprotected to eventually give the penaresidin B in one step.6h,j,o,q In summary, starting from the commercially available D-galactal, we have accomplished the formal synthesis of penaresidin B in a linear sequence of 11 steps with an overall yield of 13%.
 | ||
| Scheme 5 Formal synthesis of penaresidin B (2). | ||
Overall, we have developed a short, high-yielding synthetic approach for the formation of penaresidin B. The eight-step synthesis of azetidine aldehyde with contiguous stereogenic centers entailed our novel strategy for ready access to 3-amino-2,3-dideoxysugar via regio- and stereoselective tandem hydroamination/glycosylation of glycal as the key step. One-pot transformations have also been applied to decrease the number of isolated steps and increase the efficiency of the synthesis while the Julia–Kocienski olefination was employed to couple the azetidine and side chain subunit. As three-component reactions have been proven to be useful for large scale syntheses and are applicable to various azetidines, scalable synthesis and efficient preparations of penaresidin A, penazetidine A and other azetidine alkaloids bearing different side chains can be anticipated. The potential success of this methodology in the creation of a library of azetidine alkaloid analogues will set the foundation for in-depth studies of structure–activity relationships.
We gratefully acknowledge Nanyang Technological University (RG6/13) and the Ministry of Education, Singapore (MOE 2009-T2-1-030) for the financial support of this research.
Notes and references
- (a) T. Akihisa, S. Mafune, M. Ukiya, Y. Kimura, K. Yasukawa, T. Suzuki, H. Tokuda, N. Tanabe and T. Fukuoka, J. Nat. Prod., 2004, 67, 479–480 CrossRef CAS PubMed; (b) H. O'Dowd, J. G. Lewis, J. Trias, R. Asano, J. Blais, S. L. Lopez, G. K. Park, C. Wu, W. Wang and M. F. Gordeev, Bioorg. Med. Chem. Lett., 2008, 18, 2645–2648 CrossRef PubMed; (c) M. Bouazaoui, J. Martinez and F. Cavelier, Eur. J. Org. Chem., 2009, 2729–2732 CrossRef CAS; (d) S. Malik and U. K. Nadir, Synlett, 2008, 108–110 CrossRef CAS PubMed; (e) T. A. Brandt, S. Caron, D. B. Damon, J. Dibrino, A. Ghosh, D. A. Griffith, S. Kedia, J. A. Ragan, P. R. Rose, B. C. Vanderplas and L. Wei, Tetrahedron, 2009, 65, 3292–3304 CrossRef CAS PubMed; (f) J. A. Burkhard, C. Guérot, H. Knust, M. Rogers-Evans and E. M. Carreria, Org. Lett., 2010, 12, 1944–1947 CrossRef CAS PubMed; (g) J. A. Burkhard, B. Wagner, H. Fisher, F. Schuler, K. Müller and E. M. Carreira, Angew. Chem., Int. Ed., 2010, 49, 3524–3527 CrossRef CAS PubMed.
- For general reviews on the synthesis of azetidines, see: (a) Y. Dejaegher, N. M. Kuz'menok, A. M. Zvonok and N. De Kimpe, Chem. Rev., 2002, 102, 29 CrossRef CAS PubMed; (b) F. Couty, G. Evano and D. Prim, Mini-Rev. Org. Chem., 2004, 1, 133 CrossRef CAS; (c) F. Couty and G. Evano, Org. Prep. Proced. Int., 2006, 38, 427 CrossRef CAS; (d) A. Brandi, S. Cicchi and F. M. Cordero, Chem. Rev., 2008, 108, 3988 CrossRef CAS PubMed; (e) F. Couty, in Science of Synthesis, ed. D. Enders, Thieme, Stuttgart, 2009, vol. 40a, pp. 773–817 Search PubMed.
- For more recent reviews, see: F. Couty and G. Evano, Synlett, 2009, 3053–3064 CrossRef CAS PubMed.
- (a) J. Kobayashi, J. Cheng, M. Ishibashi, M. R. Walchli, S. Yamamura and Y. Ohizumi, J. Chem. Soc., Perkin Trans. 1, 1991, 1135–1137 RSC; (b) J. Kobayashi, M. Tsuda, J.-F. Chen, M. Ishibashi, H. Takikawa and K. Mori, Tetrahedron Lett., 1996, 37, 6775–6776 CrossRef CAS.
- K. A. Alvi, M. Jaspars and P. Crews, Bioorg. Med. Chem. Lett., 1994, 4, 2447 CrossRef CAS.
- For previous synthesis of azetidine alkaloids and analogues, see: (a) T. Hiraki, Y. Yamagiwa and T. Kamikawa, Tetrahedron Lett., 1995, 36, 4841–4844 CrossRef CAS; (b) H. Takikawa, T. Maeda and K. Mori, Tetrahedron Lett., 1995, 36, 7689–7692 CrossRef CAS; (c) H. Yoda, T. Oguchi and K. Takabe, Tetrahedron: Asymmetry, 1996, 7, 2113–2116 CrossRef CAS; (d) A. Yashima, H. Takikawa and K. Mori, Liebigs Ann., 1996, 7, 1083–1089 Search PubMed; (e) K. Mori, J. Heterocycl. Chem., 1996, 33, 1497–1517 CrossRef CAS; (f) H. Yoda, T. Oguchi and K. Takabe, Tetrahedron Lett., 1997, 38, 3283–3284 CrossRef CAS; (g) S. Knapp and Y. Dong, Tetrahedron Lett., 1997, 38, 3813–3816 CrossRef CAS; (h) H. Takikawa, T. Maeda and M. Seki, J. Chem. Soc., Perkin Trans. 1, 1997, 97–111 RSC; (i) G. Q. Lin and D. G. Liu, Heterocycles, 1998, 47, 337–348 CrossRef CAS PubMed; (j) D. G. Liu and G. Q. Lin, Tetrahedron Lett., 1999, 40, 337–340 CrossRef CAS; (k) A. Salgado, M. Boeykens, C. Gauthier, J. Declercq and N. De Kimpe, Tetrahedron, 2002, 58, 2763–2775 CrossRef CAS; (l) H. Yoda, T. Uemura and K. Takabe, Tetrahedron Lett., 2003, 44, 977–979 CrossRef CAS; (m) A. Salgado, M. Boeykens, C. Gauthier, Y. Dejaegher, G. Verniest, C. Lopin, K. A. Tehrani and N. De Kimpe, Tetrahedron, 2003, 59, 2231–2239 CrossRef CAS; (n) A. C. B. Burtoloso and C. R. D. Correia, Tetrahedron Lett., 2004, 45, 3355–3358 CrossRef CAS PubMed; (o) K. Ohshita, H. Ishiyama, Y. Takahashi, J. Ito, Y. Mikami and J. Kobayashi, Bioorg. Med. Chem., 2007, 15, 4910–4916 CrossRef CAS PubMed; (p) S. Raghavan and V. Krishnaiah, J. Org. Chem., 2010, 75, 748 CrossRef CAS PubMed; (q) B. V. S. Reddy, C. Kishore and A. S. Reddy, Tetrahedron Lett., 2014, 55, 49–51 CrossRef CAS PubMed.
- (a) F. Q. Ding, R. William, F. Wang, J. Ma, L. Ji and X.-W. Liu, Org. Lett., 2011, 13, 652 CrossRef CAS PubMed; (b) F. Q. Ding, R. William, S. M. Wang, B. K. Gorityala and X.-W. Liu, Org. Biomol. Chem., 2011, 9, 3929 RSC; (c) F. Q. Ding, R. William, S. T. Cai, J. Ma and X.-W. Liu, J. Org. Chem., 2012, 77, 5245 CrossRef CAS PubMed; (d) F. Q. Ding, R. William and X.-W. Liu, J. Org. Chem., 2013, 78, 1293–1299 CrossRef CAS PubMed; (e) F. Q. Ding, L. Ji, R. William, H. Chai and X.-W. Liu, Beilstein J. Org. Chem., 2014, 10, 1325–1332 CrossRef PubMed; (f) F. Q. Ding, R. William, B. K. Gorityala, J. Ma, S. Wang and X.-W. Liu, Tetrahedron Lett., 2010, 51, 3146–3148 CrossRef CAS PubMed; (g) F. Q. Ding, R. William, M. L. Leow, H. Chai, J. Z. M. Fong and X.-W. Liu, Org. Lett., 2014, 16, 26–29 CrossRef CAS PubMed; (h) F. Q. Ding, M. L. Leow, R. William, J. Ma, H. Liao and X.-W. Liu, Chem. – Asian J., 2014, 9, 2548–2554 CrossRef CAS PubMed . For recent review, see: ; (i) F. Q. Ding, S. Cai, R. William and X.-W. Liu, RSC Adv., 2013, 3, 13594–13621 RSC.
- See ESI† for the details of extensive NMR experiments.
- For more recent reviews, see: (a) T. Y. S. But and P. H. Toy, Chem. – Asian J., 2007, 2, 1340–1355 CrossRef CAS PubMed; (b) K. C. Swamy, N. N. Kumar, E. Balaraman and K. V. Kumar, Chem. Rev., 2009, 109, 2551–2651 CrossRef CAS PubMed . Examples of construction of azetidine by Mitsunobu reaction see: M. Bouazaoui, J. Martinez and F. Cavelier, Eur. J. Org. Chem., 2009, 2729–2732 CrossRef CAS and references cited therein.
- M. V. Sargent and S. Wangchareontrakul, J. Chem. Soc., Perkin Trans. 1, 1990, 129–132 RSC.
- G. Simon, T. Eljezi, B. Legeret, F. Charmantray, J. A. Castillo, C. Guerard-Helaine, M. Lemaire, M. Bouzon, P. Marliere, V. Helaine and L. Hecquet, ChemCatChem, 2013, 5, 784–795 CrossRef CAS.
- J. Pospíšil, Tetrahedron Lett., 2011, 52, 2348–2352 CrossRef PubMed.
- For more recent reviews, see: (a) P. R. Blakemore, J. Chem. Soc., Perkin Trans. 1, 2002, 2563–2585 RSC; (b) K. Plesniak, A. Zarecki and J. Wicha, Top. Curr. Chem., 2007, 275, 163–250 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cc09904d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |