
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of isoindolinones via a ruthenium-catalyzed cyclization of N-substituted benzamides with allylic alcohols†
Ramasamy
Manoharan
and
Masilamani
Jeganmohan
*
Department of Chemistry, Indian Institute of Science Education and Research, Pune 411021, India. E-mail: mjeganmohan@iiserpune.ac.in
First published on 7th January 2015
Abstract
N-Substituted aromatic and heteroaromatic amides reacted with substituted allylic alcohols in the presence of a ruthenium catalyst, AgSbF6 and a Cu(OAc)2·H2O oxidant, affording 3-substituted isoindolinone derivatives with diverse substituents in good to excellent yields. A possible reaction mechanism involving a five-membered ruthenacycle intermediate was proposed and strongly supported by experimental evidence.
The isoindolinone core unit is present in various natural products, biologically active molecules and pharmaceuticals (Fig. 1).1 It serves as a key synthetic intermediate for synthesizing various highly useful organic molecules and natural products.2 Particularly, the 3-substituted isoindolinone skeleton is found in various biologically active molecules.3 As a result, various synthetic methods are available in the literature to synthesize 3-substituted isoindolinone derivatives.4–7 Generally, 3-substituted isoindolinones are prepared by nucleophilic addition of metal reagents into isoindoline-1,3-diones,4a the cyclization of ortho-substituted aryllithiums with imines,4b,c or strong base-induced metalation followed by functionalization at the 3-position of isoindolinones.4d Additionally, 3-substituted isoindolinones can be prepared by metal-catalyzed cyclization of ortho-halo substituted aromatics with imines5a and tandem cyclization of ortho-halo substituted aromatics with CO and amines.5b
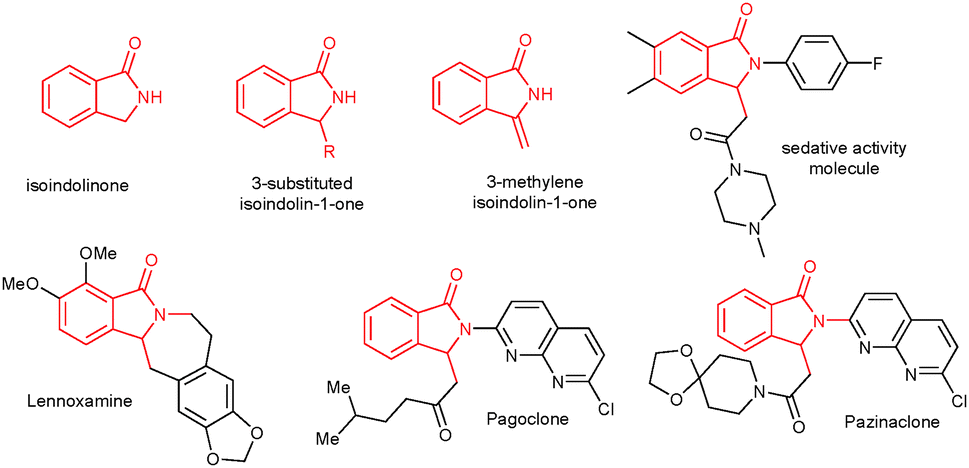 | ||
| Fig. 1 Isoindolinone core biologically active molecules. | ||
Recently, 3-substituted isoindolinones were efficiently prepared by using metal catalysts via C–H bond activation in a highly atom economical and environmentally friendly manner.6–8 Aromatic imines underwent cyclization with isocyanates in the presence of a rhenium catalyst, providing 3-substituted isoindolinones.8aN-Substituted benzamides reacted with alkenes in the presence of metal catalysts, giving isoindolinones in good to excellent yields.8b–g In the reaction, mostly activated alkenes such as acrylates, ethyl vinyl ketone, acrylamide and conjugated 1,2-diketones were used.8
Due to the vast availability, easy accessibility and simple preparation of allylic alcohols, they have been widely used as alkene partners in the coupling reaction with aromatic electrophiles or organometallic reagents in the presence of metal catalysts.9 It is important to note that in most of the catalytic reactions, allylic alcohols are chemically equivalent to α,β-unsaturated enones and aldehydes. Recently, allylic alcohols have also been efficiently used as coupling partners in the reaction with heteroatom substituted aromatics, and this transformation leads to ortho alkylated aromatics in the presence of metal catalysts via C–H bond activation.10 Herein, we report a ruthenium-catalyzed cyclization of N-substituted benzamides with allylic alcohols to give 3-substituted isoindolinone derivatives in good yields. A possible reaction mechanism involving a five-membered ruthenacycle intermediate was proposed and strongly supported by experimental evidence.
Treatment of n-benzyl 4-methoxy benzamide (1a) with 3-buten-2-ol (2a) (2.2 equiv.) in the presence of [{RuCl2(p-cymene)}2] (5.0 mol%), AgSbF6 (20 mol%) and Cu(OAc)2·H2O (2.2 equiv.) in 1,2-dichloroethane at 110 °C for 16 h gave 3-substituted isoindolinone derivative 3aa in 72% isolated yield (Scheme 1). Initially, the cyclization reaction was examined with various solvents such as MeOH, iso-PrOH, THF, DMF, 1,2-dimethoxyethane and toluene under similar reaction conditions. Among them, ClCH2CH2Cl was very effective, giving 3aa in 79% GC yield. THF, 1,4-dioxane and 1,2-dimethoxyethane were partially effective, affording product 3aa in 34%, 45% and 48% GC yields, whereas remaining solvents were totally ineffective.
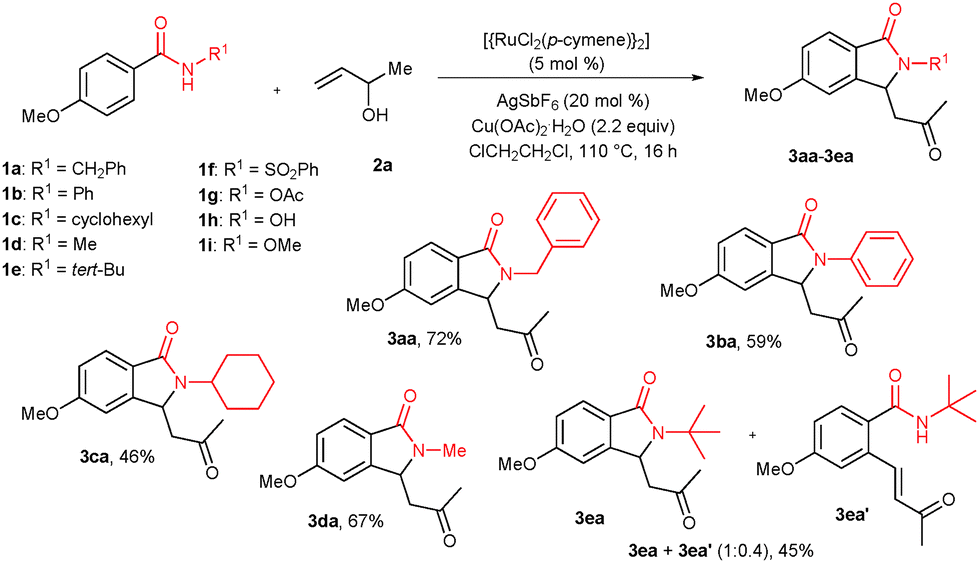 | ||
| Scheme 1 Cyclization of N-substituted benzamides with 2a. | ||
The reaction was also tested with additives such as AgSbF6, AgBF4, AgOTf and KPF6. Among them, AgSbF6 was very effective, giving product 3aa in 79% GC yield. AgBF4 and AgOTf were partially effective, yielding 3aa in 47% and 27% GC yields, respectively. KPF6 was not suitable for the reaction. The cyclization reaction was also tested with various acetate and oxidant sources such as AgOAc, CsOAc, KOAc, NaOAc, Ag2O and Cu(OAc)2·H2O. Among them, Cu(OAc)2·H2O was very effective, providing 3aa in 79% GC yield. Remaining acetate sources were not effective. The reaction was also tested with less than 50 mol% of Cu(OAc)2·H2O under an air atmosphere. However, in the reaction, product 3aa was observed only in 38% GC yield. The reaction was tested with other catalysts (5 mol%) such as Ru(COD)Cl2, Ru(PPh3)3Cl2 and RuCl3·H2O apart from [{RuCl2(p-cymene)}2]. However, no cyclization product 3aa was observed in these complexes. The amount of the [{RuCl2(p-cymene)}2] catalyst (2 mol%) and (10 mol%) was also examined. Using 2 mol% and 10 mol% of the catalyst, product 3aa was observed in 32% and 80% GC yields, respectively. Thus, 5 mol% of the catalyst amount was sufficient for the reaction. The amount of reactant 2a (1.2 equiv. and 3.0 equiv. apart from 2.2 equiv.) was also tested. In 1.2 equiv. of 2a, product 3aa was observed in 55% GC yield and in 3.0 equiv. of 2a, product 3aa was observed in 79% GC yield. The cyclization reaction was also tested at 60 °C and 80 °C apart from 110 °C. At 60 °C, no product 3aa was observed and at 80 °C product 3aa was observed in 35% GC yield. Control experiments showed that in the absence of AgSbF6 or [{RuCl2(p-cymene)}2] or Cu(OAc)2·H2O, no 3aa was obtained.
Under the optimized reaction conditions, the cyclization of other N-substituted benzamides 1b–i with 2a was tested (Scheme 1). N-Phenyl 1b and cyclohexyl 1c substituted benzamides reacted with 2a, providing cyclization products 3ba and 3ca in 59% and 46% yields, respectively. N-Methyl substituted benzamide 1d gave isoindolinone derivative 3da in 67% yield. But, N-tert butyl benzamide 1e provided a mixture of cyclic product 3ea and ortho alkenylated product 3ea′ in 45% combined yield and in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.4 ratio. In other N-substituted benzamides 1f–i, the expected cyclization product was not observed.
:0.4 ratio. In other N-substituted benzamides 1f–i, the expected cyclization product was not observed.
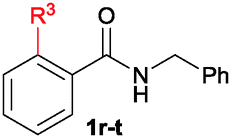
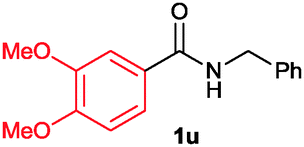
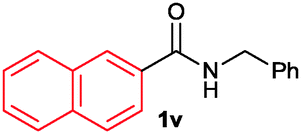
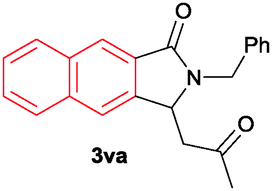
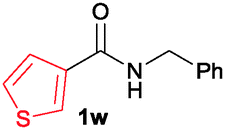
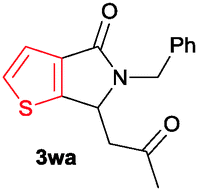
The scope of the cyclization reaction was examined using N-benzyl substituted benzamides 1j–v (Table 1). Benzamides 1j and 1k reacted efficiently with 2a, providing the cyclization products 3ja and 3ka in 69% and 60% yields, respectively (entries 1 and 2). Halogen groups such as I, Br, Cl and F substituted benzamides 1l–o reacted efficiently with 2a, affording products 3la–3oa in good to moderate yields, respectively (entries 3–6). Interestingly, electron-withdrawing groups such as CF3 and NO2 substituted benzamides 1p and 1q reacted with 2a, giving cyclization products 3pa and 3qa in 54% and 46% yields, respectively (entries 7 and 8). Apart from the para substituted benzamides, ortho OMe, Me and Br substituted benzamides 1r–t also efficiently participated in the reaction, yielding products 3ra–ta in 80%, 65% and 62% yields, respectively (entries 9–11). Unsymmetrical 3,4-dimethoxy (1u) and 2-naphthyl (1v) substituted benzamides regioselectively reacted with 2a yielding products 3ua and 3va in 53% and 58% yields, respectively (entries 12 and 13). In substrates 1u and 1v, the C–H bond activation takes place at the C-6 position of the benzene ring and at the C-3 position of the naphthalene ring selectively. Interestingly, heteroaromatic amide 1w also efficiently participated in the reaction, affording product 3wa in 60% yield (entry 14).
| Entry | 1 | Product 3 | Yieldb (%) |
|---|---|---|---|
| a All reactions were carried out using 1j–w (100 mg), ethyl-2-buten-2-ol (2a) (2.2 equiv.), [{RuCl2(p-cymene)}2] (5 mol%), AgSbF6 (20 mol%) and Cu(OAc)2·H2O (2.2 equiv.) in ClCH2CH2Cl (3.0 mL) at 110 °C for 16 h. b Isolated yield. c The reaction was carried at 110 °C for 28 h. | |||
|
|
||
| 1 | 1j: R2 = Me | 3ja: R2 = Me | 69 |
| 2 | 1k: R2 = H | 3ka: R2 = H | 60 |
| 3 | 1l: R2 = I | 3la: R2 = I | 61 |
| 4 | 1m: R2 = Br | 3ma: R2 = Br | 59 |
| 5 | 1n: R2 = Cl | 3na: R2 = Cl | 58 |
| 6 | 1o: R2 = F | 3oa: R2 = F | 47 |
| 7 | 1p: R2 = CF3 | 3pa: R2 = CF3 | 54 |
| 8 | 1q: R2 = NO2 | 3qa: R2 = NO2 | 46 |
|

|
||
| 9 | 1r: R3 = OMe | 3ra: R3 = OMe | 80 |
| 10 | 1s: R3 = Me | 3sa: R3 = Me | 65 |
| 11 | 1t: R3 = Br | 3ta: R3 = Br | 62 |
| 12 |
|

|
53 |
| 13c |
|
|
58 |
| 14 |
|
|
60c |
The scope of the cyclization reaction was also further examined using substituted allylic alcohols (Scheme 2). Treatment of pent-1-en-3-ol (2b), hex-1-en-3-ol (2c) and hept-1-en-3-ol (2d) with benzamide 1a under similar reaction conditions gave cyclization products 3ab–ad in 67%, 65% and 62% yields, respectively. 1-Phenylbut-3-en-2-ol (2e) also nicely participated in the reaction, affording the corresponding cyclization product 3ae in 68% yield. Interestingly, prop-2-en-1-ol (2f) reacted efficiently with 1a, giving a formyl substituted cyclic compound 3af in 60% yield.
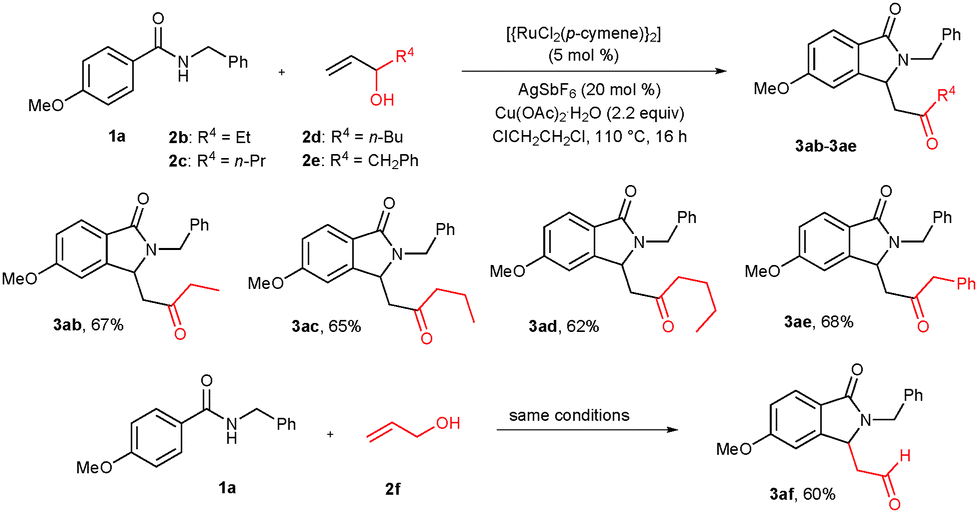 | ||
| Scheme 2 Scope of the substituted allylic alcohols. | ||
Based on the previous reports6–10 and our observation, a possible reaction mechanism is proposed in Scheme 3. Basically, a multi-step reaction is involved in the cyclization reaction. First, AgSbF6 likely removes the Cl− ligand from the [{RuCl2(p-cymene)}2] complex in the presence of Cu(OAc)2 providing a cationic ruthenium acetate species 4. Coordination of the nitrogen atom of 1 to the ruthenium species 4 followed by ortho-metalation provides ruthenacycle intermediate 5. Coordinative insertion of α,β-unsaturated enone 6 into the Ru–carbon bond of intermediate 5 gives intermediate 7. We strongly believe that the allylic alcohols 2 convert into α,β-unsaturated enones 6 in the presence of the ruthenium catalyst and Cu(OAc)2.11 β-Deprotonation of intermediate 7 by an acetate source followed by protonation of nitrogen affords ortho-alkenylated benzamide 8 and regenerates the ruthenium species 4 (proceeds viapath A).11c Later, coordination of the nitrogen atom of ortho-alkenylated benzamide 8 into ruthenium species 4 followed by intramolecular coordination of the double bond into ruthenium affords intermediate 9 and AcOH. Intramolecular coordinative insertion of the N–Ru bond of intermediate 9 into the alkene moiety followed by enolization provides ruthenium enolate intermediate 10. Protonation of intermediate 10 in the presence of AcOH provides product 3 and regenerates the active ruthenium species 4. The control of the formation of product 11 which proceeds via enolization of intermediate 7 followed by protonation is highly important to successfully carry out the present cyclization reaction (viapath B).10
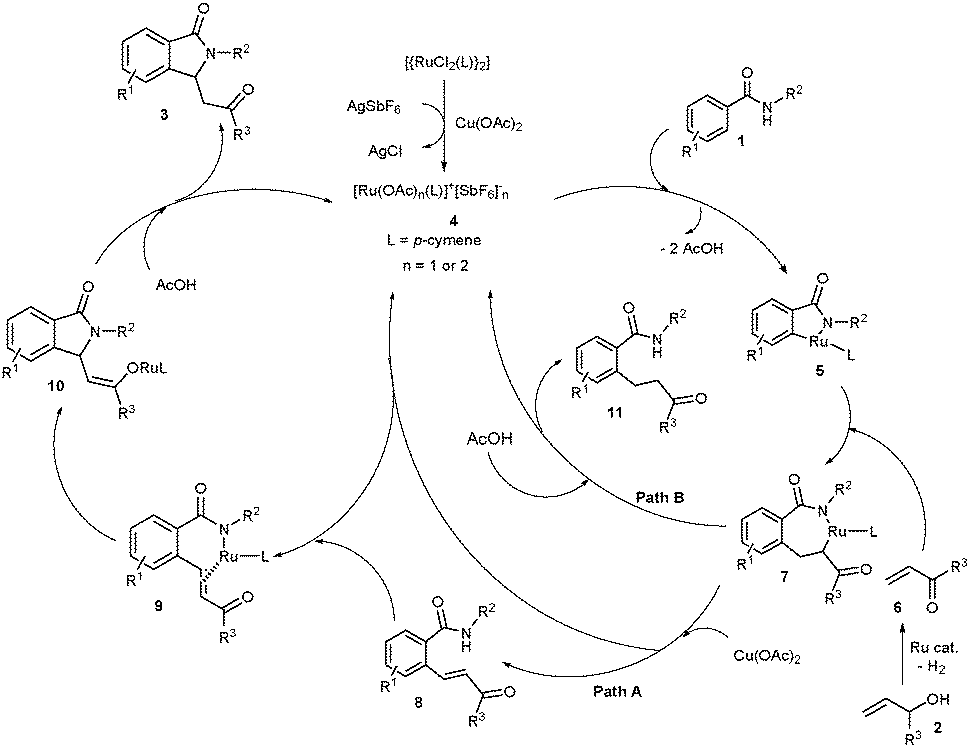 | ||
| Scheme 3 Proposed mechanism. | ||
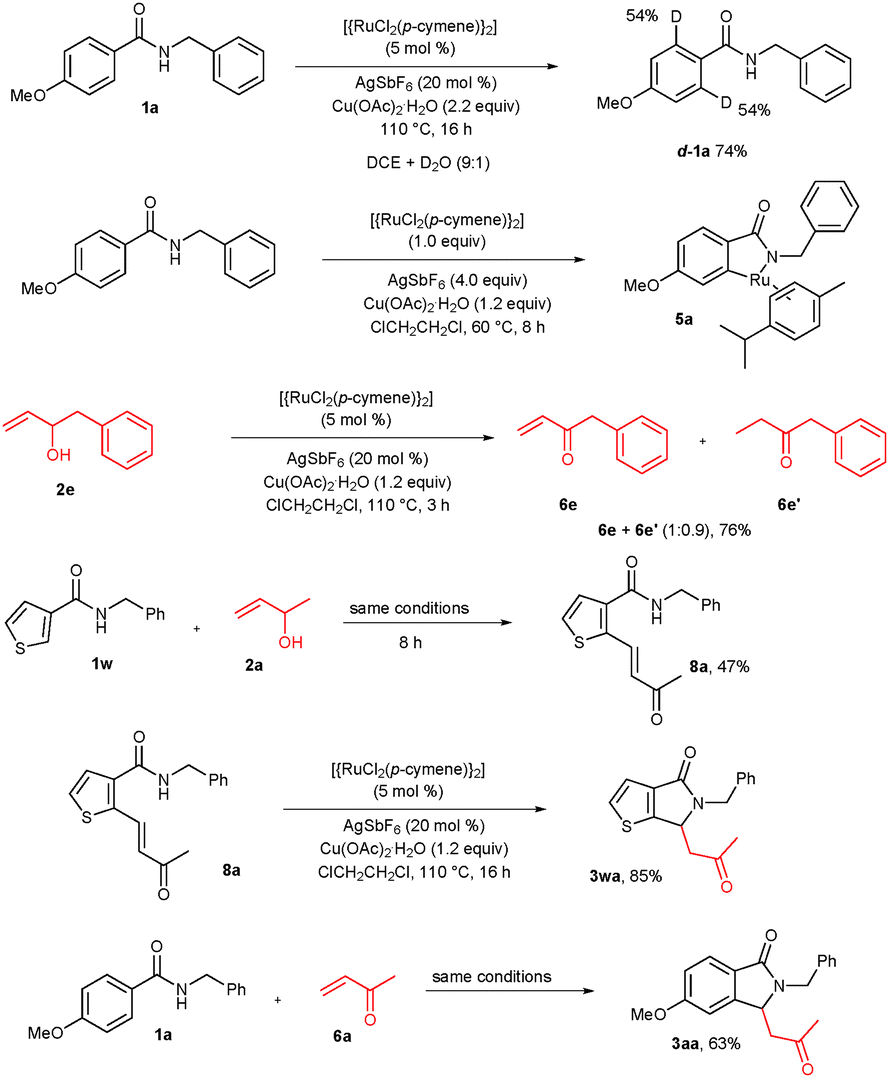
The formation of a key five-membered ruthenacycle intermediate 5 is the rate determining reversible step in the reaction. To support the reversible step, N-benzyl 4-methoxy benzamide (1a) was treated with a ruthenium catalyst, AgSbF6 and Cu(OAc)2·H2O, D2O in DCE solvent at 110 °C for 16 h. As expected, 54% deuterium incorporations were observed at both ortho carbons of benzamide d-1a in a combined 74% yield (Scheme 4). In the meantime, we have tried to isolate the key ruthenacycle intermediate 5 in the reaction of 4-methoxy benzamide 1a with a stoichiometric amount of the ruthenium complex (1.0 equiv.), AgSbF6 (4.0 equiv.) and Cu(OAc)2·H2O (1.2 equiv.) in DCE solvent at 60 °C for 8 h. In the reaction, metalacycle intermediate 5 was isolated. However, we were not able to crystallize intermediate 5. But, the complex 5 was tentatively assigned by 1H, 13C NMR, HRMS and MALDI-TOF spectroscopic techniques (see ESI†). To confirm the formation of activated alkene 6, 1-phenylbut-3-en-2-ol (2e) was treated with the ruthenium catalyst, AgSbF6 and Cu(OAc)2·H2O at 110 °C for 3 h. In the reaction, approximately a 1:1 mixture of 1-phenylbut-3-en-2-one (6e) and the reduced 1-phenylbutan-2-one (6e′) were observed in a combined 76% yield. It seems that in the cyclization reaction, initially product 6e is formed which further reacts with benzamide 1 providing the cyclization product 3. If benzamide is not present in the reaction mixture, the alkene moiety of 6e is subsequently reduced. Further, we have tried to isolate ortho alkenylated benzamide 8 in the reaction of 2-thienyl amide (1w) with 2a under the optimized reaction conditions at the shorter reaction time of 8 h. In the reaction, the expected alkenylated product 8a was observed in 47% yield. Later, ortho alkylated benzamide 8a was treated with the ruthenium catalyst, AgSbF6 and Cu(OAc)2·H2O at 110 °C for 16 h giving the expected cyclic compound 3wa in 85% yield. Further, benzamide 1a reacted with methyl vinyl ketone (6a) under the optimized reaction conditions providing the expected cyclic product 3aa in 63% yield. This experimental evidence clearly supports the proposed mechanism in Scheme 3.
 | ||
| Scheme 4 Mechanistic evidence. | ||
To successfully carry out the present cyclization reaction, suppression of the enolization of intermediate 7 into 11 is highly important. It is known that N,N-disubstituted benzamides react with allylic alcohols leading to ortho alkylated benzamides in the presence of rhodium or ruthenium complexes.10 But, in the present reaction, N-substituted benzamides reacted with allylic alcohols yielding isoindolinone derivatives 3. To know the clear mechanism, we have tried the reaction of N,N-diethyl benzamide 14 with 2a under the optimized reaction conditions (Scheme 5). In the reaction, ortho alkenylated benzamide 15 and ortho alkylated benzamide 16 were observed in combined 57% yields in a 1:1 ratio. But, in the presence of AcOH (3.0 equiv.) under similar reaction conditions, the same reaction provided a major amount of ortho alkylated benzamide 16 along with a minor amount of 15 in 69% yield in a 10:1 ratio. Similarly, the reaction of N-substituted benzamide 1a with 2a was tried in the presence of 3.0 equiv. of AcOH under the optimized reaction conditions. In the reaction, cyclization product 3aa and ortho alkylated benzamide 18 were observed in 21% and 69% yields, respectively. At this stage, we conclude that an excess amount of AcOH might increase the electrophilicity of the carbonyl group in intermediate 7via protonation. It is likely that intermediate 19 could be formed. Thus, instead of β-hydride elimination, enolization takes place effectively.10c,12
 | ||
| Scheme 5 Reaction of N,N-diethyl benzamide with 2a. | ||
In conclusion, we have demonstrated a ruthenium-catalyzed cyclization of N-substituted benzamides with allylic alcohols in the presence of a ruthenium catalyst. A possible reaction mechanism involving a five-membered ruthenacycle intermediate was proposed and strongly supported by experimental evidence.
We thank the DST (SR/S1/OC-26/2011), India for the support of this research. R. M. thanks the CSIR for a fellowship.
Notes and references
- Selected reviews: (a) B. Sener, B. Goezler, R. D. Minard and M. Shamma, Phytochemistry, 1983, 22, 2073 CrossRef CAS; (b) M. Efdi, S. Fujita, T. Inuzuka and M. Koketsu, Nat. Prod. Res., 2010, 24, 657 CrossRef CAS PubMed; (c) G. Blaskó, D. J. Gula and M. Shamma, J. Nat. Prod., 1982, 45, 105 CrossRef; (d) Y. C. Chia, F. R. Chang, C. M. Teng and Y. C. Wu, J. Nat. Prod., 2000, 63, 1160 CrossRef CAS PubMed.
- Selected references: (a) M. Anzini, A. Cappelli, S. Vomero, G. Giorgi, T. Langer, G. Bruni, M. R. Romeo and A. S. Basile, J. Med. Chem., 1996, 39, 4275 CrossRef CAS PubMed; (b) J. Wu, W. Zhang and C. Wang, Synthesis, 2009, 1821 CAS.
- (a) E. De Clercq, J. Med. Chem., 1995, 38, 2491 CrossRef CAS; (b) I. Pendrak, S. Barney, R. Wittrock, D. M. Lambert and W. D. Kingsbury, J. Org. Chem., 1994, 59, 2623 CrossRef CAS; (c) E. C. Taylor, P. Zhou, L. D. Jennings, Z. Mao, B. Hu and J.-G. Jun, Tetrahedron Lett., 1997, 38, 521 CrossRef CAS.
- (a) L. A. Paquette, R. D. Dura and I. Modolo, J. Org. Chem., 2009, 74, 1982 CrossRef CAS PubMed; (b) J. B. Campbell, R. F. Dedinas and S. A. Trumbower-Walsh, J. Org. Chem., 1996, 61, 6205 CrossRef CAS PubMed; (c) A. Couture, E. Deniau, D. Ionescu and P. Grandclaudon, Tetrahedron Lett., 1998, 39, 2319 CrossRef CAS; (d) J. B. Campbell, R. F. Dedinas and S. Trumbower-Walsh, Synlett, 2010, 3008 CrossRef CAS PubMed.
- (a) J. B. Campbell, R. F. Dedinas and S. Trumbower-Walsh, Synlett, 2010, 3008 CrossRef CAS PubMed; (b) X. Gai, R. Grigg, T. Khamnaen, S. Rajviroongit, V. Sridharan, L. Zhang, S. Collard and A. Keep, Tetrahedron Lett., 2003, 44, 7441 CrossRef CAS PubMed.
- Selected cyclization reviews: (a) P. Thansandote and M. Lautens, Chem. – Eur. J., 2009, 15, 5874 CrossRef CAS PubMed; (b) T. Satoh and M. Miura, Chem. – Eur. J., 2010, 16, 11212 CrossRef CAS PubMed; (c) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651 RSC; (d) C. Zhu, R. Wang and J. R. Flack, Chem. – Asian J., 2012, 7, 1502 CrossRef CAS PubMed; (e) P. B. Arokiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef PubMed; (f) L. Ackermann, Acc. Chem. Res., 2014, 47, 281 CrossRef CAS PubMed.
- Selected recent ruthenium papers: (a) P. B. Arockiam, C. Fischmeister, C. Bruneau and P. H. Dixneuf, Green Chem., 2011, 13, 3075 RSC; (b) Y. Hashimoto, K. Hirano, T. Satoh, F. Kakiuchi and M. Miura, Org. Lett., 2012, 14, 2058 CrossRef CAS PubMed; (c) W. Liu and L. Ackermann, Chem. Commun., 2014, 50, 1878 RSC; (d) K. Parthasarathy, N. Senthilkumar, J. Jayakumar and C.-H. Cheng, Org. Lett., 2012, 14, 3478 CrossRef CAS PubMed; (e) Y. Kommagalla, K. Srinivas and C. V. Ramana, Chem. – Eur. J., 2014, 20, 7884 CrossRef CAS PubMed; (f) C. G. Ravikiran and M. Jeganmohan, Chem. Commun., 2014, 50, 2442 RSC; (g) P. Kishor and M. Jeganmohan, Chem. Commun., 2013, 49, 9651 RSC.
- Rhenium: (a) S. Sueki, Y. Guo, M. Kanai and Y. Kuninobu, Angew. Chem., Int. Ed., 2013, 52, 11879 CrossRef CAS PubMed . Rhodium: ; (b) F. Wang, G. Song and X. Li, Org. Lett., 2010, 12, 5430 CrossRef CAS PubMed; (c) C. Zhu and J. R. Falck, Tetrahedron, 2012, 68, 9192 CrossRef CAS PubMed; (d) C. Zhu and J. R. Falck, Org. Lett., 2011, 13, 1214 CrossRef CAS PubMed; (e) F. W. Patureau, T. Besset and F. Glorius, Angew. Chem., Int. Ed., 2011, 50, 1064 CrossRef CAS PubMed . Palladium: ; (f) D.-D. Li, T.-T. Yuan and G.-W. Wang, Chem. Commun., 2011, 47, 12789 RSC; (g) J. W. Wrigglesworth, B. Cox, G. C. Lloyd-Jones and K. I. Booker-Milburn, Org. Lett., 2011, 13, 5326 CrossRef CAS PubMed . Ruthenium: ; (h) L. Ackermann, L. Wang, R. Wolfram and A. V. Lygin, Org. Lett., 2012, 14, 728 CrossRef CAS PubMed; (i) M. C. Reddy and M. Jeganmohan, Org. Lett., 2014, 16, 4866 CrossRef CAS PubMed , references therein.
- (a) E. W. Werner, T.-S. Mei, A. J. Burckle and M. S. Sigman, Science, 2012, 338, 1455 CrossRef CAS PubMed; (b) C. C. Oliveira, R. A. Angnes and C. R. D. Correia, J. Org. Chem., 2013, 78, 4373 CrossRef CAS PubMed.
- (a) L. Huang, Q. Wang, J. Qi, X. Wu, K. Huang and H. Jiang, Chem. Sci., 2013, 4, 2665 RSC; (b) J. Qi, L. Huang, Z. Wang and H. Jiang, Org. Biomol. Chem., 2013, 11, 8009 RSC; (c) Z. Shi, M. Boultadakis-Arapinis and F. Glorius, Chem. Commun., 2013, 49, 6489 RSC.
- (a) T. C. Johnson, D. J. Morris and M. Wills, Chem. Soc. Rev., 2010, 39, 81 RSC; (b) B. Li, C. Darcel and P. H. Dixneuf, Chem. Commun., 2014, 50, 5970 RSC; (c) Y. Kommagalla, V. B. Mullapudi, F. Francis and C. V. Ramana, Catal. Sci. Technol., 2015, 5, 114 RSC.
- (a) X.-Y. Shi and C.-J. Li, Org. Lett., 2013, 15, 1476 CrossRef CAS PubMed; (b) L. Yang, C. A Correia and C.-J. Li, Org. Biomol. Chem., 2011, 9, 7176 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and spectroscopic data. See DOI: 10.1039/c4cc09877c |
| This journal is © The Royal Society of Chemistry 2015 |