
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bonding situation in Be[N(SiMe3)2]2 – an experimental and computational study†
D.
Naglav
,
A.
Neumann
,
D.
Bläser
,
C.
Wölper
,
R.
Haack
,
G.
Jansen
and
S.
Schulz
*
University of Duisburg-Essen, Universitätsstr. 5-7, S07 S03 C30, 45117 Essen, Germany. E-mail: stephan.schulz@uni-due.de; Fax: +49 201 1833830; Tel: +49 201 1834635
First published on 30th January 2015
Abstract
The solid state structure of Be[N(SiMe3)2]2 (1) was determined by in situ crystallisation and the bonding situation investigated by quantum chemical calculations. The Be–N bond is predominantly ionic, but some evidence for the presence of a partial Be–N double bond character was found.
Beryllium chemistry has led a cinderella-like existence compared to the heavier group 2 elements, in particular Mg, due to the toxicity of beryllium and its compounds.1 This has changed significantly in the last decade and highly interesting compounds such as beryllium diorganyls,2 NHC-stabilized beryllium dihalides,3 diorganyls,4 and hydride (i-PrNHC-Be(Me)H),5 or the Lewis acid–base adducts [(Cy3P)2Pt-Be(Cl)X] (X = Cl, Me) have been prepared.6 Moreover, the search for Be(I) complexes and the use of BeF2 as a fluoride acceptor in liquid ammonia pushed the beryllium chemistry recently.7,8
Aside from as-described compounds, there has been steady interest in Be–N chemistry since the Be–N bond in homoleptic, monomeric beryllium diamides Be(NR2)2 is expected to show π-bonding character due to the high electrophilic nature of the Be center and its small size. Comparable findings were reported for monomeric aminoboranes R2BNR′2, containing a B![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond, and monomeric iminoboranes RBNR′, showing B
N double bond, and monomeric iminoboranes RBNR′, showing B![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bonding.9 Alternatively, a high ionic contribution was discussed for the bonding in beryllium amides due to the high electronegativity difference. Therefore, beryllium diamides have been synthesized and structurally characterized, but they were found to almost exclusively adopt dimeric or trimeric structures in the solid state.10 Only Be[N(SiMe3)2]21 and a very few beryllium amides as reported by Nöth and Schlosser were found to be monomeric in solution,11,12 but their solid state structures remain unknown. However, vapor phase studies supported the presence of π-bonding in Be[N(SiMe3)2]21.13 The Be–N bond length of 1.562(24) Å is significantly shorter than the calculated value of a Be–N single bond (1.73 Å),14 but slightly longer than the calculated BeN double bond (1.50 Å),15 respectively. In addition, Power et al. observed the shortest Be–N bond (1.519(4) Å) in the heteroleptic beryllium amide (2,6-Mes2-C6H3BeNSiMe3)2, to the best of our knowledge the only structurally characterized beryllium amide containing a twofold-coordinated Be atom in a linear arrangement.16 Our increasing interest in beryllium chemistry17 prompted us to determine the solid state structure of Be[N(SiMe3)2]21 and to analyze its bonding situation by quantum chemical calculations.
N triple bonding.9 Alternatively, a high ionic contribution was discussed for the bonding in beryllium amides due to the high electronegativity difference. Therefore, beryllium diamides have been synthesized and structurally characterized, but they were found to almost exclusively adopt dimeric or trimeric structures in the solid state.10 Only Be[N(SiMe3)2]21 and a very few beryllium amides as reported by Nöth and Schlosser were found to be monomeric in solution,11,12 but their solid state structures remain unknown. However, vapor phase studies supported the presence of π-bonding in Be[N(SiMe3)2]21.13 The Be–N bond length of 1.562(24) Å is significantly shorter than the calculated value of a Be–N single bond (1.73 Å),14 but slightly longer than the calculated BeN double bond (1.50 Å),15 respectively. In addition, Power et al. observed the shortest Be–N bond (1.519(4) Å) in the heteroleptic beryllium amide (2,6-Mes2-C6H3BeNSiMe3)2, to the best of our knowledge the only structurally characterized beryllium amide containing a twofold-coordinated Be atom in a linear arrangement.16 Our increasing interest in beryllium chemistry17 prompted us to determine the solid state structure of Be[N(SiMe3)2]21 and to analyze its bonding situation by quantum chemical calculations.
1 was synthesized by reaction of BeCl2 and KN(SiMe3)2.111H, 13C and 29Si NMR spectra of 1 showed the expected resonances due to the trimethylsilyl groups, and the 9Be NMR spectrum showed a resonance at 12.3 ppm in C6D6 (9.6 ppm in THF-d8/12.4 ppm in Tol-d8).
A single crystal of 1 was grown on the diffractometer at a temperature of 150 K using a miniature zone melting procedure with focused infrared-laser-radiation,18,19 which we previously used for the structural characterization of small inorganic and organometallic molecules.20,211 crystallises in the monoclinic space group P21/n with the molecule placed on the general positions (Fig. 1). The central Be atom shows an almost perfect linear coordination environment (N1–Be1–N2 178.73(16)°) and the N atoms adopt trigonal-planar coordination spheres. The flat arrangement of the N atoms and their respective neighbours (max. deviation from best plane N1: 0.0224(10), N2: 0.0012(10) Å) as well as their bond angles point to an sp2 hybridisation, a prerequisite for the formation of Be–N π-bonding. The short Be–N bond lengths (Be1–N1 1.525(2), Be1–N2 1.519(2) Å), which are comparable to that observed in the gas phase of 1 (1.562(24) Å)13 are identical to that reported for 2,6-Mes2-C6H3BeN(SiMe3)2 (1.519(4) Å),16 and the Si–N⋯N–Si torsion angles of approx. 90° further support the presence of BeN double bonds. However, analogous structural findings would also be expected for a predominantly ionic contribution and are individually considered with no experimental proof for the presence of π-bonding character in the Be–N bond.
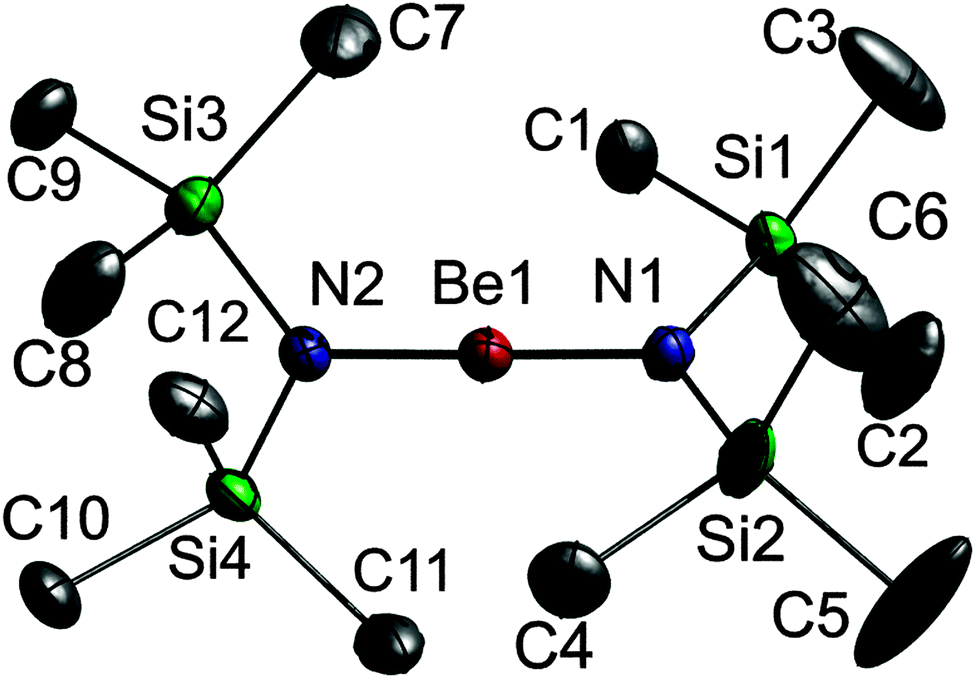 | ||
| Fig. 1 Solid state structure of 1; non-H-atoms are shown as thermal ellipsoids at 50% probability levels. H atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: Be1–N1 1.525(2), Be1–N2 1.519(2), N1–Si1 1.7286(12), Si1–N2 1.7251(12); N1–Be1–N2 178.73(16), Si1–N1–Si2 127.20(7), Si1–N1–Be1 115.55(10), Si2N1–Be1 116.64(10); Si1–N1⋯N2Si3 −92.12(8), Si1–N1⋯N2Si4 88.32(8), Si2–N1⋯N2Si3 84.03(8), Si2–N1⋯N2Si4 −95.53(8). | ||
Theoretical computations with dispersion corrected density functional theory confirm this picture.22 Geometry optimizations lead to a S4-symmetrical structure 1a with a Be–N bond length of 1.525 Å and a perfectly planar environment of the N atoms (Si–N–Si angle 128.3°). The Si–N–N–Si torsion angle is 90°.
Geometry optimizations of Be(NR2)2 (R = SiH32, Me 3, H 4) followed by frequency calculations resulted in D2d symmetrical minimum structures with torsion angles of 90°. The nitrogen charge and the R–N–R bond angles strongly depend on the organic ligands (cf.Table 1), which is in line with their electronegativity and steric demand. In contrast, the charge on the Be atom in 1a–4 is about 1.7 e for all cases according to the theory of atoms in molecules (AIM).23 Compared with other BeX2 compounds (X = Me, OH, F; cf.Table 1) we note that (i) the Be charge is fairly independent of X, (ii) the density at the bond critical point ρ(rc) is inversely proportional to the Be–X distance, (iii) the Laplacian Δρ(rc) correlates with the expected bond polarity, and (iv) the energy density H(rc) is positive for 7, indicating the expected ionic interaction, while it is negative for 5, which in combination with the positive value of Δρ(rc) is indicative of mixed closed shell/shared interaction cases. While 6 has a nearly vanishing H(rc), the bond critical point parameters of 1a–4 resemble more closely that of the metal organic BeMe25 (cf.Table 1). Accepting the resulting picture of strongly polar covalent Be–N bonds, the bond ellipticities ε (0.07 (1a)–0.13 (4)) together with the dumbbell-shaped localization regions of the electron localization function (ELF)24 between the Be and N atoms (cf.Fig. 2 and Fig. S2–S4, ESI†) indicate a certain double bond character.
| Be–N [Å] | R–N–R [°] | q(Be) [e] | q(N) [e] | ρ(rc) [au] | Δρ(rc) [au] | H(rc) [au] | |
|---|---|---|---|---|---|---|---|
| 1a | 1.525 | 128.3 | 1.69 | −2.35 | 0.116 | 0.705 | −0.018 |
| 2 | 1.528 | 123.0 | 1.70 | −2.36 | 0.114 | 0.703 | −0.016 |
| 3 | 1.517 | 112.5 | 1.67 | −1.44 | 0.118 | 0.766 | −0.015 |
| 4 | 1.522 | 109.4 | 1.71 | −1.56 | 0.114 | 0.752 | −0.010 |
| Be–X | q(Be) [e] | q(X) [e] | ρ(rc) [au] | Δρ(rc) [au] | H(rc) [au] | ||
|---|---|---|---|---|---|---|---|
| 5 | 1.692 | 1.69 | −0.92 | 0.098 | 0.312 | −0.030 | |
| 6 | 1.441 | 1.72 | −1.44 | 0.126 | 1.057 | 0.001 | |
| 7 | 1.392 | 1.74 | −0.87 | 0.133 | 1.271 | 0.007 |
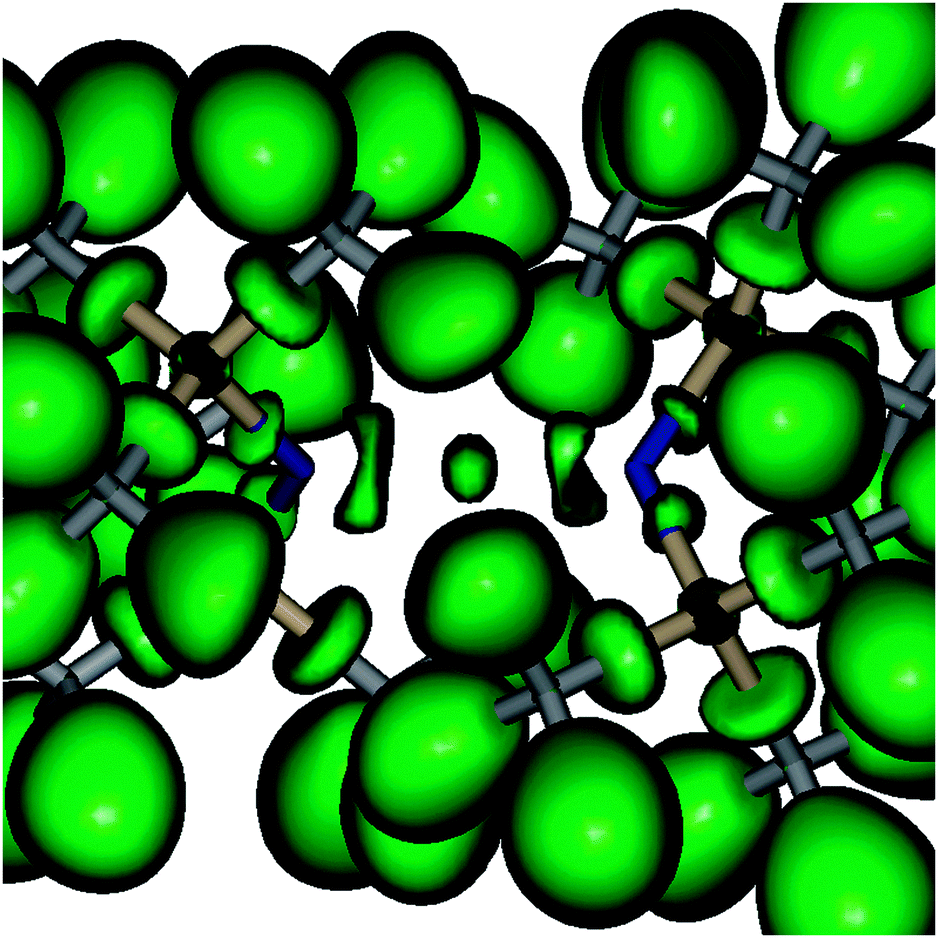 | ||
| Fig. 2 ELF isosurfaces of Be[N(SiMe3)2]2 (isovalue 0.850, Be atom in the centre). | ||
The solid state structure of Be[N(SiMe3)2]21 was determined using an in situ crystallisation approach. Quantum chemical calculations on different beryllium diamides Be(NR2)2 with different steric and electronic properties prove that the Be–N bond is strongly polarized and should therefore be described as predominantly ionic. However, some evidence for the presence of a partial Be–N double bond character in 1 was also found.
We thank one of the referees for fruitful discussions and hinting on the possibility of an S4 minimum structure of 1a.
Notes and references
- T. Handa, S. Nagai, M. Kitaichi, K. Chin, Y. Ito, T. Oga, K. Takahashi, K. Watanabe, M. Mishima and T. Izumi, Sarcoidosis Vasc. Diffuse Lung Dis., 2009, 26, 24 Search PubMed
; C. Strupp, Ann. Occup. Hyg., 2011, 55, 43 CrossRef CAS PubMed
- S. C. Chmely, T. P. Hanusa and W. W. Brennessel, Angew. Chem., Int. Ed., 2010, 49, 5870 CrossRef CAS PubMed
- R. J. Gilliard, M. Y. Abraham, Y. Wang, P. Wei, Y. Xie, B. Quillian, H. F. Schaefer, P. v. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2012, 134, 9953 CrossRef CAS PubMed
- J. Gottfriedsen and S. Blaurock, Organometallics, 2006, 25, 3784 CrossRef CAS
- M. S. Hill, M. Arrowsmith, G. Kociok-Köhn, D. J. MacDougall and M. F. Mahon, Angew. Chem., Int. Ed., 2012, 51, 2098 CrossRef PubMed
- H. Braunschweig, K. Gruss and K. Radacki, Angew. Chem., Int. Ed., 2009, 48, 4239 CrossRef CAS PubMed
- S. A. Couchman, N. Holzmann, G. Frenking, D. J. D. Wilson and J. L. Dutton, Dalton Trans., 2013, 42, 11375–11384 RSC
- F. Kraus, S. Baer, M. R. Buchner and A. J. Karttunen, Chem. – Eur. J., 2012, 18, 2131 CrossRef CAS PubMed
- P. Paetzold, C. v. Plotho, G. Schmid, R. Boese, B. Schrader, D. Bougeard, U. Pfeiffer, R. Gleiter and W. Schäfer, Chem. Ber., 1984, 117, 1089 CrossRef CAS
-
M. F. Lappert, A. Protchenko, P. P. Power and A. Seeber, Metal Amide Chemistry, J. Wiley & Sons Ltd, 2009 Search PubMed
- H. Bürger, C. Forker and J. Goubeau, Monatsh. Chem., 1965, 96, 597 CrossRef
- H. Nöth and D. Schlosser, Inorg. Chem., 1983, 22, 2700 CrossRef
- A. H. Clark and A. Haaland, Acta Chem. Scand., 1970, 24, 3024 CrossRef CAS
- P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 186 CrossRef PubMed
- P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 12770 CrossRef PubMed
- M. Niemeyer and P. Power, Organometallics, 1997, 36, 4688 CAS
- D. Naglav, D. Bläser, C. Wölper and S. Schulz, Inorg. Chem., 2014, 53, 1241 CrossRef CAS PubMed
-
R. Boese and M. Nussbaumer, In Situ Crystallisation Techniques, in Organic Crystal Chemistry, ed. D. W. Jones, Oxford University Press, Oxford, England, 1994, p. 20 Search PubMed
- Bruker D8 Kappa II diffractometer with APEX2 detector (MoKα radiation, λ = 0.71073 Å). The structure was solved by Direct Methods (SHELXS-97, G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467 CrossRef CAS
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 472 reflections measured, 7557 independent reflections (Rint = 0.0507). The final R1 values were 0.0471 (I > 2σ(I)) and 0.0588 (all data). The final wR(F2) values were 0.1213 (I > 2σ(I)) and 0.1326 (all data). The goodness of fit on F2 was 1.024.
472 reflections measured, 7557 independent reflections (Rint = 0.0507). The final R1 values were 0.0471 (I > 2σ(I)) and 0.0588 (all data). The final wR(F2) values were 0.1213 (I > 2σ(I)) and 0.1326 (all data). The goodness of fit on F2 was 1.024. - S. Schulz, B. Lyhs, G. Jansen, D. Bläser and C. Wölper, Chem. Commun., 2011, 47, 3401 RSC
- A. Kuczkowski, S. Heimann, A. Weber, S. Schulz, D. Bläser and C. Wölper, Organometallics, 2011, 30, 4730 CrossRef CAS
- All geometries were fully optimized using tightened convergence criteria and improved integration grids at the density functional theory level, employing the BP86 exchange–correlation functional [
(a) A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS
-
(a)
R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, Oxford, 1990 Search PubMed
-
(a) A. D. Becke and K. E. Edgecombe, J. Chem. Phys., 1990, 92, 5397 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure and characterization of 1 as well as computational details of 1a–7. CCDC 1003578 (1). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc09732g |
| This journal is © The Royal Society of Chemistry 2015 |