
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesigning nanomolar antagonists of DC-SIGN-mediated HIV infection: ligand presentation using molecular rods†
Stefania
Ordanini
a,
Norbert
Varga
a,
Vanessa
Porkolab
bcd,
Michel
Thépaut
bcd,
Laura
Belvisi
ae,
Andrea
Bertaglia
a,
Alessandro
Palmioli
a,
Angela
Berzi
f,
Daria
Trabattoni
f,
Mario
Clerici
gh,
Franck
Fieschi
bcd and
Anna
Bernardi
*ae
aUniversità degli Studi di Milano (UniMI), Dip. Chimica, via Golgi 19, 20133, Milano, Italy. E-mail: anna.bernardi@unimi.it; Tel: +39 02 50314092
bUniv. Grenoble Alpes, Inst. de Biologie Structurale, Grenoble, France
cCNRS, IBS, F-38044, Grenoble, France
dCEA, IBS, F-38044 Grenoble, France
eCNR-ISTM, Inst. of Molecular Science and Technologies, Milano, Italy
fUniMI, Dip. Scienze Biomediche e Cliniche “L. Sacco”, Milano, Italy
gUniMI, Dip. Fisiopatologia Medico-Chirurgica e Trapianti, Segrate, Italy
hFondazione Don Carlo Gnocchi IRCCS, Milano, Italy
First published on 28th January 2015
Abstract
DC-SIGN antagonists were designed combining one selective monovalent glycomimetic ligand with trivalent dendrons separated by a rigid core of controlled length. The design combines multiple multivalency effects to achieve inhibitors of HIV infection, which are active in nanomolar concentration.
Carbohydrate–protein interactions in biological systems mostly occur among multivalent partners. This increases the potency of sugar ligands that in monovalent form would be too weak to have biological relevance. To interfere with such interactions, multivalent antagonists have been designed, which include a variety of scaffolds.1 Potency enhancement in multivalent ligands can result from different mechanisms, including clustering of soluble partners (Fig. 1A), chelation (Fig. 1B) and statistical rebinding (or proximity, Fig. 1C) effects, which can be exploited to different degrees by different ligand/target pairs.2
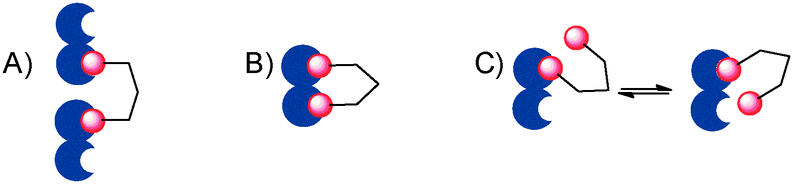 | ||
| Fig. 1 (A) A multivalent ligand (red) clustering a soluble protein (blue); (B) a multivalent ligand chelating a divalent protein; (C) a multivalent ligand binding to a protein exploiting the statistical rebinding (proximity) effect. | ||
The ability of a multivalent ligand to chelate (i.e. to bind simultaneously) more than one binding site of a target critically depends on the spacer separating the ligand subunits. The design of effective spacers requires a delicate balance between rigidity, which favors association by decreasing the entropic cost, and flexibility, which helps the ligand system to adapt to the protein, being itself a flexible, dynamic target. Most systems reported so far have been built using flexible spacers, which are both synthetically more accessible, and more tolerant of design imperfections.3,4
Together with the Pieters group, we have recently reported the synthesis of rod-like rigid spacers 1–3 (Scheme 1 and Scheme SI-2 in the ESI†) of variable length, based on phenylene-ethynylene units and their incorporation into divalent ligands for P. aeruginosa lectin LecA.5 We now show how these rod-like spacers can be used in the modular design of hexavalent dendrimers with a rigid core of defined length, targeted against DC-SIGN, a tetrameric C-type lectin receptor of dendritic cells hijacked by many pathogens in their infection cycle.6 Antagonists of DC-SIGN have been proposed as inhibitors of various viral infections, including HIV, Ebola and Dengue.7
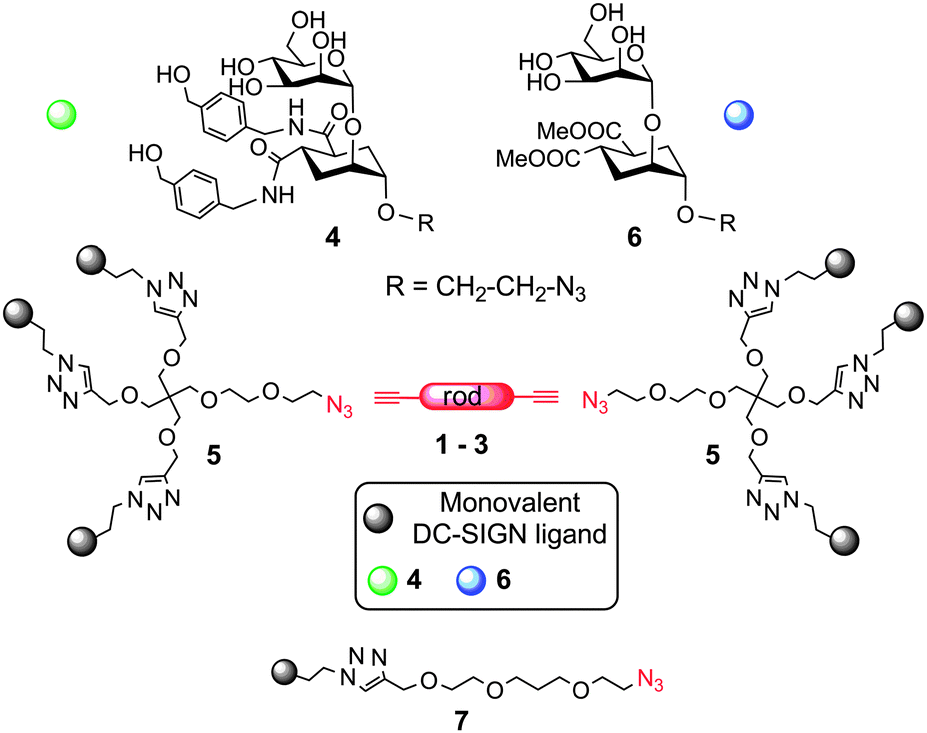 | ||
| Scheme 1 Strategy for the design of the rod-based dendrimers. | ||
The general strategy we describe here to target DC-SIGN strives to exploit both chelation and statistical rebinding effects by spacing two appropriate trivalent ligands in a controlled fashion, so as to achieve efficient binding with relatively low valency of the construct. The design of the dendrimers (Scheme 1) includes 4, a mannose-based monovalent ligand of moderate affinity (0.3 mM) and good selectivity for DC-SIGN over Langerin,8 and dendron 5, which presents three copies of 4 and is equipped with an azido group at the focal point for conjugation to the rods.9 Depending on the length of the rod spacer (see ESI†), the new constructs were predicted to span the required distance for chelation of contiguous binding sites in DC-SIGN (∼4 nm).10 Additionally, dendron 5 has a PEG-like structure, which grants the flexibility required for optimal adaptation.11 Controls bearing the lower affinity mannoside 6 allowed assessing the role of the monovalent ligand in determining the overall activity of the construct. Divalent structures were also synthesized, either by direct conjugation of 4 and 6 to the rods, or using 7, to afford dimers of comparable length (Scheme 1). Analysis of the binding efficiency and comparison of hexavalent and divalent structures allowed dissecting the relative importance of chelation and statistical rebinding for this interaction. Optimal combination of rod length, valency and monovalent ligand yielded a hexavalent dendrimer which inhibits DC-SIGN mediated transmission of HIV infection in nanomolar concentrations. Here we report our results.
Dendron 59 and rods 1–35 were prepared as previously described. A copper(I) catalyzed dipolar cycloaddition (CuAAC) between 5 and the appropriate diynes 1–3 yielded hexavalent dendrimers of increasing dimensions, bearing either 4 or 6 as the ligand (Table 1). Divalent controls were synthesized using the same approach using ligands 7.4 and 7.6 (with a linker length equivalent to the length of the dendron chain), or conjugating 4 and 6 directly to 3. The compounds prepared are shown in Table 1. All compounds were water soluble (≥150 μM). They were purified by either reverse phase chromatography (C18 silica) or gel filtration (Sephadex LH-20) and characterized by NMR and MS analysis. Details of the synthesis and characterization are reported in the ESI.† Hexavalent dendrimers 8.4 and 8.6, known to operate only by statistical rebinding and clustering effects,9 were used as reference.
|
Structure
|
Ligand 6 | Ligand 4 |
|---|---|---|

|
1.5.6 | 1.5.4 |
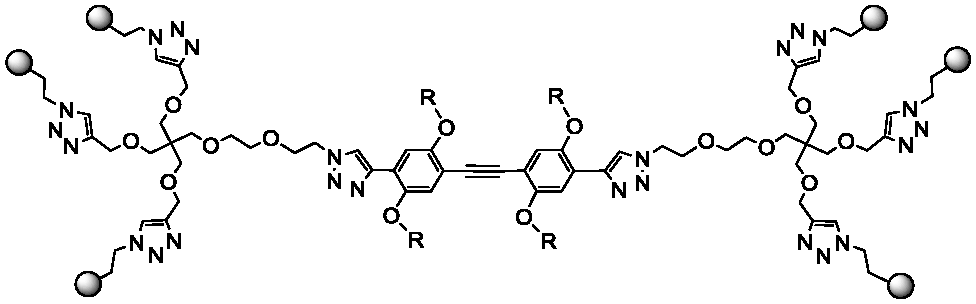
|
2.5.6 | 2.5.4 |

|
3.5.6 | 3.5.4 |

|
1.7.6 | 1.7.4 |

|
2.7.6 | 2.7.4 |

|
3.7.6 | 3.7.4 |

|
3.6 | 3.4 |

|
8.6 | 8.4 |
The activity of all compounds was tested by SPR in the previously established competition assay that measures their ability to inhibit binding of the DC-SIGN extracellular domain (ECD) to mannosylated bovine serum albumin (Man-BSA) immobilized onto the SPR sensor surface.8 The results are shown in Fig. 2 and Table 2. Sensorgrams and inhibition curves are collected in the ESI† (Fig. SI-7 and SI-8). Clear trends are shown by the constructs bearing the lower activity pseudo-dimannoside ligand 6 (Fig. 2, blue bars; Table 2, ligand 6 row). In the hexavalent series (8.6, 1.5.6, 2.5.6 and 3.5.6), affinity and relative inhibitory potency β12 increase regularly with the length of the rod, reaching IC50 = 9 μM and β = 17 with 3.5.6 (Table 2).
 | ||
| Fig. 2 DC-SIGN inhibition activities of glycodendrimers bearing ligand 6 (blue bars) or 4 (green bars). DC-SIGN (20 μM) and compounds at increasing concentration were co-injected over a CM4 chip where Man-BSA (Manα1–3[Manα1–6]Man-BSA, 15 trimannose residues, on average) has been immobilized. IC50 are expressed as dendrimer concentration (μM). | ||
| IC50 (μM) (β-factor)a | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Scaffold | |||||||||
| Ligand | Mono | 8 | 1.5 | 2.5 | 3.5 | 1.7 | 2.7 | 3.7 | 3 |
| a Valency corrected β-factor12 (relative inhibitory potency). b Lower limit of the assay was reached. | |||||||||
| 6 | 901 (1) | 39 (4) | 34 (4) | 25 (6) | 9 (17) | 295 (2) | 175 (3) | 67 (7) | 36 (13) |
| 4 | 271 (1) | 5b (10) | 6b (8) | 7b (7) | 5b (9) | 61 (2) | 34 (4) | 19 (7) | 8b (17) |
Similarly, in the group of divalent ligands 1.7.6, 2.7.6 and 3.7.6 the IC50 values decrease regularly as the rod length increases and the β-factor increases from 2 to 7 (Fig. 2, divalent rods, blue bars). For these compounds, data also show an important effect of the length of the linker connecting the rod to the active ligand. The short linker dimer 3.6 with an IC50 of 36 μM is twice as active as the corresponding long linker derivative 3.7.6 (IC50 67 μM), as a result of entropy loss associated with a long flexible linker. Comparison of β-factors for hexavalent and divalent constructs of a similar core reveals the positive effect of the local increase of ligand concentration generated by the trivalent dendron in 1.5.6, 2.5.6 and 3.5.6, which corresponds to an increase of the relative potency by at least a factor of two. As expected, all dendrimers bearing the higher affinity monovalent ligand 4 are more active than the corresponding ones based on 6 (compare blue bars and green bars in Fig. 2). In the divalent series 1.7.4, 2.7.4 and 3.7.4, activities and β-factors increase regularly with the rod length. Again, optimal affinity values are obtained with a long rod and a short flexible linker (3.4, IC50 = 8 μM), indicating a good preorganization of the antagonist.3 No apparent trend is revealed in the hexavalent series (8.4, 1.5.4, 2.5.4 and 3.5.4): all compounds show similar IC50 in the low μM range (Fig. 2, hexavalent rod panel) and similar β-factor (approximately 10, Table 2). With an apparent IC50 of 5 μM, these compounds have achieved the intrinsic affinity of DC-SIGN ECD for the Man-BSA functionalized surface, thus the lower limit of the assay. The inhibition curves observed only mirror the interaction curve of the reporter system, which has become limiting (Fig. SI-8, ESI†).
Selected compounds were therefore tested in a more sensitive assay, using a cellular model of HIV-1 infection. B-THP-1 cells expressing DC-SIGN were used as a model of DCs and inhibition of trans infection of CD4+ T-lymphocytes was analyzed (see ESI† for full method details). The results of these trans infection studies (Fig. 3, top) showed a clear dose–response effect for all the tested inhibitors 8.4, 1.5.4, 3.5.4, 3.7.4 and 3.4. An IC50 around 1 μM was confirmed for 8.4, as previously reported,9 and a similar value was obtained for divalent 3.7.4. The hexavalent elongated structures of 1.5.4 and 3.5.4 and the divalent short linker version 3.4 display an activity improved by one order of magnitude or more and dependent on the rod length and the overall valency of the system. In a second set of experiments, dose–response was measured at lower concentrations for 1.5.4, 3.5.4 and 3.4 (Fig. 3, bottom). Data fitting to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 hyperbolic decay afforded IC50 values of 67 nM, 24 nM and 161 nM, respectively. Thus, these experiments confirmed that also in the cellular model inclusion of a rigid spacer at the dendrimer core has a strong positive effect on the antagonist activity. Dendrimer 3.5.4 (IC50 24 nM) is 40 times more active than 8.4 and among the most potent DC-SIGN antagonists reported.13 The data also confirm the impressive activity of the divalent ligand 3.4, but, as in the SPR study, suggest that higher local concentration of the ligand is beneficial to effectively block the infection. It is worth noting that SPR assays are performed using DC-SIGN solubilised in the medium and therefore they measure effects derived also from the protein aggregation mechanism (Fig. 1A). In contrast, in cellular studies the protein is immobilized in the cell membrane. Nonetheless, both assays clearly show that both the length of the rod and the overall valency of the material have an influence on the IC50 values.
:1 hyperbolic decay afforded IC50 values of 67 nM, 24 nM and 161 nM, respectively. Thus, these experiments confirmed that also in the cellular model inclusion of a rigid spacer at the dendrimer core has a strong positive effect on the antagonist activity. Dendrimer 3.5.4 (IC50 24 nM) is 40 times more active than 8.4 and among the most potent DC-SIGN antagonists reported.13 The data also confirm the impressive activity of the divalent ligand 3.4, but, as in the SPR study, suggest that higher local concentration of the ligand is beneficial to effectively block the infection. It is worth noting that SPR assays are performed using DC-SIGN solubilised in the medium and therefore they measure effects derived also from the protein aggregation mechanism (Fig. 1A). In contrast, in cellular studies the protein is immobilized in the cell membrane. Nonetheless, both assays clearly show that both the length of the rod and the overall valency of the material have an influence on the IC50 values.
 | ||
| Fig. 3 Trans infection experiments. Compounds were tested at the concentrations indicated and compared with the effect of the medium (MED). Experiments were performed on CD4+ T-lymphocytes isolated from 3 different healthy donors. The concentration of p24 in co-culture supernatants reflects the level of infection. Values represent the mean ± SD (top) 8.4, 1.5.4, 3.5.4, 3.7.4, and 3.4 in the concentration range 1–50 μM; (bottom) 1.5.4, 3.5.4 and 3.4 in the concentration range 0.01–10 μM. | ||
In order to interpret the results on a structural basis, the shape and size of the ligands were simulated by molecular dynamics, using the 6 series as models. For the sake of simplicity, the PEG chains on the rods were replaced with methyl ethers. Starting structures were obtained by simulated annealing and then subjected to multiple Stochastic Dynamics (SD) simulation cycles for a total SD time ranging from 125 to 150 ns (OPLSA-2005 force field, GB/SA water solvation model; controls were synthesized also using an explicit water solvation model, details are provided in the ESI†). Mannose is known to use O3 and O4 to coordinate the Ca2+ ion in the DC-SIGN binding site and the distance between two adjacent sites in the tetrameric protein was estimated to be at least 38 Å by SAXS.10 Thus, the potential of multivalent compounds to simultaneously coordinate two Ca2+ sites was evaluated by monitoring the distance between Man-O3 of two distal sugar residues during the simulation. The time course of the distances is shown in Fig. SI-1/SI-4 of the ESI.† Average values 〈dO3–O3〉 and values at the maximum extension Max dO3–O3 are reported in Table SI-1 (ESI†), together with the dendrimers' gyration radius. The simulations confirm that the hexavalent dendrimer 8.6 (Max dO3–O3 35.4 Å) is not likely to bridge between two DC-SIGN binding sites, even at its maximum extension.9 All dendrimers that include a rod at their core, even the shortest one 1, can comfortably reach across in their most extended conformations. However, the flexibility of the long linker in 1.7.6, 3.7.6 and 3.5.6 allows the sugars to fold over the aromatic cores, producing much more compact conformations that represent over 95% of the sampled population. Representative folded conformations of 3.7.6 and 3.5.6 are shown in Fig. 4.
 | ||
| Fig. 4 (A) Structures of the most represented conformations sampled during the dynamic simulations of 3.7.6. Two types of folds (type I and type II) are observed. (B) Representative structures from the simulation of 3.5.6: type II folds are favored. | ||
The most extended average structure was calculated for the short linker dimer 3.6, which features Max dO3–O3 and 〈dO3–O3〉 of 43.3 Å and 31.7 Å, respectively. Most importantly, 30% of the structures sampled during the simulations display dO3–O3 larger than 35 Å and are therefore likely to be productive as protein chelating agents. This supports the notion that dimer 3.6 is optimally preorganized for DC-SIGN binding. Indeed, docking it within two adjacent binding sites of a DC-SIGN tetramer yielded a complex (Fig. 5) that was stably bound during dynamic simulations performed using an implicit GB/SA water model (25 ns). Similar complexes generated for the long linker dimers 3.7.6 (Fig. SI-6B, ESI†) and 1.7.6 (Fig. SI-6C, ESI†) also showed stable chelation over the course of 25 ns simulations, accompanied by extensive dynamics of the flexible linkers.
 | ||
| Fig. 5 Docked complex of divalent ligand 3.6 on the DC-SIGN tetramer showing that the compound can reach across to adjacent binding sites. | ||
It was recognized early on that multivalent presentations of native ligands could improve recognition by DC-SIGN14 and the concept was used with many different constructs.1 It is still debated whether designing ligands for accurate fitting of polyvalent targets is worth the effort, as compared to “brute force” high valency presentation of weak ligands (often easily accessible: e.g. monosaccharides). Of course this has implications beyond carbohydrate–protein interactions and the final response will have to account also for additional considerations, such as costs (siding against design) and regulatory problems (siding against poorly controlled and ill characterized polyvalent materials). Additionally, the optimal presentation format may vary in different situations depending, most notably, on whether the multivalent target is soluble or embedded in membranes, which controls the occurrence of aggregation effects. It is well-established and confirmed by the present study that the design of the monovalent ligand has a measurable impact on the affinity of constructs of moderate valence. Probably, the most important consequence of monovalent ligand design is the ability to achieve selectivity.8 The work described here shows that rod-like rigid spacers can be used to control the size of multivalent constructs to match the target dimension, thus maximizing the impact of the system valency. Extended spacers of “perfect” length as in 3.4 generate excellent divalent binders. Addition of a flexible linker causes entropic loss upon binding and reduces the activity (compare 3.4 and 3.7.4 in Fig. 3, top). However, inclusion of even a slight increase in local concentration of the ligand, such as that provided by the two trivalent dendrons in 3.5.4, can make up for under-optimization of the linker and is a good trade-off between the two concepts of fitting by design and affinity by avidity.
The design of DC-SIGN antagonists is actively explored as an approach to block mucosal pathogens that are still a challenge for the development of vaccines. Compound 3.5.4 was designed as a rational combination of three elements: an effective (and selective) monovalent ligand, a rigid core of appropriate length and two trivalent dendrons. The material obtained inhibits DC-SIGN mediated HIV transmission with an IC50 in the nM range, a result that compares well with known DC-SIGN antagonists (polymers, dendrimers or gold nanoparticles) of much higher valency.13a,c,d
This work was supported with funds from ETN-Carmusys (PITN-GA-2008-213592) and CM1102 COST Action.
Notes and references
- A. Bernardi, J. Jimenez-Barbero, A. Casnati, C. De Castro, T. Darbre, F. Fieschi, J. Finne, H. Funken, K. E. Jaeger, M. Lahmann, T. K. Lindhorst, M. Marradi, P. Messner, A. Molinaro, P. V. Murphy, C. Nativi, S. Oscarson, S. Penades, F. Peri, R. J. Pieters, O. Renaudet, J. L. Reymond, B. Richichi, J. Rojo, F. Sansone, C. Schaffer, W. B. Turnbull, T. Velasco-Torrijos, S. Vidal, S. Vincent, T. Wennekes, H. Zuilhof and A. Imberty, Chem. Soc. Rev., 2013, 42, 4709 RSC.
- R. J. Pieters, Org. Biomol. Chem., 2009, 7, 2013 CAS.
- E. T. Mack, P. W. Snyder, R. Perez-Castillejos, B. Bilgicer, D. T. Moustakas, M. J. Butte and G. M. Whitesides, J. Am. Chem. Soc., 2012, 134, 333 CrossRef CAS PubMed.
- V. M. Krishnamurthy, L. A. Estroff and G. M. Whitesides, in Fragment based approaches in drug discovery, ed. W. Jahnke and D. A. Erlanson, Wiley-VCH, 2006, pp. 11–53 Search PubMed.
- F. Pertici, N. Varga, A. van Duijn, M. Rey-Carrizo, A. Bernardi and R. J. Pieters, Beilstein J. Org. Chem., 2013, 9, 215 CrossRef CAS PubMed.
- Y. van Kooyk and T. B. H. Geijtenbeek, Nat. Rev. Immunol., 2003, 3, 697 CrossRef CAS PubMed.
- A. Bavdek, R. Kostanjsek, V. Antonini, J. H. Lakey, M. D. Serra, R. J. C. Gilbert and G. Anderluh, FEBS J., 2012, 279, 126 CrossRef CAS PubMed.
- N. Varga, I. Sutkeviciute, C. Guzzi, J. McGeagh, I. Petit-Haertlein, S. Gugliotta, J. Weiser, J. Angulo, F. Fieschi and A. Bernardi, Chem. – Eur. J., 2013, 19, 4786 CrossRef CAS PubMed.
- N. Varga, I. Sutkeviciute, R. Ribeiro-Viana, A. Berzi, R. Ramdasi, A. Daghetti, G. Vettoretti, A. Amara, M. Clerici, J. Rojo, F. Fieschi and A. Bernardi, Biomaterials, 2014, 35, 4175 CrossRef CAS PubMed.
- G. Tabarani, M. Thepaut, D. Stroebel, C. Ebel, C. Vives, P. Vachette, D. Durand and F. Fieschi, J. Biol. Chem., 2009, 284, 21229 CrossRef CAS PubMed.
- S. Menon, K. Rosenberg, S. A. Graham, E. M. Ward, M. E. Taylor, K. Drickamer and D. E. Leckband, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 11524 CrossRef CAS PubMed.
- M. Mammen, S.-K. Chio and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2755 CrossRef CAS.
- (a) O. Martinez-Avila, L. M. Bedoya, M. Marradi, C. Clavel, J. Alcami and S. Penades, ChemBioChem, 2009, 10, 1806 CrossRef CAS PubMed; (b) K. C. A. Garber, K. Wangkanont, E. E. Carlson and L. L. Kiessling, Chem. Commun., 2010, 46, 6747 RSC; (c) J. Luczkowiak, S. Sattin, I. Sutkeviciute, J. J. Reina, M. Sanchez-Navarro, M. Thepaut, L. Martinez-Prats, A. Daghetti, F. Fieschi, R. Delgado, A. Bernardi and J. Rojo, Bioconjugate Chem., 2011, 22, 1354 CrossRef CAS PubMed; (d) C. R. Becer, M. I. Gibson, J. Geng, R. Ilyas, R. Wallis, D. A. Mitchell and D. M. Haddleton, J. Am. Chem. Soc., 2010, 132, 15130 CrossRef CAS PubMed.
- D. A. Mitchell, A. J. Fadden and K. Drickamer, J. Biol. Chem., 2001, 276, 28939 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of all compounds; modeling methods and results; SPR experiments: materials, methods and primary data; infection studies and cytotoxicity: materials and methods. See DOI: 10.1039/c4cc09709b |
| This journal is © The Royal Society of Chemistry 2015 |