
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stacking interactions by two Phe side chains stabilize and orient assemblies of even the minimal amphiphilic β-sheet motif†
Zarzhitsky
Shlomo
ab,
T. P.
Vinod
bc,
Raz
Jelinek
bc and
Hanna
Rapaport
*ab
aAvram and Stella Goldstein-Goren Department of Biotechnology Engineering, Ben-Gurion University of the Negev, Beer-Sheva 84105, Israel. E-mail: hannarap@bgu.ac.il
bIlse Katz Institute for Nanoscale Science and Technology, Ben-Gurion University of the Negev, Beer-Sheva 84105, Israel
cDepartment of Chemistry, Ben-Gurion University of the Negev, Beer-Sheva 84105, Israel
First published on 12th January 2015
Abstract
Here we demonstrate that the smallest possible motif of the amphiphilic and pleated β-strand structure can be generated using tri-peptides stabilized by π–π stacking interactions. Monitoring the early stages of Phe-Glu-Phe fibril formation revealed unique angular orientations. Phe-Glu-Phe fibrils were further exploited as adsorbing templates for metal ions.
Deciphering the rules relating function and structure of naturally occurring biomolecules could pave the way for the development of novel functional biomaterials that are also biocompatible and biodegradable. Polynucleotides and proteins have been shown to self-organize into hierarchically functional structures.1–4 In recent years, there has been growing interest in utilizing peptides as building blocks in certain secondary structures capable of assembling into nanostructures, for chemical, biological and medicinal applications.5,6 Tailoring the exact sequence of amino acids in designed peptide assemblies allows tuning of the kinetics of the molecular arrangements, and their mechanical and chemical properties.
Peptides sharing the amphiphilic motif of alternating hydrophilic and hydrophobic amino acids tend to form β-sheet structures and assemble as monolayers, sheets and as elongated bilayer fibrils in bulk solutions.7–9 Under appropriate conditions of concentrations, ionic strength and pH, these peptide fibrils in aqueous solutions are stabilized by hydrophobic interactions between the layers and cross strand hydrogen bonds along each layer. Segregation of the hydrophobic side chains from both layers makes them point towards each other and exposes the hydrophilic side chains to the surrounding solution. A network of such fibrils can stabilize a hydrogel phase.10–13 Studies of short amyloid peptide assemblies revealed the dominant role of aromatic π–π stacking interactions in driving the formation of highly stable fibrils.14 Phe-Phe dipeptides and derivatives thereof were shown to form various nanostructures14–16 and more recently even the single amino acid Phe was found to form fibrils in aqueous solutions.17 Nanotubes formed by Phe-Phe dipeptides were found to be highly stable18 yet in order to generate hydrogels based on this motif, the dipeptide had to be extended by an additional hydrophobic and aromatic residue19 or by the Fmoc (fluorenylmethyloxycarbonyl chloride) moiety.16,20 Fmoc-dipeptides20,21 as well as Fmoc derivatives of single amino acids were found to generate hydrogels22 highlighting the intrinsic tendency of this group to assemble into fibrils. Based on these studies it may be deduced that the intermolecular interactions between non-aromatic dipeptides are insufficient to drive their assembly into fibrils whereas with all hydrophobic tripeptides β-sheet fibrils and their hydrogels can be obtained. Based on this recent evidence concerning the unique stabilizing effect of phenyl–phenyl interactions on peptide assemblies,9,23,24 here we investigated whether the smallest amphiphilic β-pleated motif, with the general sequence Phe-X-Phe, bearing a central X hydrophilic amino acid, could form X-functionalized β-sheet fibril assemblies, as well as hydrogels.
The peptide Phe-Glu-Phe was found, at concentrations up to 20 mM, to completely dissolve in basic solutions (pH > 10.5) and precipitate out of solution under acidic conditions, favoring the protonated state of the Glu side chains. As the assembly into fibrils is highly dependent on the characteristic pKa value of the central Glu residue,21 pH measurements through titrating with hydrochloric acid were carried out with three peptide solutions, 1, 2.5 and 5 mM (Fig. 1a); the pH of the three Phe-Glu-Phe solutions dropped sharply to ∼9.2 and then moderately decreased towards ∼7.5 indicating a pKa at ∼8.3, which can be associated with the peptides' amine termini. This apparent pKa region was indeed not observed in the titrations of the termini-protected peptide analogue Ac-Phe-Glu-Phe-NH2 (Fig. S1, ESI†). The Phe-Glu-Phe solutions showed an additional pKa region between pH ∼ 6 and 4 attributed to the Glu side chains. Moreover, the 2.5 and 5 mM Phe-Glu-Phe solutions showed a positive jump in pH along this pKa region, accompanied by the appearance of turbidity (similar behavior was detected for the 5 mM Ac-Phe-Glu-Phe-NH2, Fig. S1, ESI†).
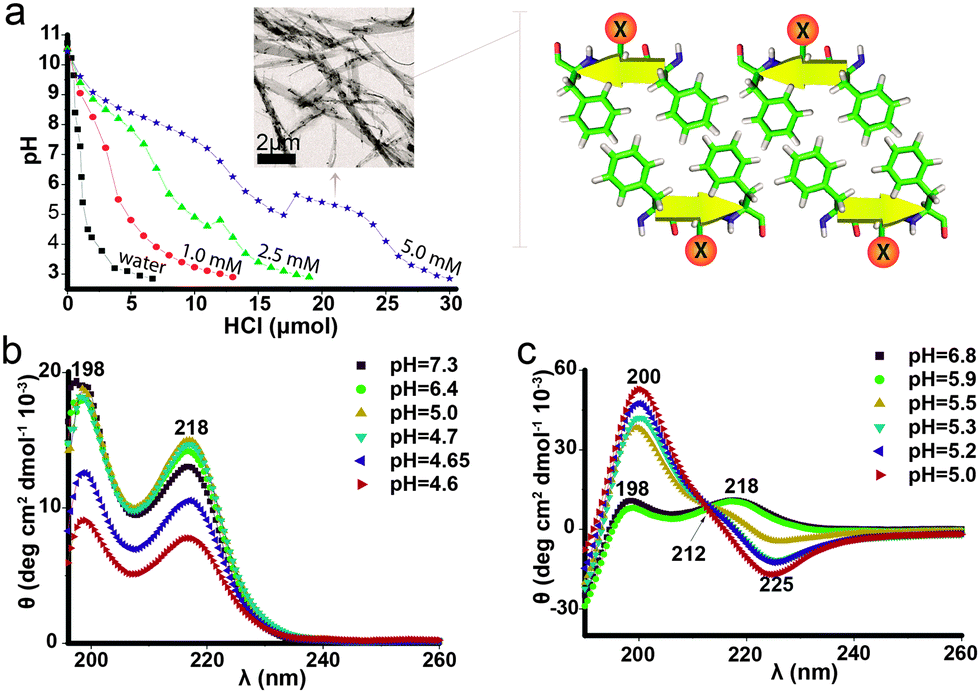 | ||
| Fig. 1 Secondary structure analysis as a function of pH. (a) Titration curves of basified Phe-Glu-Phe, 1, 2.5 and 5 mM solutions and as a reference of deionized water. Inset: TEM micrograph of the 5 mM solution sampled at pH = 5.3 showing assembled peptide ribbons composed of peptide bilayers assembled in an anti-parallel β-sheet (four peptides composing a repeat unit in the bilayer fibrils illustrated schematically on the right). CD spectra of (b) 5 mM Phe-Glu-Phe and (c) 1 mM Phe-(Glu-Phe)2. | ||
Phe-Glu-Phe 5 mM samples at pH 7, below the amine apparent pKa, visualized by TEM (transmission electron microscopy), showed no detectable molecular assemblies, yet at pH 5.3, at the Glu apparent pKa region, elongated ribbons several micrometres in length and up to hundreds of nanometres in width were detected (Fig. 1a inset). Hence, the instantaneous increase in pH, at the apparent Glu pKa region, arises from fast fibrilar growth and elongation accompanied by the withdrawal of protons from the solution to neutralize the charged Glu residues. Phe-Glu-Phe 5 mM solutions were further characterized by CD (circular dichroism) measurements at a range of pH values below and above this apparent pKa. All spectra were found to be highly consistent with two positive peaks at 198 and 218 nm (Fig. 1b) reported previously for Phe amino acid,25 the dipeptide Phe-Phe18 and poly-Phe.26 These two characteristic peaks were attributed to π–π* and n–π* transitions, respectively, as the direct outcome of phenyl rings π-stacking.27,28 Interestingly, the Phe-Glu-Phe CD spectra indicate that such Phe–Phe stacking interactions are maintained both above and below the apparent Glu pKa, where fibril assemblies are already present. As stacking interactions were not observed in previously reported CD measurements of 13 residue peptides that exhibit this dyad motif,29 we were interested in determining whether the five residue version of this motif, i.e. Phe-(Glu-Phe)2, would exhibit the π-stacking interactions. The CD spectra of this peptide showed a transition at pH 5.5–5.9 (at its pKa region, Fig. S1, ESI†), from the Phe–Phe stacking absorption to that of a β-sheet structure (Fig. 1c). Hence, this heptapeptide at high pH shows π-stacking interactions indicating that the charged peptide folds in a manner that allows the stacking of phenyl rings. Then at the pKa region these interactions yielded the cross strand β-sheet hydrogen bonds. This two-state conformational transition is also supported by an isodichroic point at 212 nm (black arrow Fig. 1c). FTIR (Fourier transform infrared spectroscopy) measurements of both the tri- and the hepta-peptides that were first assembled into fibrils and then dried showed spectra with the amide I peak at 1636 and its shoulder at 1694 cm−1, indicative of the anti-parallel β-sheet packing (Fig. S2, ESI†). Previously reported Phe-Phe and Phe-Phe-Phe peptides also showed the FTIR peak at 1630 cm−1, which was attributed to sheet-like conformation.30,31 Phe-Glu-Phe, which was further characterized in solution for β-sheet formation by ThT (thioflavin T) binding assay, already showed increased fluorescence intensity for the lower concentration solutions ∼1 mM which in titration measurements showed almost no detectable pKa region. In contrast the non-aromatic Val-Glu-Val peptide did not show any increase in fluorescence, hence no β-sheet structure at this concentration (Fig. S3, ESI†). These results emphasize the unique contribution of the Phe–Phe interactions detected by the CD measurements of Phe-Glu-Phe, to the assembly of this amphiphilic tripeptide in β-sheet fibrils.
The formation of fibrillar structures by Phe-Glu-Phe at low pH solutions has led us to examine whether at increased peptide concentrations hydrogels could be obtained. Titrations of the peptide solutions to low pH, however, did not result in hydrogel formation but rather in turbidity indicative of peptide aggregation and precipitation. Nonetheless, when peptide Phe-Glu-Phe was dissolved in pure HFIP (hexafluoroisopropanol) and next supplemented with deionized water, stable hydrogels did form (determined by flipping over test tubes and observing flow cessation as well as by rheology measurements, see Fig. S4, ESI†). The HFIP dissolved the peptide and with the addition of water as a co-solvent, the more hydrophilic conditions drove the peptides to form a sufficiently extensive fibril network that stabilized a hydrogel. The hydrogels were obtained at concentrations as low as 0.1% w/v (2.2 mM) in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 (v/v) HFIP:deionized water (pH ∼ 4). This exceptional ability of such a small amphiphilic peptide to form fibrils that stabilize hydrogels points to the prominent contribution of π-stacking interactions between the Phe side chains.
:9 (v/v) HFIP:deionized water (pH ∼ 4). This exceptional ability of such a small amphiphilic peptide to form fibrils that stabilize hydrogels points to the prominent contribution of π-stacking interactions between the Phe side chains.
To assess the contribution of the central hydrophilic amino acid as well as the peptide termini to hydrogel formation, the peptides Phe-Thr-Phe, Ac-Phe-Glu-Phe-NH2 and Phe-Cys-Phe were examined under various conditions to determine whether they form hydrogels. Indeed, these peptides did form hydrogels, although they did so at a higher concentration of 4% w/v. In addition, the positively charged peptide Phe-Lys-Phe was found to completely dissolve under the aforementioned conditions. Yet, by dissolving > 5% of the peptide in 0.1 M KCl at ∼70 °C, followed by cooling the solution to room temperature a hydrogel was stabilized. Observations made by electron microscopy for all these Phe-X-Phe peptides in conditions favoring the assembly and gelation showed a broad range of fibrillar structures. In contrast, the tripeptide Val-Glu-Val did not form hydrogels under any of the conditions described above, emphasizing the essential contribution of the phenylalanine's π–π interactions to the stabilization of the hydrogel assemblies. Further support for the existence of π-stacking interactions in fibrils formed by Phe-Glu-Phe was obtained by fluorescence measurements of 5 mM in 1:1 v/v HFIP:deionized water, which showed the peaks at 279 and 327 nm and their red-shift with time indicative of fibril growth (Fig. S5, ESI†).
To provide further insight into fibril structures, a sample of Phe-Glu-Phe peptide solution was placed on a mica surface and allowed to completely dry before scanning with AFM (atomic force microscopy). High-resolution tip scans (see experimental) acquired in regions between micron-size fibers (Fig. S6a, ESI†) revealed domains of aligned fibrils with persistence lengths extending over hundreds of nanometres (Fig. 2). The smooth edges of the fibril domains appeared in close contact with neighboring domains of similarly aligned fibrils. Interestingly, the fibrils in the two neighboring domains appeared aligned at a close to normal angle relative one another. The average width of the fibrils was found by Fourier analysis and by averaging topography profiles (Fig. S7, ESI†) to be 2.3 nm, suggesting that the width of each fibril is composed of two peptides. In addition, the scanned height 0.9 nm, corresponded well to the dimensions of one layer of the peptide in the pleated conformation (Fig. 2). Such a single layer of the peptides can be induced by the hydrophilic mica substrate. The normally aligned fibril domains may point to π–π stacking interactions between Phe side chains on the edge of one domain that cooperatively affect also the neighboring domain. This type of normally aligned fibril domain was also observed for Phe-Glu-Phe on Si wafer and in samples of the Phe-Thr-Phe peptide on mica (Fig. S6d and e, ESI†).
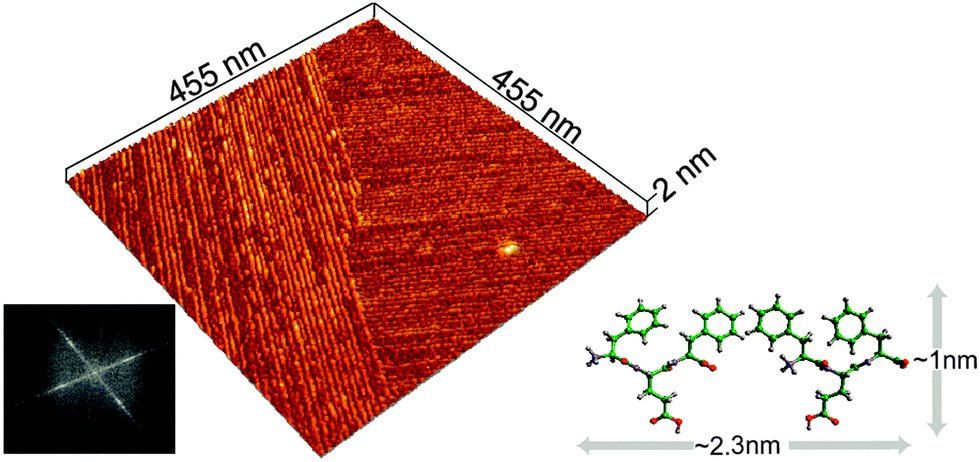 | ||
| Fig. 2 Assembly on a solid surface. AFM scan of dried 2.2 mM Phe-Glu-Phe on mica dissolved in 1:9 (v/v) HFIP:deionized water, showing peptides in fibrillar domains aligned at 85° as determined by the Fourier transform image analysis (bottom left). Cross-sectional topography analysis of the assembled peptides fibrils yielded height = 0.9 and width = 2.3 nm (Fig. S7, ESI†). These fibril dimensions correspond to the height of Phe-Glu-Phe in a β-pleated conformation and to two peptides packed along each fibril's width (illustrated schematically with dimensions for this hypothetical arrangement measured by the molecular modeling program Cerius2, bottom right). | ||
Further evidence for the preferred alignment between fibrils was also obtained by cryo-TEM images acquired in the early stages of Phe-Glu-Phe assembly. To this end, we utilized 0.1% w/v Phe-Glu-Phe in 1:9 v/v HFIP:deionized water solution which was found to form a hydrogel rather slowly, over the course of 1 h. The sample taken immediately after the addition of deionized water to the Phe-Glu-Phe in HFIP solution, (t = 0 min), showed peptide fibrils, micrometres long and 8.7 ± 1.5 nm in width (representing 3 laterally condensed fibril units detected by AFM). Already at this early stage of the assembly process, distinct alignment angles between the fibrils were detected, pointing to π–π stacking interactions extending across interacting fibrils. Detailed analysis of the angles formed between intersecting and stemming fibrils (white and black arrows on Fig. 3a, respectively) indicated a higher tendency to relative orientations at close to the right angle. The 15 minute samples overall showed an even larger number of inter-fibril alignments highly biased towards 80°–90° (Fig. 3b). The termini protected analog Ac-Phe-Glu-Phe-NH2 showed no evidence for inter-fibril alignment but rather packed into wide and thick ribbons indicating that the packing of this peptide, in which the Phe side chains on neighboring peptides along the backbone direction are further apart compared to the free termini peptide, resulted in loss of the cross-fibril stacking interactions (see Fig. S8, ESI†).
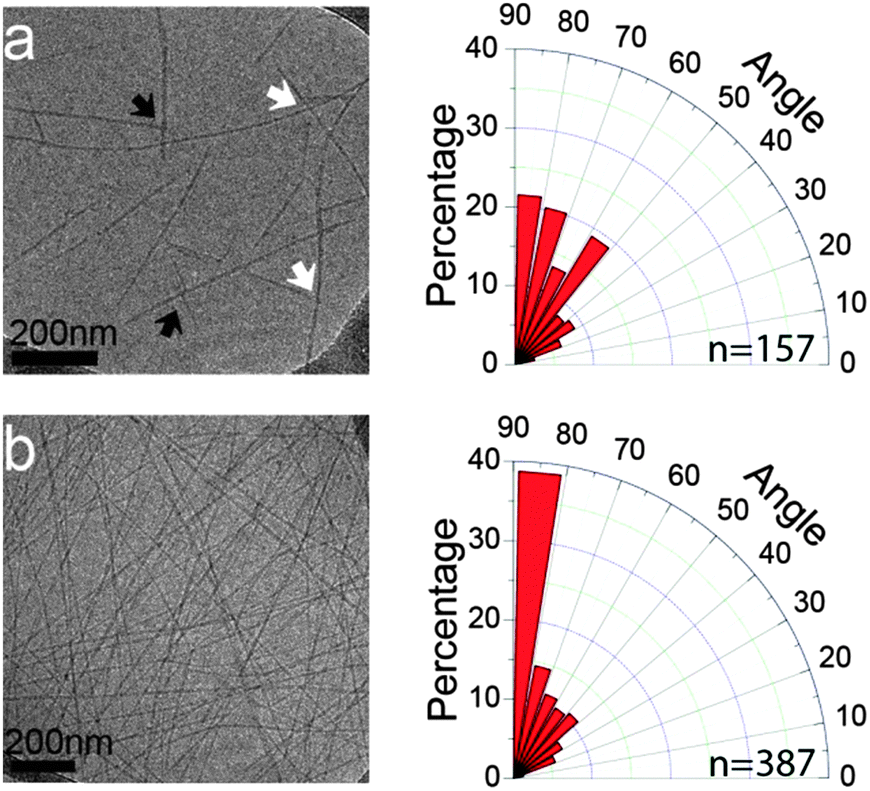 | ||
| Fig. 3 Time dependent assembly as observed by Cryo-TEM. Phe-Glu-Phe assemblies, sampled at 0 and 15 min (a and b, respectively) with the corresponding histogram (to the right of each micrograph) illustrating the angular distribution of interacting (stemming and intersecting, black and white arrows, respectively) fibrils. The number of measured angles for each sample indicated on the angular plot. | ||
The tendency of Phe decorated interacting fibrils to align at close to right angles although not addressed in previous publications can be observed in electron microscopy images of the single Phe fibrils,17 di-phenylalanine nanotubes32 and tri-phenylalanine ribbons.31
The amphiphilic tripeptide Phe-Glu-Phe fibrils exposing the Glu anionic side chains to the surrounding solution were further exploited as adsorbing templates for metal ions. Silver cations could be adsorbed onto the peptide fibrils and be reduced by ascorbic acid to form metallic structures (see Fig. S9a, ESI†) coating the peptide nanofibrils. In addition it was found that gold nanoparticles (citrate stabilized, d = 40 nm) which were first dispersed in a solution of preassembled peptide fibrils became preferentially adsorbed onto fibril edges (see Fig. S9b, ESI†). The gold anchored peptide fibrils were next used to adsorb the silver ions that were further reduced to the metallic form. In this sequential templating procedure single crystalline silver metal nanorods were found to nucleate with their long axes lying parallel to the peptide fibril long axes (Fig. 4a and b).
 | ||
| Fig. 4 Gold embedded silver nanorod formation on peptide templates. (a) TEM image of gold decorated fibrils that were supplemented with silver ion solution and further reduced with an equimolar solution of the reducing agent. (b) STEM image of the sample in (a) with the corresponding electron diffraction pattern. Gold nanoparticles, determined by energy dispersive spectroscopy (EDS), could be detected (darker round shapes) impregnated within single crystalline silver rods elongated in the 〈200〉 direction. | ||
The amphiphilic tripeptides Phe-X-Phe can support π–π interactions between phenylalanine side chains that are interacting across antiparallel neighboring β-strands, to the extent that enables the formation of fibrillar assemblies as well as hydrogels. In a recent publication by the groups of Ulijn and Tuttle the amphiphilic and positively charged tripeptides Lys-Tyr-Phe, Lys-Tyr-Tyr, Lys-Phe-Phe and Lys-Tyr-Trp were shown to form hydrogels following molecular modeling analysis of all naturally occurring tripeptides (203 combinations). Especially it is interesting that the Phe-Lys-Phe peptide reported here that was found to form hydrogel showed identical aggregation propensity values (see Table S5, ESI† in ref. 33) as the Lys-Phe-Phe peptide that also formed hydrogel.33 In the Phe-X-Phe family the π–π interactions were found to traverse the fibrils and adopt preferred orientations between intersecting fibrils and ordered fibril domains. Highly stable fibrils were obtained using Phe-Glu-Phe tripeptides stabilized by the stacking interactions in concert with the hydrogen bonds between the Glu side chains. This peptide exhibited a unique platform of highly ordered and stable fibrils amenable to subsequent functionalization.
Notes and references
- P. W. K. Rothemund, Nature, 2006, 440(7082), 297 CrossRef CAS PubMed.
- S. Zhang, Nat. Biotechnol., 2003, 21(10), 1171 CrossRef CAS PubMed.
- M. Sarikaya, C. Tamerler, A. K. Jen, K. Schulten and F. Baneyx, Nat. Mater., 2003, 2(9), 577 CrossRef CAS PubMed.
- C. P. Barnes, S. A. Sell, E. D. Boland, D. G. Simpson and G. L. Bowlin, Adv. Drug Delivery Rev., 2007, 59(14), 1413–1433 CrossRef CAS PubMed.
- X. Yan, P. Zhu and J. Li, Chem. Soc. Rev., 2010, 39(6), 1877 RSC.
- R. V. Ulijn and A. M. Smith, Chem. Soc. Rev., 2008, 37(4), 664 RSC.
- A. Birman, K. Kjaer, Y. Prior, I. Nevo and L. Leiserowitz, Angew. Chem., Int. Ed., 2010, 49(13), 2354 CrossRef CAS PubMed.
- H. Isenberg, K. Kjaer and H. Rapaport, J. Am. Chem. Soc., 2006, 128(38), 12468 CrossRef CAS PubMed.
- H. Rapaport, Supramol. Chem., 2006, 18(5), 445 CrossRef CAS.
- S. Zhang, F. Gelain and X. Zhao, Semin. Cancer Biol., 2005, 15(5), 413 CrossRef PubMed.
- R. V. Rughani, D. A. Salick, M. S. Lamm, T. Yucel, D. J. Pochan and J. P. Schneider, Biomacromolecules, 2009, 10(5), 1295 CrossRef CAS PubMed.
- A. Aggeli, I. A. Nyrkova, M. Bell, R. Harding, L. Carrick, T. C. McLeish, A. N. Semenov and N. Boden, Proc. Natl. Acad. Sci. U. S. A., 2001, 98(21), 11857 CrossRef CAS PubMed.
- S. Zhang, D. M. Marini, W. Hwang and S. Santoso, Curr. Opin. Chem. Biol., 2002, 6(6), 865 CrossRef CAS.
- E. Gazit, FASEB J., 2002, 16(1), 77 CrossRef CAS PubMed.
- M. Reches and E. Gazit, Science, 2003, 300(5619), 625 CrossRef CAS PubMed.
- A. M. Smith, R. J. Williams, C. Tang, P. Coppo, R. F. Collins, M. L. Turner, A. Saiani and R. V. Ulijn, Adv. Mater., 2008, 20(1), 37–41 CrossRef CAS.
- L. Adler-Abramovich, L. Vaks, O. Carny, D. Trudler, A. Magno, A. Caflisch, D. Frenkel and E. Gazit, Nat. Chem. Biol., 2012, 8(8), 701 CrossRef CAS PubMed.
- L. Adler-Abramovich, M. Reches, V. L. Sedman, S. Allen, S. J. B. Tendler and E. Gazit, Langmuir, 2006, 22(3), 1313 CrossRef CAS PubMed.
- S. Ray, A. K. Das and A. Banerjee, Chem. Commun., 2006, 2816 RSC.
- A. Mahler, M. Reches, M. Rechter, S. Cohen and E. Gazit, Adv. Mater., 2006, 18(11), 1365 CrossRef CAS.
- C. Tang, A. M. Smith, R. F. Collins, R. V. Ulijn and A. Saiani, Langmuir, 2009, 25(16), 9447 CrossRef CAS PubMed.
- S. Sutton, N. L. Campbell, A. I. Cooper, M. Kirkland, W. J. Frith and D. J. Adams, Langmuir, 2009, 25(17), 10285 CrossRef CAS PubMed.
- H. Rapaport, K. Kjaer, T. R. Jensen, L. Leiserowitz and D. A. Tirrell, J. Am. Chem. Soc., 2000, 122(50), 12523 CrossRef CAS.
- H. Rapaport, H. Grisaru and T. Silberstein, Adv. Funct. Mater., 2008, 18(19), 2889 CrossRef CAS.
- A. J. Adler, N. J. Greenfield and G. D. Fasman, Methods Enzymol., 1973, 27, 675 CAS.
- E. Peggion, Bioorg. Chem., 1974, 3(2), 125 CrossRef CAS.
- E. T. Pashuck and S. I. Stupp, J. Am. Chem. Soc., 2010, 132(26), 8819 CrossRef CAS PubMed.
- M. Gupta, A. Bagaria, A. Mishra, P. Mathur, A. Basu, S. Ramakumar and V. Chauhan, Adv. Mater., 2007, 19(6), 858 CrossRef CAS.
- S. Zarzhitsky, H. Edri, Z. Azoulay, I. Cohen, Y. Ventura, A. Gitelman and H. Rapaport, Pept. Sci., 2013, 100(6), 760 CrossRef PubMed.
- M. Reches and E. Gazit, Science, 2003, 300(5619), 625 CrossRef CAS PubMed.
- P. Tamamis, L. Adler-Abramovich, M. Reches, K. Marshall, P. Sikorski, L. Serpell, E. Gazit and G. Archontis, Biophys. J., 2009, 96(12), 5020 CrossRef CAS PubMed.
- A. Handelman, S. Lavrov, A. Kudryavtsev, A. Khatchatouriants, Y. Rosenberg, E. Mishina and G. Rosenman, Adv. Opt. Mater., 2013, 11(1), 875 CrossRef.
- W. J. M. FrederixPim, G. G. Scott, Y. Abul-Haija, D. Kalafatovic, C. G. Pappas, N. Javid, N. T. Hunt, R. V. Ulijn and T. Tuttle, Nat. Chem., 2015, 7(1), 30 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and Fig. S1–S9. See DOI: 10.1039/c4cc09673h |
| This journal is © The Royal Society of Chemistry 2015 |