
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Towards a methanol economy based on homogeneous catalysis: methanol to H2 and CO2 to methanol
E.
Alberico
*ab and
M.
Nielsen
*cd
aIstituto di Chimica Biomolecolare, CNR, tr. La Crucca 3, 07100 Sassari, Italy. E-mail: elisabetta.alberico@cnr.it
bLeibniz-Institut für Katalyse an der Universität Rostock, Albert-Einstein-Strasse 29a, 18059 Rostock, Germany
cDepartment of Chemistry and Chemical Biology, Harvard University, 12 Oxford Street, Cambridge, Massachusetts 02138, USA
dCentre for Catalysis and Sustainable Chemistry, Department of Chemistry, Technical University of Denmark, Kemitorvet 207, 2800 Kgs. Lyngby, Denmark. E-mail: marnie@kemi.dtu.dk
First published on 16th February 2015
Abstract
The possibility to implement both the exhaustive dehydrogenation of aqueous methanol to hydrogen and CO2 and the reverse reaction, the hydrogenation of CO2 to methanol and water, may pave the way to a methanol based economy as part of a promising renewable energy system. Recently, homogeneous catalytic systems have been reported which are able to promote either one or the other of the two reactions under mild conditions. Here, we review and discuss these developments.
 E. Alberico | Elisabetta Alberico obtained her degree in Chemistry (Laurea) from the University of Sassari, Italy, and her PhD under the supervision of Prof. Albrecht Salzer at the Rheinisch-Westfälische Technische Hochschule (RWTH), Aachen, Germany. Since 2001 she holds a permanent position as researcher at the Institute of Biomolecular Chemistry of the National Research Council in Sassari. She is currently a visiting researcher at the Leibniz Institute for Catalysis (LIKAT), Rostock, in the group of Prof. Matthias Beller. Her research interests are in the fields of organometallic chemistry, asymmetric homogeneous hydrogenation and transfer hydrogenation and application of catalytic methods to the synthesis of molecules endowed with biological activity. |
 M. Nielsen | Martin Nielsen obtained his PhD at Aarhus University in Denmark under the supervision of Prof. Karl Anker Jørgensen. He then worked as a postdoc on a Alexander von Humboldt Research Fellowship at the Leibniz Institute for Catalysis (LIKAT), Rostock, in the group of Prof. Matthias Beller. Hereafter, he moved to Harvard University, USA, where he did his next postdoc with Prof. Theodore A. Betley on a Danish Council for Independent Research Fellowship followed by a Marie Curie International Outgoing Fellowship. Currently, he is a senior researcher at the Technical University of Denmark. His research interests include catalysis and sustainable chemistry. |
Introduction
More than 80% of the total worldwide energy consumption is today based on fossil fuels,1 resulting in an increase of anthropogenic CO2 in the atmosphere. Moreover, it is well known that oil sources are likely to be near-depleted by the end of this century if we maintain or increase the present rate of their consumption. Hence, there is an urgency to develop techniques for exploiting alternative energy resources such as wind, sunlight, and biomass in order to generate electricity and/or H2, the latter being equivalent to chemically stored electrical energy. The capability of storing electricity is important because of the intermittent supply of most renewable energy sources.2–4Hydrogen is an ideal fuel as its combustion releases energy and water as the sole by-product. However, mainly because of its physical and chemical properties, hydrogen is not an ideal energy vector: it is a gas with a limited volumetric energy density which, as fuel, especially in the field of automotive applications, has to be either compressed at very high pressure (350–700 bars) or liquefied at very low temperature (−253 °C). It is flammable and can diffuse through several metals and materials.
The chemical storage of hydrogen in solid or liquid compounds from which it can be released as gas through a suitable (and ideally fully reversible) dehydrogenation reaction has been intensively investigated as a possibility to overcome some of these limitations.5 Several criteria have to be considered when evaluating the potential of a chemical substance as hydrogen carrier, among others, its storage capacity (H2 wt%), its volumetric hydrogen content (kg H2 m−3 carrier) and the energy efficiency of the whole process of carrier hydrogenation–dehydrogenation, from hydrogen production to final hydrogen utilization. Methanol belongs to the broader class of liquid storage compounds which includes liquid organic hydrogen carriers (LOHC i.e. methylcyclohexane and N-ethylperhydrocarbazole),6a alcohols6b–d and formic acid.6d,e It is a key platform chemical for existing fuel and chemical infrastructures and contains 12.6% w/w hydrogen, which can be released through aqueous reforming.7 Heterogeneous catalysts, which promote this reaction7 and the reverse one, the conversion of CO2 and H2 to MeOH,8 operate at high temperatures (>200 °C) and/or applied pressures (>25 bar). Indeed the high temperature required for dehydrogenation is one of the main factors which so far has made MeOH less suitable as energy carrier in the field of portable applications. Therefore, developing milder routes for these two conversions is highly desirable in order to reach a viable H2 energy system based on a CO2–MeOH cycle. Moreover, the carbon dioxide released in the process and from any other source might be hydrogenated back to methanol using hydrogen obtained from renewable sources, ideally from water electrolysis powered by solar energy, thus completing a carbon neutral cycle.
The advantages and limitations of a methanol based economy, how far this is from being implemented, especially in relation to technologies currently available for carbon dioxide capture and sequestration and water electrolysis for hydrogen production, have been reviewed in excellent monographs and articles and the reader is referred to them for an in-depth acquaintance with the topic.9
In this review we would like to draw the readers' attention to recent reports concerning the development and application of homogenous catalysts for MeOH dehydrogenation (≤95 °C and atmospheric pressure) and CO2 hydrogenation to MeOH (≤145 °C and ≤60 bars) which are active under comparatively mild conditions.
Looking into the mechanism of a homogeneously catalysed CO2/MeOH hydrogenation/dehydrogenation system, it becomes evident that a few organic intermediates must exist alongside this stepwise transformation. As shown in Scheme 1, the generally proposed MeOH dehydrogenation pathway by homogeneous catalysis commences with the formation of formaldehyde, which is then converted to formic acid promoted by a H2O molecule. The final step is CO2 production from formic acid. In each of these three steps, a H2 molecule is liberated. The CO2 hydrogenation pathway is envisioned to follow the reversed sequence of the same reaction steps.
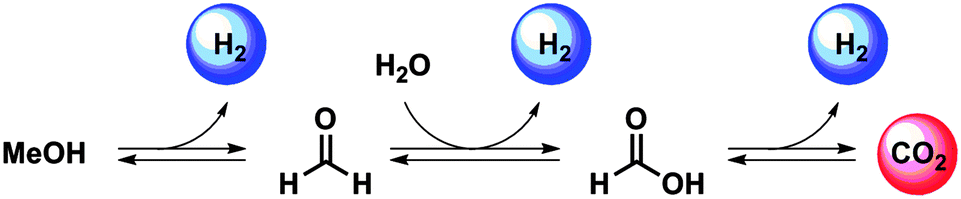 | ||
| Scheme 1 Schematic representation of the steps that constitute the reversible MeOH dehydrogenation and CO2 hydrogenation promoted by homogeneous catalysts. | ||
In the absence of water and in the presence of suitable catalysts, methanol may decompose to hydrogen and formaldehyde (the latter may further react to afford other products, depending on the catalyst and the reaction conditions) (eqn (1)) or be fully dehydrogenated to hydrogen and carbon monoxide (eqn (2)). The catalysts which were found able to promote such processes in the absence of a hydrogen acceptor in almost all cases were applied with much greater success to the dehydrogenation of ethanol and iso-propanol, the latter being often the substrate of choice to test new catalysts, because of the more favourable thermodynamics and the possibility of a higher operational temperature.6b–d,10,11
 | (1) |
 | (2) |
Methanol dehydrogenation
Partial methanol conversion
The first examples of homogeneously catalysed methanol dehydrogenation were published approximately 30 years ago. However, because reactions were carried out in the absence of water, the possibility for the full conversion of methanol to CO2 and three H2 was precluded. Yet they will be briefly discussed here as they set the very early stage for future developments towards the successful sustainable production of hydrogen from alcohols.Saito could show that catalyst [Ru(OAc)(Cl)(PEtPPh2)3], prepared in situ from [Ru2(OAc)4(Cl)] and PEtPPh2, is competent for the dehydrogenation of methanol to formaldehyde at reflux with an initial TOF of 0.96 h−1 and a total TON of 34 over 90 hour reaction time (Table 1, entry 1).12a,c An optimum of two equivalents of acetic acid to ruthenium is required to achieve this result as compared to TOF 0.60 h−1 without any. While formaldehyde was the only product in the early stage of the reaction, at higher conversion both methylformate (the product of a Tishchenko-like dimerization of formaldehyde) and dimethoxymethane (formaldehyde dimethyl acetal) could eventually be detected in solution. No carbon monoxide or methane contaminants were detected in the gas phase.
| Entry | Catalyst (mmol) | MeOH (mL) | Additive (mmol) | T (°C) | Time (h) | H2 (mmol) | Yielda (%) | TOFb (h−1) | TONc | Products | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| n.r. not reported.a The yield is referred to methanol and is calculated assuming that each mmole of converted methanol has been dehydrogenated to formaldehyde and hydrogen.b Average TOF referred to overall reaction time.c TON is referred to hydrogen production.d Because a range of catalyst concentrations is given, it is not possible, based on reported TOF and TON, to estimate the amount of hydrogen evolved and yield thereof.e Initial TOF. | |||||||||||
| 1 | [Ru(OAc)(Cl)(PEtPPh2)3] | 400 | CH3COOH | 65 | 90 | 3.4 | 0.034 | 0.38 | 34 | HCHO, HCOOCH3, H2C(OCH3)2 | 12a and c |
| 0.1 | 0.2 | ||||||||||
| 2 | [Ru(Cl)2(PPh3)3] | 10 | — | 150 | 18 | 5.2 | 2.1 | 3.6 | 65 | HCOOCH3, H2C(OCH3)2 | 13 |
| 0.08 | |||||||||||
| 3 | [Ru(H)2(N2)(PPh3)3] | 5 | NaOH | 150 | 2 | 6.4 | 12.8 | n.r. | 15 | ||
| 1–5 × 10−3 | 5 | ||||||||||
| 4 | [Ru(H)2(PPh3)4] | 5 | NaOH | 150 | 2 | 7.5 | 15 | n.r. | 15 | ||
| 1–5 × 10−3 | 5 | ||||||||||
| 5 | RuCl3·3H2O | 200 | MeONa | 79 | n.r. | — | — | 1.68e | — | HCOONa | 12d |
| 1 | 610 | ||||||||||
Almost at the same time Maitlis reported the dehydrogenation of methanol to methylformate and dimethoxymethane catalysed by [Ru(Cl)2(PPh3)3] at 150 °C achieving a total TON = 65 for hydrogen evolution over 18 hours, at which time activity ceased likely due to formation of inactive [(PPh3)2Ru(CO)(μ-H)(μ-Cl)2(CO)(PPh3)2], which was isolated from the reaction mixture (Table 1, entry 2).13
Cole-Hamilton showed that the rate of hydrogen evolution from methanol over 2 h at 150 °C could be greatly improved by performing the reaction in the presence of a 10 fold excess of base (NaOH) as to the catalyst when using preformed [Ru(H)2(N2)(PPh3)3] (TOF = 6.4 h−1, Table 1, entry 3) or [Ru(H)2(PPh3)4] (TOF = 7.5 h−1, Table 1, entry 4).15 Notably, RuCl3 with no ancillary ligand is able to promote methanol dehydrogenation if sodium methoxide is added to the reaction mixture, thus showing the critical role played by the base.12d With 20% sodium methoxide the solution boiling point was raised from 65 °C to 79 °C bringing about an 84 fold increase of the initial turnover rate from 0.02 h−1 without base to 1.68 h−1 (Table 1, entry 5). The accelerating effect was however mostly ascribed to the high base concentration, since the catalytic activities at sodium methoxide concentrations below 5% (boiling point of the solution 66.8 °C) were far lower than those extrapolated from the Arrhenius plot.
Table 1 summarizes and compares the activity of complexes which were able to promote, although with poor activity, the thermal decomposition of methanol.
Other ruthenium based catalysts, namely {[Ru(SnCl3)5(PPh3)](Et4N)3},16a {Ru(SnCl3)2[P(OMe)3]3},16b and [(η5-C5H5)Ru(SnF3)(PPh3)2]16c–e are also able to dehydrogenate methanol with liberation of hydrogen. These catalysts are not included in Table 1 as the main focus was, in these cases, on the production of acetic acid, or rather its methyl ester, from sole methanol, thus circumventing the use of iodide and carbon monoxide as otherwise required by the Monsanto process.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 MeOH/acetone solution at reflux under photoirradiation with a 400 high-pressure mercury lamp affording a maximum TOF of 156 and 130 h−1 respectively.18 Neither catalyst was active in the dark and the presence of acetone, acting as sensitizer, boosted the activity of both. [IrH(SnCl3)3]3−, either preformed or generated in situ under photoirradiation from IrCl3·3H2O, SnCl2·2H2O and LiCl, could also catalyse the dehydrogenation of methanol to hydrogen (with trace amounts of methane) and formaldehyde dimethylacetal at reflux, upon exposure to a high pressure mercury lamp.19
:1 MeOH/acetone solution at reflux under photoirradiation with a 400 high-pressure mercury lamp affording a maximum TOF of 156 and 130 h−1 respectively.18 Neither catalyst was active in the dark and the presence of acetone, acting as sensitizer, boosted the activity of both. [IrH(SnCl3)3]3−, either preformed or generated in situ under photoirradiation from IrCl3·3H2O, SnCl2·2H2O and LiCl, could also catalyse the dehydrogenation of methanol to hydrogen (with trace amounts of methane) and formaldehyde dimethylacetal at reflux, upon exposure to a high pressure mercury lamp.19
Aqueous methanol dehydrogenation to hydrogen and carbon dioxide: methanol reforming
In 2013, the first examples of homogeneously catalysed complete methanol dehydrogenation to CO2 and three molecules of H2 were disclosed.11 | (3) |
 | (4) |
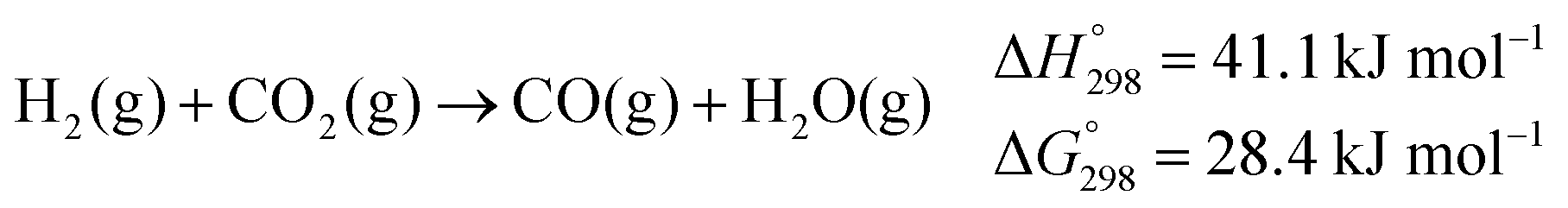 | (5) |
Lower operational temperatures are highly desirable, especially in view of the application of methanol as fuel in Reformed Methanol Fuel Cells (RMFC) for mobile applications: here methanol reforming takes place on board to directly fuel the cell with hydrogen. Because of the high temperature required by heterogeneous catalysts, the overall efficiency of state of the art devices does not exceed 40%. Furthermore high temperatures favour formation of CO20 which, already at few ppm concentration, may poison the precious electrocatalyst at the cell anode. It should be mentioned that another possible source of CO contamination under the conditions of steam reforming is the reverse water gas shift reaction (eqn (5)).
A very first report concerning the possibility of applying an homogeneous catalyst to the thermal dehydrogenation of alcohols in the presence of water was published by Cole-Hamilton and relied on the use of [Rh(bipy)2]Cl as catalyst.21 The rationale behind the use of water is the possibility to open a new decomposition path according to eqn (3) and Scheme 1, which is thermodynamically less demanding than simple dehydrogenation as to eqn (1) or (2). Reactions were carried out using a 95:5 v/v solution of alcohol/H2O, in the presence of NaOH (0.25–5 × 10−3 mol) with [Rh(bipy)2]Cl (5 × 10−6 mol) at 120 °C in a closed system for 3 h. Alcohols other than methanol, such as ethanol, isopropanol and butane-2,3-diol, afforded almost a 100 fold higher rate of hydrogen production compared to methanol, which had a TOF = 7 h−1 (Table 2, entry 1). No detail though was provided for the gas- and liquid-phase composition of the system after reaction; however, in the case of ethanol, beside H2 in the gas phase and ethanal and its condensation products in the liquid phase, CH4 and CO2 in a 1:1 ratio were detected (the latter after acid addition to the solution) showing the ability of the catalyst to promote intermediate aldehyde decarbonylation and formation of CO2. 25 years later, almost at the same time, two groups, Grützmacher's22 and Beller's,23a reported the efficient aqueous dehydrogenation of methanol to hydrogen and carbon dioxide below 100 °C promoted by homogenous ruthenium catalysts (Fig. 1, 1 and 2, respectively), with almost no trace of CO contamination, setting the stage for a foreseeable application in RMFC. Very recently Milstein has shown that also ruthenium catalyst 3 (Fig. 1) is able to promote the same transformation.24 A common feature of these catalysts is that they possess multidentate ligands which confer improved thermal stability to the resulting metal complexes while blocking three (like in 2 and 3) or four (like in 1) of the possible metal coordination sites; this is particular useful in order to hamper the possible decarbonylation of formaldehyde arising from methanol dehydrogenation.
| Entry | Catalyst (10−3 mmol; ppm to MeOH) | MeOH (mmol) | H2O (mmol) | Solvent (mL) | Additive (mmol) | T (°C) | Time (h) | H2 (mmol) | Yielda (%) | TOFb (h−1) | TON | Conv. (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n.r. not reported.a The yield is referred to methanol and is calculated assuming that each mmol of converted methanol has been fully dehydrogenated providing three mmoles of hydrogen.b The TOF is the average value measured over the entire reaction time.c Data extracted from ref. 21, Table 1, experiment performed in a closed vessel.d Data extracted from ref. 22, SI pag. 14, base-free experiment.e Data extracted from ref. 23a, SI pag. 21, long-term experiment with focus on catalyst stability.f Because no internal standard was used, it is not possible to provide conversions. For each experiment it is however possible to provide yields relative to the combined amount of water and base, which are the limiting reagent, according to supplementary equation 4 in ref. ref. 23a: entry 3, yield 27%; entry 4, yield 20%; entry 5, yield 1%; entry 6, yield 3.4%.g Data extracted from ref. 23b, Table 1, entry 11.h Data extracted from ref. 23c, SI pag. 6, long-term experiment.i Data extracted from ref. 24, SI pag. 2 Table S1.j The TON is the total one over three consecutive uses (recycling) of the organic phase containing the catalyst; likewise, the yield has been calculated based on the total amount of methanol, 3 × 20 mmol, used over the three experiments. | ||||||||||||
| 1 | [Rh(bipy)2]Clc | 111 | 27.8 | — | NaOH | 120 | 3 | 0.105 | 0.03 | 7 | 21 | n.r. |
| 5; 45 | 5 | |||||||||||
| 2 | [K(dme)2][RuH(trop2dad)] 1d | 2 | 2.6 | THF | — | 90 | 10 | 5.0 | 84.0 | 54 | 540 | 90 |
| 10; 500 | 1 | |||||||||||
| 3 | [Ru(H)(Cl)(CO)(PNPiPr)] 2be | 890 | 222 | — | KOH | 91 | 576 | 311 | 11.6 | 613 | 353409 |
|
| 0.88; 1 | 320 | |||||||||||
| 4 | [Ru(H)(Cl)(CO)(PNPiPr)] 2b | 890 | 222 | — | KOH | 91 | 24 | 231 | 26 | 64 | 192 | |
| 150; 150 | 320 | |||||||||||
| 5 | [Ru(H)(Cl)(CO)(PNPPh)] 2a | 198 | 111 | — | NaOH | 72 | 2 | 3.4 | 0.57 | 34 | 68 | |
| 49.3; 250 | 1 | |||||||||||
| 6 | [Fe(H)(BH4)(CO)(PNPiPr)] 2cg | 222 | 55.5 | — | KOH | 91 | 46 | 9.8 | 1.5 | 214 | 9834 | |
| 1; 4.5 | 80 | |||||||||||
| 7 | [Ru(H)(BH4)(CO)(PNPPh)] 2dh | 222 | 55.5 | Triglyme | — | 93.5 | 192 | 42.8 | 6.4 | 22 | 4286 | 6.4 |
| 5; 22.5 | 4 | |||||||||||
| [Ru(H)2(dppe)2] | ||||||||||||
| 5; 22.5 | ||||||||||||
| 8 | [Ru(H)(Cl)(CO)(NNPtBu)] 3i | 20 | 111 | Toluene | KOH | 100–105 | 648 | 131 | 72.8j | 44 | 28661j |
79 |
| 5; 250 | 2 | 40 | ||||||||||
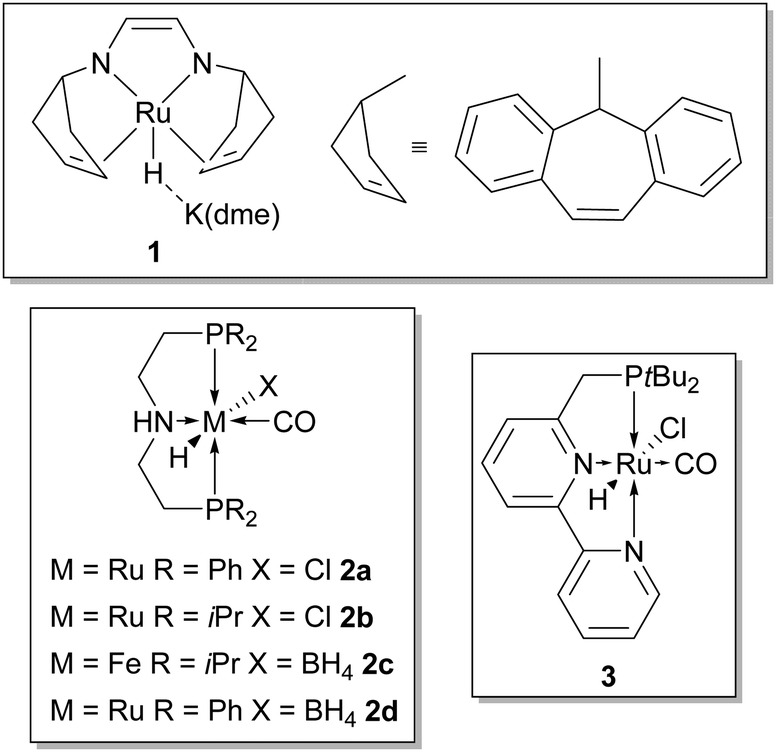 | ||
| Fig. 1 Homogeneous catalysts which promote the aqueous dehydrogenation of methanol to hydrogen and carbon dioxide. | ||
Most importantly, the abstraction of hydrogen from the alcohol relies on a cooperative mechanism involving both the metal and the ligand, the latter being therefore termed “non-innocent”.25 Experimental evidence suggests that, when catalysed by such complexes, exhaustive dehydrogenation of methanol might proceed through three consecutive steps: dehydrogenation of methanol to hydrogen and formaldehyde (eqn (1)), dehydrogenation of the gem-diol arising from the reaction of formaldehyde with water (eqn (6)),26 final dehydrogenation of the resulting formic acid to hydrogen and carbon dioxide (eqn (7)). This sequence is identical to the general reaction pathway shown in Scheme 1.11
 | (1) |
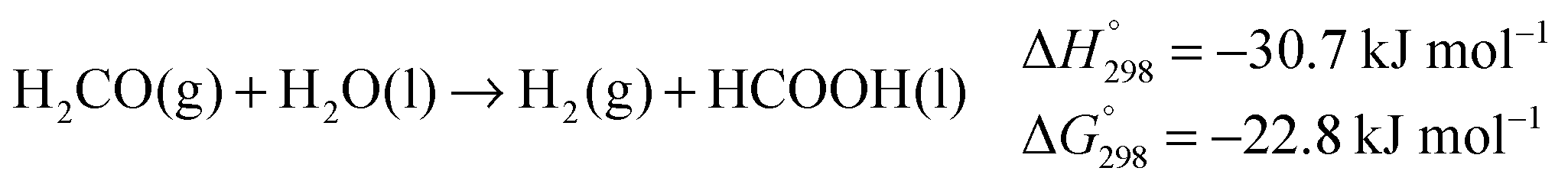 | (6) |
 | (7) |
 | (8) |
000 h−1. Based on the isolation and characterisation of some relevant intermediates and following investigation into their reactivity, the authors have suggested the catalytic cycle illustrated in Scheme 2. The hydride 1 does not react appreciably with alcohols but it does with H2O (or carboxylic acid) to generate a very reactive neutral Ru species 4 (characterized by NMR), with release of one molecule of H2 and one equivalent of base. This is therefore considered to be the first step of the catalytic cycle. Addition of the OH bond of a substrate molecule, either methanol (this case is illustrated in Scheme 2), gem-diol (arising from hydration of formaldehyde), or formic acid, across the Ru–N bond affords the enamido complex 5. This activates the α-CH of the coordinated substrate which is then transferred to the ligand to give an imino–amino Ru(0) complex 6 after release of the dehydrogenation product. 6 can reiterate this process with a second substrate molecule to provide diamino complex 7 from which one equivalent of hydrogen is then released, aided by base and heat, to regenerate 1. Intermediates 7 and its adduct with benzaldehyde have been synthesized via stoichiometric reactions and characterized by X-ray analysis. Species 4 and 6 are competent for dehydrogenation of either MeOH, formaldehyde gem-diol or formic acid (in Scheme 2 the dehydrogenation of formic acid as by 4 to afford 6 and CO2 is shown in grey). This is an example of cooperative catalysis in which the ligand is both redox- and chemically active, supporting the metal shuttle between oxidation states 2 and 0 and adding the hydrogen termini abstracted from the substrate which are then released as hydrogen gas.
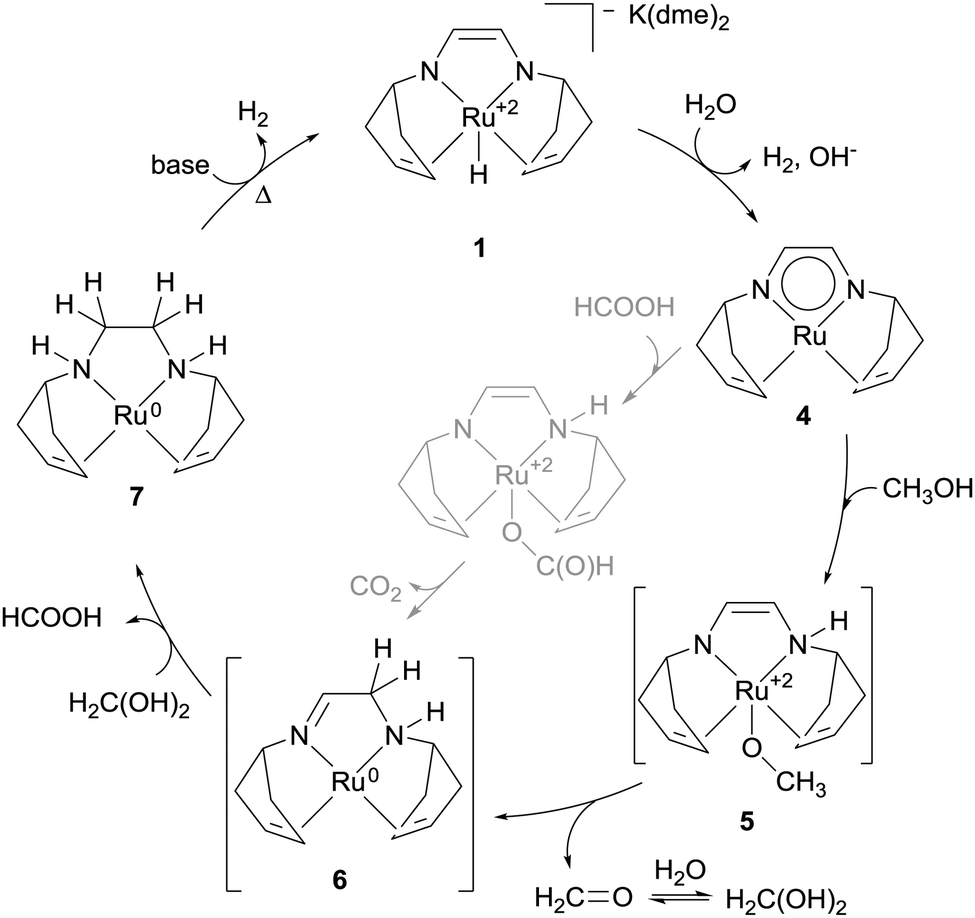 | ||
| Scheme 2 Proposed catalytic cycle for the aqueous dehydrogenation of MeOH promoted by [K(dme)2][RuH(trop2dad)] 1. | ||
Ruthenium catalysts [Ru(H)(Cl)(CO)(PNPR)] (PNPR = bis[(2-diphenylphosphino)ethyl]amine if R = Ph; bis[(2-diisopropyl-phosphino)ethyl]amine if R = iPr) 2a and 2b, respectively supported by aliphatic PNP ligands, originally developed for ester hydrogenation27a (2a) and acceptorless dehydrogenation of alcohols other than methanol27b,c (2b), were identified as competent catalysts for the aqueous reforming of methanol under basic conditions.23a Several parameters were found to affect the activity and productivity of the catalysts: type and amount of base, temperature and water content. The reaction turned out to be favoured by a high base content with KOH as base of choice, high temperature and low water amount, yet activity was observed already at 75 °C at 0.1 M base concentration and even in the presence of 90% v/v of water. Under optimised conditions, neat MeOH (10 mL), KOH = 8 M, catalyst 2b (1.58 μmol, 6.4 ppm), at 95 °C, an average TOF = 4723 h−1 was achieved over three hours. The evolved gas was pure hydrogen with almost no trace of CO contamination (below 1 ppm) while CO2 was trapped as carbonate. By performing a longer term experiment in the presence of less base and a higher amount of catalyst (MeOH 8 mL, H2O 2 mL, NaOH = 0.1 M, catalyst 2a 49.3 μmol, 250 ppm, 72 °C) a steady-state condition was achieved within hours when CO2 could escape the reaction medium and the expected H2/CO2 3:1 ratio was observed. Complex 2b proved extremely robust as hydrogen evolution continued for three weeks affording a TON = 350000 with an average TOF = 200 h−1 (using 1.0 ppm 2b in a 9:1 ratio of MeOH/H2O), measured over the last 24 h (Table 2, entry 3). Parallel to base consumption and the ensuing decrease of boiling temperature, in fact, a decrease in activity was observed.
Compound 2b is merely a catalyst precursor which needs one equivalent of base to be converted into amido complex 8 (Scheme 3). The amido complex has been reported to react with iso-propanol or hydrogen gas to afford dihydride 9:27b the protic hydrogen of the –OH moiety in the alcohol would add to the basic nitrogen of the ligand in 8 whereas the hydride of the α-CH would be transferred to the metal centre. It was therefore suggested that 8 and 9 are the two key species between which dehydrogenation of methanol, formaldehyde gem-diol(ate) and formate (which was detected in solution under catalytic-like conditions) occurs with ensuing hydrogen evolution.23a
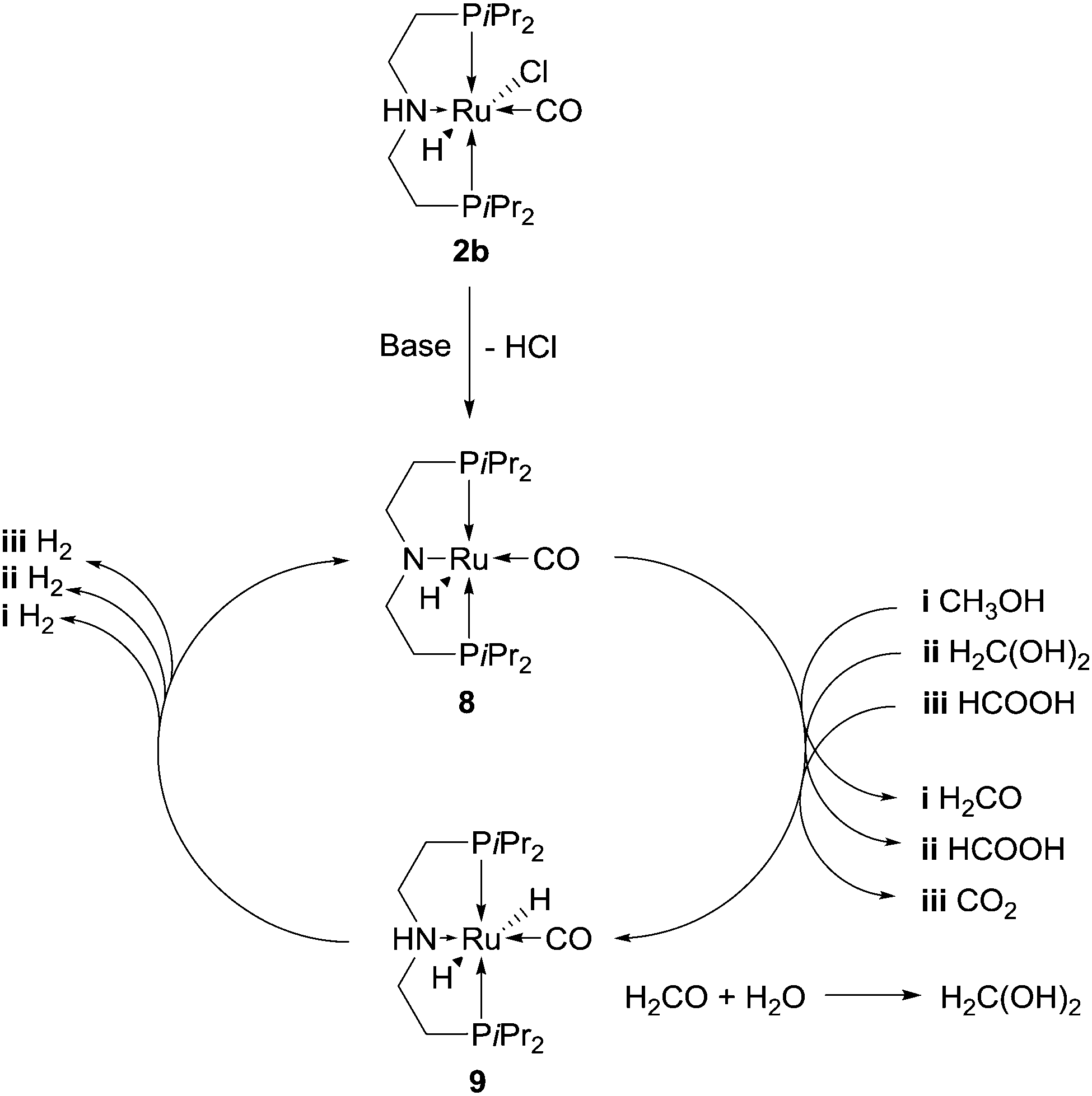 | ||
| Scheme 3 Oversimplified proposed catalytic cycle for the aqueous dehydrogenation of MeOH promoted by [Ru(H)(Cl)(CO)(PNPiPr)] 2b. | ||
DFT calculations have provided support to the proposed metal–ligand cooperative mechanism, highlighting the decisive role of the protic solvent, either water or methanol, in relaying a proton shuttle from the ligand nitrogen to the ruthenium hydride for the formation of a ruthenium dihydrogen complex from which hydrogen is eventually released.28 However it has been suggested, based on calculations, that formate decomposition to hydrogen and carbon dioxide could take place through both inner- and outer-sphere mechanisms.28b According to the calculated free-energy landscape of the overall reaction, dehydrogenation of gemdiol(ate) has the lowest rate constant and dehydrogenation of formate the highest among the three steps,28b which is in line with the experimental observation that free formate, but neither formaldehyde nor gem-diolate, was observed in solution The actual rate of each step would however depend on the relative concentration of the species involved (“steady-state regime”).
The outer-sphere, cooperative mechanism does not require a change in the formal oxidation state of the central metal to trigger substrate reactivity and is hence open to 1st row transition metals. These metals usually prefer one-electron redox changes, making it more challenging to stabilise and maintain the catalyst functionality, should the reaction proceed according to classical oxidative addition, insertion and reductive elimination. Indeed the analogous iron complex 2c was synthesized and proved effective in aqueous methanol reforming.23b,29 Under optimised conditions, MeOH/H2O 9/1 v/v (10 mL), KOH = 8 M, catalyst 2c (4.18 μmol, 18.8 ppm to MeOH), at 91 °C, a TOF = 702 h−1 was achieved in the first hour, and a TON = 6270 over 43 hours. By adding 5 equivalents of free ligand to the solution it was possible to extend the catalyst lifespan over almost five days, affording, under otherwise identical conditions, a TON = 9184 (111 h) (Table 2, entry 6). Although based on a far cheaper and abundant metal, the iron system is clearly less active than the ruthenium one, as correctly anticipated by theoretical calculations.28a However if the turnover activity per unit cost, recently suggested by Crabtree as a further “green” parameter to judge a catalyst,30 then the advantage brought about by iron is easily apparent and should stimulate further efforts towards the development of 1st row transition metal based catalysts.
One drawback associated with the use of catalysts 2 in methanol reforming is the need for a high base concentration. The role of the base is many-fold: on the one hand it allows, by means of the salt effect, to increase the operational temperature and thus both the thermodynamic driving force and the rate of reaction; it provides an additional free energy advantage because of the acid–base reaction with the generated CO2; it is likely necessary to regenerate the catalytic active species from dormant resting states.31 Furthermore, because free formate is actually detected in solution when using catalysts 2 under basic conditions, it was suspected that its dehydrogenation might be slow at low methanol conversion. Therefore it was envisioned that either the addition of a higher catalyst amount, of a second catalyst which had proved effective in formic acid decomposition or of a high-boiling solvent might contribute to achieve methanol reforming even in the absence of base. Indeed, after catalyst and solvent screening, it was found that the combined use of catalyst Ru–MACHO–BH42d, which does not need an exogenous base to be activated, and [Ru(H)2(dppe)2] (dppe = 1,2-bis(diphenylphosphino)ethane) 10, in the presence of triglyme (MeOH 9 mL, H2O 1 mL, triglyme 4 mL, 2d 5 μmol, 10 5 μmol) allows to dehydrogenate methanol at 93.5 °C with an average TOF = 93 h−1 over 7 hours.23c The catalyst system stayed active for 8 days affording an overall TON of 4286 (Table 2, entry 7). Interestingly the two catalysts operate in a synergistic manner, as their combined activity is superior to the sum of the activities displayed by each of them alone in the same transformation.
Very recently Milstein et al. have added catalyst 3 (Fig. 1) to those amenable to methanol reforming: under optimised conditions, MeOH 20 mmol, 0.8 mL, H2O 111 mmol, 2 mL, toluene 2 mL, KOH 40 mmol, catalyst 3 5 × 10−3 mmol, 250 ppm to MeOH, Tset = 115 °C (effective T = 100–105 °C), a 70% yield of hydrogen was achieved over 9 day reaction time, corresponding to an average TOF = 45 h−1.24 The base is necessary in order to generate the true catalytic species 11 from 3 (1 equivalent to the catalyst) and, according to the authors, to generate formate from methanol. In fact, while no hydrogen is produced from methanol in the absence of base, conversion of formic acid to hydrogen and CO2 with the same catalyst is instead possible. Toluene is likely necessary to increase catalyst solubility as no activity was detected in the absence of an organic solvent. Furthermore, the presence of a biphasic system allows for the easy recovery and recycling of the catalyst: the organic phase of the experiment under optimised conditions was separated and used twice for the reforming of new batches of methanol, showing undiminished activity and affording, after an overall reaction time of almost 1 month, a remarkable TON of about 29000 (Table 2, entry 8). Like in the cases of catalysts 1 and 2, catalyst 3, or rather its activated form 11 (Scheme 4), relies on ligand–metal cooperation to abstract hydrogen from the substrate, either methanol, formaldehyde gem diolate or formate.25f The dearomatised complex 11, resulting from deprotonation at the pyridinylmethylenic carbon, can in fact regain aromatisation at the pyridine moiety by interacting with the substrate, adding a “proton-like” hydrogen to the side-arm of the ligand and a “hydride-like” hydrogen to ruthenium (Scheme 4). The resulting dihydride 12 then releases hydrogen.
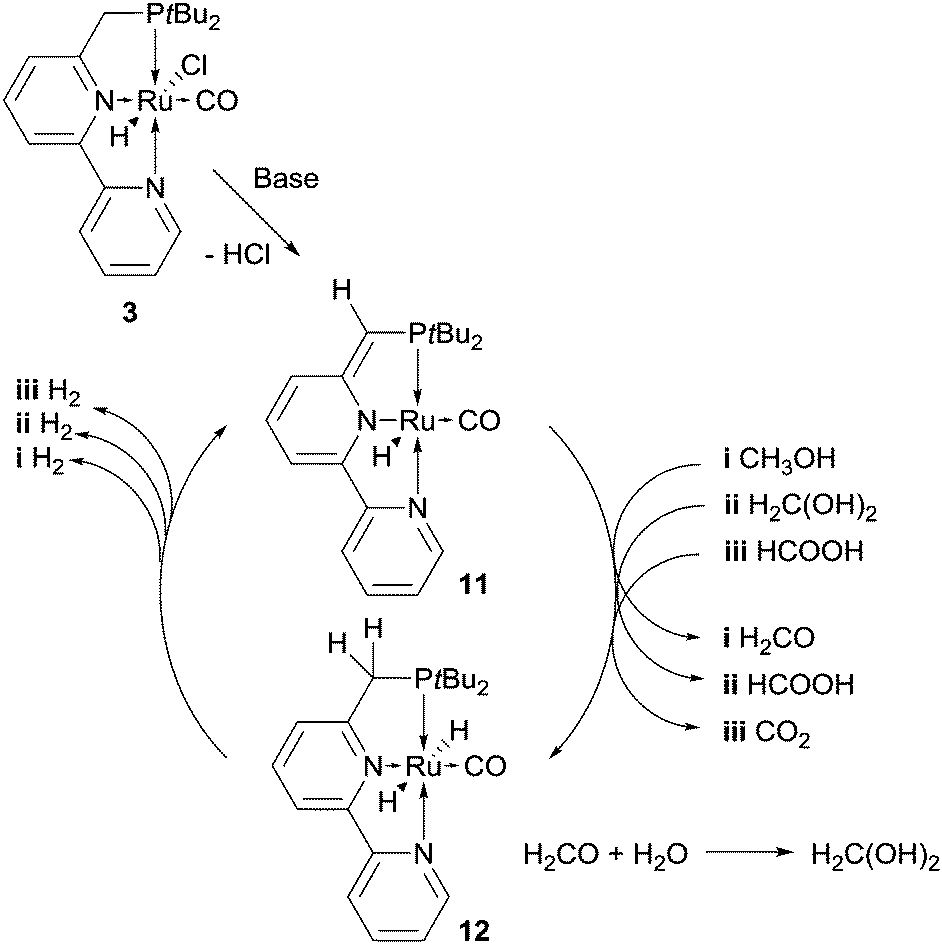 | ||
| Scheme 4 Proposed key-intermediates in the aqueous dehydrogenation of MeOH promoted by 3, through aromatisation–dearomatisation of the pyridine pincer based ligand. | ||
Very recently Crabtree et al. have reported the acceptorless dehydrogenation of methanol using homogeneous iridium bis(N-heterocyclic) carbene catalysts: under optimised conditions (dry MeOH, 3 mL, KOH 6.7 M, [Ir] 1 μmol, 91 °C) using a bis(N-heterocyclic) carbene complex stabilised with CO ligands an overall TON of 2900 over 15 hours was achieved which could be increased to 8000 over 40 hours.32 However, by measuring the relative amounts of HCOOK and K2CO3 produced, it turned out that only 5% of the converted MeOH had been exhaustively dehydrogenated to CO2 (trapped as carbonate) and H2, the rest being transformed into formate. Yet the catalysts are noteworthy in that they can operate in air with undiminished efficiency and rely on simpler and cheaper ancillary ligands as to catalyst 1–3.
For the sake of comparison, selected data concerning the homogeneous catalysts able to promote aqueous reforming of methanol are collected in Table 2. For catalyst 2b, two further experiments with a higher catalyst loading (Table 2, entries 4 and 5) are reported.
Comparison between data reported in Table 1 for methanol decomposition and in Table 2 for aqueous methanol dehydrogenation clearly show that ruthenium catalysts bearing “non-innocent” pincer ligands (Table 2) have proved so far more active in methanol dehydrogenation than those whose ancillary ligands are not directly involved in substrate transformation (Table 1). This is probably due to the higher energy demanding events taking place at the metal in the latter case, like labilisation of a coordinated ligand or change in the oxidation state which is not necessary for the former, or if so, like in the case of catalyst 1 developed by Grützmacher et al., is supported by the ligand.22
Among homogeneous catalysts for aqueous methanol dehydrogenation, catalyst 2b stands out for the high average TOF of 613 h−1 over 23 days (although the average TOF during the last 24 hours of the reaction was 200 h−1, which is however still higher than the average TOF for catalysts 1 and 3). Yet yield is quite limited if compared to those recorded with 1 and 3: use of a higher catalyst loading serves to improve it although at the expenses of activity (TOF) (compare entries 3 and 4 in Table 2). Unlike others in Table 2, catalysts 2 do not require any added solvent to allow for better catalyst solubilisation and are tolerant of various methanol/water ratios. The robustness of aliphatic PNP pincer ruthenium catalysts has been further demonstrated in the dehydrogenation of higher alcohols, such as biomass derived ethanol33 and glycerol,34 a by-product of biodiesel production, selectively affording, in the last case, valuable lactic acid. Catalyst 2a could be applied directly to industrial batches of bioethanol and glycerol with undiminished, if not superior, efficiency compared to solutions of the pure substrates.
The main drawback of catalysts 2 and 3 is the requirement of high base loads, whereas catalyst 1 operates smoothly without. At the present stage of development, it has been pointed out that the capturing of CO2 from small and widespread sources may be technically feasible but economically prohibitive and CO2 should be eventually captured from the atmosphere itself.9b On the other hand, the high base concentration required by catalysts 2 and 3 might be advantageously exploited to sequestrate CO2 for its later recycle, for example by hydrogenation of the resulting bicarbonate to formate salts,35 while awaiting for a fully reversible charge–discharge CH3OH ↔ CO2 process.
Aqueous methanol dehydrogenation actually combines three catalytic cycles running at the same time, each of which might require a different catalyst for optimal efficiency. Entry 7 in Table 2 shows the feasibility of such approach, without the need for catalyst confinement. The possibility of replacing precious metals with non-noble ones has been demonstrated with catalyst 2c. Designing phosphorus-free ligands would help reducing catalyst cost further.
The activities and productivities of the homogeneous catalysts developed so far for methanol reforming lie far behind those required for practical applications and, at present, do not compare favourably with those achieved with supported metal catalysts.36 Yet, they operate under milder conditions and are thus more selective (no CO contamination). In addition, the mechanism of a catalytic process promoted by a homogeneous catalysts is in general more easily disclosed and this opens the possibility to tailor an optimized catalyst by modifying its structural electronic and steric properties. This will certainly stimulate further intense effort to develop improved systems for methanol reforming.
Carbon dioxide hydrogenation
Numerous reports have focussed on partial hydrogenation of CO2, especially to formic acid, a topic which has been recently reviewed in depth by several authors37 and readers are referred to them for detailed descriptions on this topic.For selective reduction to MeOH–H2O, activated hydrides such as boranes38 or silanes39 have usually been employed. However, examples of homogeneously catalysed reduction of CO2 to MeOH/H2O using molecular H2 as reducing agent have until recently remained elusive.40 In this account, the direct hydrogenation of CO2 to MeOH using H2 as reductant and homogeneous catalysts will be discussed. In addition, procedures that employ CO2-derivatives will be mentioned.
Sanford et al. presented the first successful example of homogeneous catalytic CO2 hydrogenation to MeOH and H2O in 2011.41 This was achieved through cascade catalysis using a combination of three catalytic systems in order to accommodate the sequential reduction as illustrated in Scheme 5.
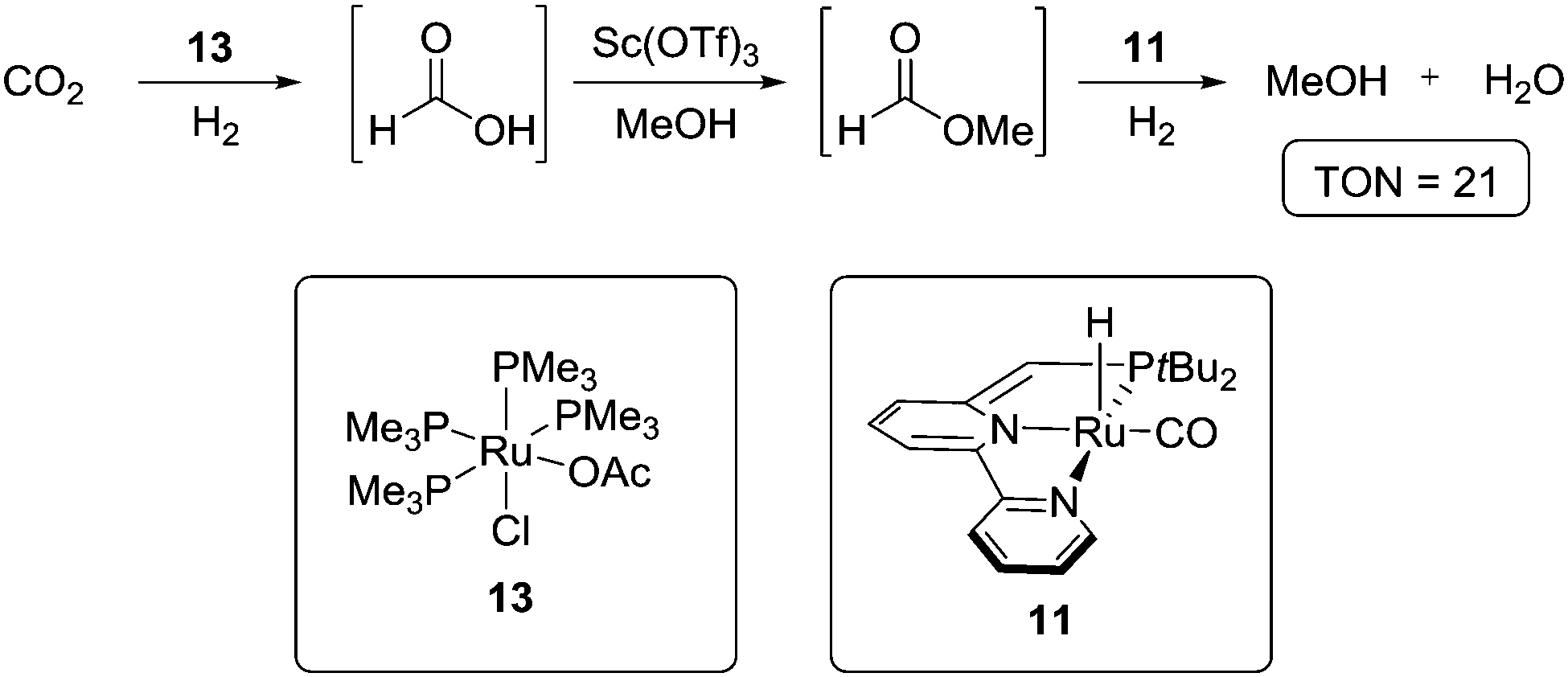 | ||
| Scheme 5 Sanford approach to CO2 hydrogenation to MeOH/H2O using a cascade catalytic system. | ||
The first step is the hydrogenation of CO2 to formic acid using catalyst 13,42 which is followed by a Sc(OTf)3 catalysed esterification to methyl formate. Overall, it was found that, in a one-pot procedure, methyl formate could be formed from CO2 with a TON of 40 after 16 hours at 135 °C. For the further hydrogenation of the ester to MeOH, complex 11, one of Milstein's catalysts,25a,f,43 was employed. This led to an optimised system, which produced MeOH with a TON of 2.5 after 16 hours at 135 °C under 30/10 bars of H2/CO2 pressure. A low CO2 pressure was necessary in order to avoid inactivation of 11 due to covalent bond formation between CO2 and the complex.44 A further problem was the inactivation of 11 by Sc(OTf)3, which explained the low overall TON of CO2 conversion to MeOH. These issues were partially circumvented by setting up a two-compartments system in which catalysts 13 and 11 are confined in one compartment, and Sc(OTf)3 in the other. This led to an improved TON of 21.
The system described by Sanford et al. illustrates the possibility for homogeneous catalytic CO2 hydrogenation. When comparing this procedure with the general hydrogenation approach shown in Scheme 1, catalyst 13 is responsible for the first reduction, Sc(OTf)3 is present as a Lewis acid for activating formic acid to form methyl formate, and 11 promotes the other two reduction steps towards methanol. Therefore, developing a catalytic system capable of hydrogenating formic acid directly would allow to dispense with Sc(OTf)3. Moreover, finding a catalyst that not only tolerates CO2, but also effectively hydrogenates it all the way to MeOH is of obvious interest. This would allow to use a single catalyst for the whole procedure and to adjust more freely the applied CO2 pressure.
Almost simultaneously with Sanford, Milstein et al. showed that the CO2-derivative dimethyl carbonate can be hydrogenated to MeOH using a single catalyst (Scheme 6).45 Under optimised conditions, a TON of 4400 could be achieved using catalyst 11, whereas a TON of 2500 was obtained with 14. Yields of 88% and >99% were obtained, respectively. In addition, when using 11 as catalyst (0.01 mol%), the reaction could be performed neat at 100 °C under 10.1 bar of H2 pressure leading to a quantitative yield of MeOH after 8 hours with a TON > 990. Hence, this procedure represents a mild, solvent- and waste free procedure. However, the use of dimethyl carbonate reduces the overall gravimetric H2 capacity of the system. In addition, no long term reactivity was provided.
 | ||
| Scheme 6 Milstein approach to hydrogenation of the CO2-derivative dimethyl carbonate using either catalyst 11 or 14. | ||
A mechanism for the catalytic cycle has been proposed based on experimental evidence45 and calculations.46 All studies suggest methyl formate and formaldehyde as intermediates, which supports the general pathway shown in Scheme 1. There is, however, some dispute as to how rupture of the OCH3 group on the resulting ruthenium alkoxide is mediated. One suggestion is that one hydrogen from the ligand arm is transferred to afford CH3OH in a cooperative metal–ligand fashion. An alternative lower energy lying route has been put forward where a CH3O group is actually transferred to the metal after transfer of the Ru–H hydride to the carbonyl group (Ru–H/Ru–OCH3 metathesis).46c
Ding et al. showed that 2a (Fig. 1), is also capable of reducing carbonates to MeOH.47 At 140 °C, a catalyst loading of 0.001–0.1 mol% suffices to form MeOH in 84–99% yield from a range of carbonate substrates. An impressive TON up to 87000 and TOF of 1200 h−1 were obtained.
Another indirect approach, where CO2 is initially transformed into an urea derivative before being hydrogenated to MeOH and amines, was also suggested by Milstein et al.48 When using 2 mol% of catalyst 11, yields up to 94% were obtained; however, with 1 mol% of 11 the yields did not exceed 63%, limiting the applicability of the system. Moreover, as opposed to the system shown in Scheme 6, using ureas as CO2-derivatives inevitably creates undesired amine side products. Even if these amines became part of a cycle encompassing urea to MeOH–amine, they would still represent unnecessary ballast compared to the optimal CO2–MeOH hydrogenation–dehydrogenation system.
In 2012, Klankermayer and Leitner et al. demonstrated for the first time that a single catalyst is capable of hydrogenating CO2 to MeOH (Scheme 7).49a This was achieved by mixing the precursors of catalyst 1549b in THF at 140 °C with 60/20 bars of pressurised H2 and CO2, respectively. In addition, EtOH (400 eq. with respect to 15) and an acid additive (MSA or HNTf2, 1.0–1.5 eq. with respect to 15) were required in order to achieve TON values exceeding 10. The precursors of 15 can be either a mixture of [Ru(acac)3] and triphos or 16. When employing [Ru(acac)3]/triphos a TON up to 135 could be achieved after 24 hours. With 16, the TON could reach 221. Moreover, it was shown that the system was active for at least 3 days, demonstrating the robustness of the system. EtOH could be replaced by MeOH but at the expense of the efficiency. Under otherwise identical reaction conditions, TONs of 52 and 24 were observed after 24 hours with EtOH and MeOH, respectively. The role of the alcohol was suggested to be ester formation by reaction with formic acid. Using the double amount of ethanol did not result in an increased reaction rate, which the authors ascribed to the fact that ethyl formate formation is not rate-limiting. However, the efficiency in the absence of an alcohol additive, under otherwise identical reaction conditions, was not reported.
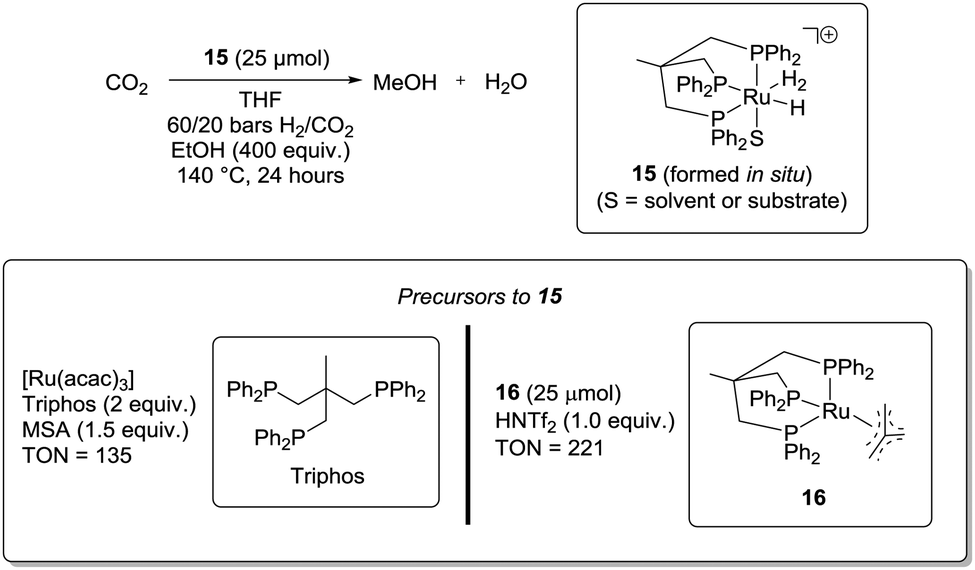 | ||
| Scheme 7 Klankermayer and Leitner approach to CO2 hydrogenation to MeOH using a ruthenium–phosphine catalytic system. The stated equivalents are with respect to ruthenium. | ||
Clear advantages of this system are that CO2 is directly hydrogenated to MeOH and H2O by a single catalyst and that the system is stable on the scale of days. As such, this system is a proof of principle that a single molecular organometallic complex can encompass all three hydrogenation steps. Moreover, avoiding THF as solvent would increase the gravimetric H2 yield of the produced MeOH.
Two years later, Klankermayer and Leitner reported that the same system is capable of performing the full hydrogenation of CO2 to MeOH without the need of an alcohol additive.50a In fact, the conditions that provides a TON of 221 in the presence of 400 equivalents of EtOH (Scheme 7, using 16) gave a very similar TON of 228 without any alcohol additive. Using half the amount of 16 (12.5 μmol) led to an initial TOF of 79 h−1, which is comparable to state-of-the-art heterogeneous Cu/ZnO catalysts.51 Moreover, re-pressurising the reaction mixture with CO2 and H2 after 16 and 32 hours allowed for a total TON of 603 after 48 hours.
Exchanging THF with 2-MeTHF (2-methyltetrahydrofurane) led to a biphasic system, which allowed for easy separation of the aqueous product phase (H2O and MeOH) from the organic phase containing the catalyst, though a small decrease in reactivity compared to THF was observed (TON of 186 after 24 hours). A total TON of 769 could be achieved after 4 cycles of CO2 hydrogenation when using 12.5 μmol of 16, showing the potential of this reaction system. However, the reactivity of the fourth cycle was merely 50% compared to the first cycle (TON of 110 compared to 247).
Due to the fact that this system represents the first example of a homogeneous catalytic system capable of catalysing the hydrogenation of CO2 to MeOH, the mechanism proposed by Klankermayer and Leitner is of interest. The mechanistic aspects of all three hydrogenation steps were studied in depth by NMR and in silico experiments. Based on this, a very detailed mechanism was proposed, which is presented here in a highly simplified manner in Scheme 8 for the sake of brevity.
 | ||
| Scheme 8 Highly truncated representation of the Klankermayer and Leitner proposal for the mechanism of the total hydrogenation of CO2 to MeOH and H2O catalysed by 15 (shown without Triphos). Acetic acid is used to model a carboxylic unit. S = solvent (THF). | ||
Initially, the solvent (S = THF) in 15 is replaced by CO2 forming complex A. After migratory insertion of CO2 into the ruthenium–hydride bond a solvent molecule coordinates to form B. It is proposed that a H2 molecule is released and re-attached during this process. At this stage, the central CO2-carbon atom has reached the oxidation state of formic acid.
A heterolytic cleavage of the Ru-bound H2 followed by a hydride transfer to the carbon atom leads to C. To facilitate these events, a H2 molecule is replacing the solvent and eventually detaches again (not shown). Moreover, the catalytic cycle has at this point reached the stage where the carbon atom has the oxidation state equivalent to that of formaldehyde.
Complex C is then converted to D by collapse of the ruthenium methanediolate to a ruthenium–formaldehyde adduct assisted by protonation by a carboxylic acid, which acts as proton shuttle. Acetic acid was used as model for the calculations in this instance. The presence of a D like intermediate was corroborated by in situ NMR studies. Furthermore, it was found that the acid-mediated path is energetically favoured over the water-assisted pathway and D represents a low energy intermediate for the formation of E. As for the step from B to C, intermediate F is formed from Evia a transient bound H2 (which replaces the solvent), and transfer of a hydride to the carbon atom, rendering it in the oxidation state of a methanol.
As for the collapse of methanediol (C to E), a low energy pathway towards H is found when introducing a carboxylic acid as assisting unit. Hence, intermediate G has a similar function for the F to H transformation as D has it for C to E. Finally, 15 is regenerated by ligation of H2 and extrusion of MeOH.
As shown in the catalytic cycle, a single complex is proposed to perform all three hydrogenation stages. Moreover, both formic acid and formaldehyde were shown to be hydrogenated by 15, supporting the notion of a single complex catalysed hydrogenation of CO2 to MeOH.
Overall, an exergonic reaction is predicted by the calculations, which is in accordance with (reverse) eqn (8).
Because of the limited number of reports on direct hydrogenation of CO2 to MeOH by a single molecular catalyst, the work by Klankermayer and Leitner represents a guideline for future studies in this area. Developing more active catalysts for this process is of obvious necessity in order to meet any demands for a practical application of hydrogen storage in a cycle CO2–MeOH based on homogeneous catalysis. Furthermore, it would be interesting to test the activity of the Klankermayer and Leitner catalyst for the opposite directed reaction, MeOH dehydrogenation.
Complex 16 has also been shown to be active for the hydrogenation of dimethyl carbonate.50b Using 1 mol% and 1.5 equivalent of HNTf2 under 50 bar H2 at 140 °C in dioxane allowed for 99% conversion with 94% selectivity towards MeOH after 16 hours.
Conclusions
Very recently, methanol dehydrogenation to carbon dioxide and the reverse reaction catalysed by homogeneous catalysis have witnessed great progress. Whereas several efficient catalytic systems comprising a single catalyst have already been devised for the former reaction, only a single example of the latter has been published to date. Moreover, much more efficient systems need to be developed for both directions in order to approach practical applicability.To date, there are no examples of any homogeneous catalytic system capable of catalysing both directions of the CO2/MeOH cycle. Perhaps combining a system from the dehydrogenation direction that would work under the Klankermayer and Leitner system, such as the Grützmacher system, could show some insight to this intriguing endeavour.
Considering the sudden increase of homogeneous catalytic systems capable of performing MeOH dehydrogenation or CO2 hydrogenation within the last very few years, it will be very interesting to follow the further developments to come in this area in the near future.
Acknowledgements
M.N. thanks the Marie Curie FP7 International Outgoing Fellowship (IOF) for financial support. E.A. and M.N. thank Professor Matthias Beller for fruitful discussions.Notes and references
- N. Armaroli and V. Balzani, Chem. – Asian J., 2011, 6, 768 CrossRef CAS PubMed.
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474 CrossRef CAS PubMed.
- F. Schüth, Chem. Ing. Tech., 2011, 83, 1984 CrossRef.
- A. Manthiram, P. N. Kumta, S. K. Sundaram and G. Ceder, Materials for Electrochemical Energy Conversion and Storage, John Wiley & Sons, 2012 Search PubMed.
- (a) A. F. Dalebrook, W. Gan, M. Grasemann, S. Moreta and G. Laurenczy, Chem. Commun., 2013, 49, 8735 RSC; (b) U. Eberle, M. Felderhoff and F. Schüth, Angew. Chem., Int. Ed., 2009, 48, 6608 CrossRef CAS PubMed.
- (a) D. Teichmann, W. Alert and P. Wasserscheid, Int. J. Hydrogen Energy, 2012, 37, 18118 CrossRef CAS PubMed; (b) M. Trincado, D. Banerjeea and H. Grützmacher, Energy Environ. Sci., 2014, 7, 2464 RSC; (c) C. Gunanathan and D. Milstein, Science, 2013, 341, 249 CrossRef CAS PubMed; (d) A. Boddien, F. Gartner, M. Nielsen, S. Losse and H. Junge, Hydrogen Generation from Formic Acid and Alcohols, Elsevier, Oxford, 2013, pp. 587–603 Search PubMed; (e) M. Grasemann and G. Laurenczy, Energy Environ. Sci., 2012, 5, 8171 RSC.
- (a) D. R. Palo, R. A. Dagle and J. D. Holladay, Chem. Rev., 2007, 107, 3992 CrossRef CAS PubMed; (b) R. M. Navarro, M. A. Peña and J. L. G. Fierro, Chem. Rev., 2007, 107, 3952 CrossRef CAS PubMed; (c) S. Sá, H. Silva, L. Brandãoa, J. M. Sousa and A. Mendes, Appl. Catal., B, 2010, 99, 43 CrossRef PubMed; (d) A. Iulianelli, P. Ribeirinha, A. Mendes and A. Basile, Renewable Sustainable Energy Rev., 2014, 29, 355 CrossRef CAS PubMed.
- L. C. Grabow and M. Mavrikakis, ACS Catal., 2011, 1, 365 CrossRef CAS.
- (a) G. A. Olah, A. Goeppert and G. K. S. Prakash, Beyond Oil and Gas: The Methanol Economy, Wiley-VCH, Weinheim, 2nd edn, 2009 Search PubMed; (b) G. A. Olah, G. K. S. Prakash and A. Goeppert, J. Am. Chem. Soc., 2011, 33, 12881 CrossRef PubMed; (c) G. A. Olah, Angew. Chem., Int. Ed., 2013, 52, 104 CrossRef CAS PubMed and references therein.
- A. J. Esswein and D. G. Nocera, Chem. Rev., 2007, 107, 4022 CrossRef CAS PubMed.
- Thermochemical data have been calculated based on the Nist Chemistry Webbook at http://webbook.nist.gov/chemistry/.
- (a) S. Shinoda, H. Itagaki and Y. Saito, J. Chem. Soc., Chem. Commun., 1985, 860 RSC; (b) H. Itagaki, Y. Saito and S. Shinoda, J. Mol. Catal., 1987, 41, 209 CrossRef CAS; (c) H. Itagaki, S. Shinoda and Y. Saito, Bull. Chem. Soc. Jpn., 1988, 61, 2291 CrossRef CAS; (d) T. Fujii and Y. Saito, J. Mol. Catal., 1991, 67, 185 CrossRef CAS; (e) H. Itagaki, N. Koga, K. Morokuma and Y. Saito, Organometallics, 1993, 12, 1648 CrossRef CAS.
- T. A. Smith, R. P. Aplin and P. M. Maitlis, J. Organomet. Chem., 1985, 291, C13 CrossRef CAS.
- L.-C. Yang, T. Ishida, T. Yamakawa and S. Shinoda, J. Mol. Catal. A: Chem., 1996, 108, 87 CrossRef CAS.
- D. Norton and D. J. Cole-Hamilton, J. Chem. Soc., Chem. Commun., 1988, 1154 Search PubMed.
- (a) S. Shinoda and T. Yamakawa, J. Chem. Soc., Chem. Commun., 1990, 1511 RSC; (b) T. Yamakawa, M. Hiroi and S. Shinoda, J. Chem. Soc., Dalton Trans., 1994, 2265 RSC; (c) H. Einaga, T. Yamakawa and S. Shinoda, J. Coord. Chem., 1994, 32, 117 CrossRef CAS; (d) H. Einaga, T. Yamakawa and S. Shinoda, J. Mol. Catal. A: Chem., 1995, 97, 35 CrossRef CAS; (e) P. A. Robles-Dutenhefner, E. M. Moura, G. J. Gama, H. G. L. Siebald and E. V. Gusevskaya, J. Mol. Catal. A: Chem., 2000, 164, 39 CrossRef CAS.
- E. Delgado-Lieta, M. A. Luke, R. F. Jones and D. J. Cole-Hamilton, Polyhedron, 1982, 11–12, 839 Search PubMed.
- (a) H. Yamamoto, S. Shinoda and Y. Saito, J. Mol. Catal., 1985, 30, 259 CrossRef CAS; (b) T. Takahashi, S. Shinoda, S. Shinoda and Y. Saito, J. Mol. Catal., 1985, 31, 301 CrossRef CAS.
- (a) K. Nomura, Y. Saito and S. Shinoda, J. Mol. Catal., 1989, 50, 303 CrossRef CAS; (b) K. Makita, K. Nomura and S. Shinoda, J. Mol. Catal., 1994, 89, 143 CrossRef CAS.
- K. M. K. Yu, W. Tong, A. West, K. Cheung, T. Li, G. Smith, Y. Guo and S. C. E. Tsang, Nat. Commun., 2012, 3, 1230, DOI:10.1038/ncomms2242.
- D. Morton and D. J. Cole-Hamilton, J. Chem. Soc., Chem. Commun., 1987, 248 RSC.
- R. E. Rodriguez-Lugo, M. Trincado, M. Vogt, F. Tewes, G. Santiso-Quinones and H. Grützmacher, Nat. Chem., 2013, 5, 342 CrossRef CAS PubMed.
- (a) M. Nielsen, E. Alberico, W. Baumann, H.-J. Drexler, H. Junge, S. Gladiali and M. Beller, Nature, 2013, 495, 85 CrossRef CAS PubMed; (b) E. Alberico, P. Sponholz, C. Cordes, M. Nielsen, H.-J. Drexler, W. Baumann, H. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 14162 CrossRef CAS PubMed; (c) A. Monney, E. Barsch, P. Sponholz, H. Junge, R. Ludwig and M. Beller, Chem. Commun., 2014, 50, 707 RSC.
- P. Hu, Y. Diskin-Posner, Y. Ben-David and D. Milstein, ACS Catal., 2014, 4, 2649 CrossRef CAS.
- Selected references: (a) C. Gunanathan and D. Milstein, Chem. Rev., 2014, 114, 12024 CrossRef CAS PubMed; (b) V. T. Annibale and D. Song, RSC Adv., 2013, 3, 11432 RSC; (c) O. R. Luca and R. H. Crabtree, Chem. Soc. Rev., 2013, 42, 1440 RSC; (d) S. Schneider, J. Meiners and B. Askevold, Eur. J. Inorg. Chem., 2012, 412 CrossRef CAS; (e) V. Lyaskovskyy and B. de Bruin, ACS Catal., 2012, 2, 270 CrossRef CAS; (f) C. Gunanathan and D. Milstein, Acc. Chem. Res., 2011, 44, 588 CrossRef CAS PubMed.
- L. E. Heim, N. E. Schlörer, J.-H. Choi and M. H. G. Prechtl, Nat. Commun., 2014, 5, 3621, DOI:10.1038/ncomms4621.
- (a) W. Kuriyama, T. Matsumoto, O. Ogata, Y. Ino, K. Aoki, S. Tanaka, K. Ishida, T. Kobayashi, N. Sayo and T. Saito, Org. Process Res. Dev., 2012, 16, 166 CrossRef CAS; (b) M. Bertoli, A. Choualeb, A. J. Lough, B. Moore, D. Spasyuk and D. G. Gusev, Organometallics, 2011, 30, 3479 CrossRef CAS; (c) M. Nielsen, A. Kammer, D. Cozzula, H. Junge, S. Gladiali and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 9593 CrossRef CAS PubMed.
- (a) X. Yang, ACS Catal., 2014, 4, 1129 CrossRef CAS; (b) M. Lei, Y. Pan and X. Ma, Eur. J. Inorg. Chem., 2015, 794 CrossRef CAS.
- The synthesis and characterization of catalyst 2c was independently reported also by Schneider and Hazari. 2c was found extremely active in the Lewis acid co-catalyzed decomposition of formic acid: (a) I. Koehne, T. J. Schmeier, E. A. Bielinski, C. J. Pan, P. O. Lagaditis, W. H. Bernskoetter, M. K. Takase, C. Würtele, N. Hazari and S. Schneider, Inorg. Chem., 2014, 53, 2133 CrossRef CAS PubMed; (b) E. A. Bielinski, P. O. Lagaditis, Y. Zhang, B. Q. Mercado, C. Würtele, W. H. Bernskoetter, N. Hazari and S. Schneider, J. Am. Chem. Soc., 2014, 136, 10234 CrossRef CAS PubMed.
- O. Eisenstein and R. H. Crabtree, New J. Chem., 2013, 37, 21 RSC.
- R. J. Hamilton and S. H. Bergens, J. Am. Chem. Soc., 2006, 128, 13700 CrossRef CAS PubMed.
- J. Campos, L. S. Sharninghausen, M. G. Manas and R. H. Crabtree, Inorg. Chem., 2015 DOI:10.1021/ic502521c.
- P. Sponholz, D. Mellmann, C. Cordes, P. G. Alsabeh, B. Li, Y. Li, M. Nielsen, H. Junge, P. Dixneuf and M. Beller, ChemSusChem, 2014, 7, 2419 CrossRef CAS PubMed.
- (a) Y. Li, M. Nielsen, B. Li, P. H. Dixneuf, H. Junge and M. Beller, Green Chem., 2015, 17, 193 RSC; (b) The selective dehydrogenation of glycerol to lactic acid was likewise attained with homogeneous N-heterocylic carbene iridium complexes, L. S. Sharninghausen, J. Campos, M. G. Manas and R. H. Crabtree, Nat. Commun., 2014, 5, 5084, DOI:10.1038/ncomms6084.
- Selected recent examples: (a) S. Enthaler, A. Brückü, A. Kammer, H. Junge, E. Irran and S. Gülak, ChemCatChem, 2015, 7, 65 CrossRef CAS; (b) Q. Liu, L. Wu, S. Gülak, N. Rockstroh, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 7085 CrossRef CAS PubMed; (c) C. Ziebart, C. Federsel, P. Anbarasan, R. Jackstell, W. Baumann, A. Spannenberg and M. Beller, J. Am. Chem. Soc., 2012, 134, 20701 CrossRef CAS PubMed; (d) C. Federsel, R. Jackstell, A. Boddien, G. Laurenczy and M. Beller, ChemSusChem, 2010, 3, 1048 CrossRef CAS PubMed.
- S. D. Davidson, H. Zhang, J. Sun and Y. Wang, Dalton Trans., 2014, 43, 11782 RSC.
- (a) C. Maeda, Y. Miyazaki and T. Ema, Catal. Sci. Technol., 2014, 4, 1482 RSC; (b) M. Beller and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2014, 53, 4527 CrossRef CAS PubMed; (c) C. Federsel, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 6254 CrossRef CAS PubMed; (d) T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365 CrossRef CAS PubMed; (e) P. G. Jessop, F. Foó and C.-C. Tai, Coord. Chem. Rev., 2004, 248, 2425 CrossRef CAS PubMed; (f) X. Yin and J. R. Moss, Coord. Chem. Rev., 1999, 181, 27 CrossRef CAS; (g) W. Leitner, Angew. Chem., Int. Ed. Engl., 1995, 34, 2207 CrossRef CAS.
- S. Chakraborty, J. Zhang, J. A. Krause and H. Guan, J. Am. Chem. Soc., 2010, 132, 8872 CrossRef CAS PubMed.
- F. Huang, G. Lu, L. Zhao, H. Li and Z.-X. Wang, J. Am. Chem. Soc., 2010, 132, 12388 CrossRef CAS PubMed.
- For other reviews on CO2 hydrogenation to methanol, see: (a) Y.-N. Li, R. Ma, L.-N. He and Z.-F. Daio, Catal. Sci. Technol., 2014, 4, 1498 RSC; (b) Y. Li, K. Junge and M. Beller, ChemCatChem, 2013, 5, 1072 CrossRef CAS.
- C. A. Huff and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 18122 CrossRef CAS PubMed.
- P. Munshi, A. D. Main, J. C. Linehan, C.-C. Tai and P. G. Jessop, J. Am. Chem. Soc., 2002, 124, 7963 CrossRef CAS PubMed.
- D. Milstein, Top. Catal., 2010, 53, 915 CrossRef CAS.
- (a) C. A. Huff, J. W. Kampf and M. S. Sanford, Organometallics, 2012, 31, 4643 CrossRef CAS; (b) M. Vogt, M. Gargir, M. A. Iron, Y. Diskin-Posner, Y. Ben-David and D. Milstein, Chem. – Eur. J., 2012, 18, 9194 CrossRef CAS PubMed.
- E. Balaraman, C. Gunanathan, J. Zhang, L. J. W. Shimon and D. Milstein, Nat. Chem., 2011, 3, 609 CrossRef CAS PubMed.
- (a) H. Li, M. Wen and Z.-W. Wang, Inorg. Chem., 2012, 51, 5716 CrossRef CAS PubMed; (b) X. Yang, ACS Catal., 2012, 2, 964 CrossRef CAS; (c) F. Hasanayn, A. Baroudi, A. A. Bengali and A. S. Goldman, Organometallics, 2013, 32, 6969 CrossRef CAS.
- Z. Han, L. Rong, J. Wu, L. Zhang, Z. Wang and K. Ding, Angew. Chem., Int. Ed., 2012, 51, 13041 CrossRef CAS PubMed.
- E. Balaraman, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2011, 50, 11702 CrossRef CAS PubMed.
- (a) S. Wesselbaum, T. vom Stein, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2012, 51, 7499 CrossRef CAS PubMed; (b) F. M. A. Geilen, B. Engendahl, M. Hölscher, J. Klankermayer and W. Leitner, J. Am. Chem. Soc., 2011, 133, 14349 CrossRef CAS PubMed.
- (a) S. Wesselbaum, V. Moha, M. Meuresch, S. Brosinski, K. M. Thenert, J. Kothe, T. vom Sten, U. Englert, M. Hölscher, J. Klankermayer and W. Leitner, Chem. Sci., 2015, 6, 693–704 RSC; (b) T. vom Stein, M. Meuresch, D. Limper, M. Schmitz, M. Hölscher, J. Coetzee, D. J. Cole-Hamilton, J. Klankermayer and W. Leitner, J. Am. Chem. Soc., 2014, 136, 13217 CrossRef CAS PubMed.
- M. Behrens, F. Studt, I. Kasatkin, S. Kühl, M. Hävecker, F. Abild-Petersen, S. Zander, F. Girgsdies, P. Kurr, B.-L. Kniep, M. Tovar, R. W. Fischer, J. K. Nørskov and R. Schlögl, Science, 2012, 336, 893 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |