
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A novel fluoro-chromogenic click reaction for the labelling of proteins and nanoparticles with near-IR theranostic agents†‡
Oriol
Planas
 ,
Thibault
Gallavardin
and
Santi
Nonell
*
,
Thibault
Gallavardin
and
Santi
Nonell
*
Institut Químic de Sarrià, Universitat Ramon Llull, Via Augusta, 390, 08017, Barcelona, Spain. E-mail: santi.nonell@iqs.url.edu
First published on 11th December 2014
Abstract
Reaction of porphycene isothiocyanates with primary and secondary amines leads to the formation of thiazolo[4,5-c]porphycenes, with a substantial shift in the absorption and fluorescence spectra. The conjugates show fluorescence in the near-infrared and are capable of photosensitizing the production of the cytotoxic species singlet oxygen.
Theranostic nanomedicine is an emerging therapeutic strategy that combines a contrast agent and a drug on a single nanoplatform for enhanced diagnosis and treatment of localised diseases.1a Of particular interest are photoactivatable theranostic agents that allow simultaneous fluorescence imaging and localised generation of cytotoxic species.1b,c Current research efforts are directed to the development of theranostic labels that show absorption and emission in the red and near-infrared spectral range,2a as well as strong and selective binding to proteins and nanoparticles.2b Several click reactions have been developed to this end, employing reactive groups such as the isothiocyanate (ITC) for conjugation to free amino residues,2c,d maleimide for cysteine-reduced thiol groups,2e,f and carbodiimide for carboxyl groups.2g
A common and severe drawback of current labels is that the optical and fluorescence properties of the conjugates are not too different from those of the unbound probes, which makes it very difficult to differentiate between covalent adducts and non-specific complexes.2c The latter pose a serious problem to bioconjugate-based biological assays and therapies as they can be released into the biological milieu, altering their results.
In the domain of fluorescence, the conventional solution to this problem is the development of turn-on labels that increase their fluorescence intensity upon binding.2h,i However, since the spectra of the bound and unbound probes are the same and only the intensity changes, good contrast cannot be generally achieved.2j A more robust solution would be the use of probes that change their absorption and fluorescence spectra upon covalent binding since this would allow to selectively excite and monitor the bound and unbound tags. This concept has been recently demonstrated for the labelling of proteins with pyrilium fluorescent dyes that change their emission from red to blue.3
However, to date no single theranostic agent has been reported, which undergoes a similar spectral change upon conjugation. In the course of our ongoing research on photoimmunotherapy4 we observed marked spectral changes when porphycenes containing a 9-ITC group (9-ITPPos) were reacted with primary and secondary amines (Fig. 1). Porphycenes, discovered by Vogel in 1986,5a are porphyrin isomers of lower symmetry and unique properties.5b–i Porphycenes are currently being explored as photodynamic therapy agents due to their excellent optical, fluorescent and photosensitising properties.6
 | ||
| Fig. 1 Structural and absorption (solid line) and fluorescence (dashed line) spectral changes upon reaction of 9ITPPo with primary and secondary amines. The spectra shown correspond to ATAZPo2 in acetone. | ||
It is well known that fluorophores containing ITC groups produce thioureas upon clicking to terminal amino residues of biomolecules or amino-functionalised nanoparticles.2f An archetypical example of such tags is the widely used fluorescein-ITC;7 however, unlike the porphycene-ITCs, its spectrum does not appreciably change upon formation of the thiourea bridge.
Isolation of the reaction products of porphycene-ITCs with amines revealed that the underlying cause for the colour change was the spontaneous evolution of the originally-formed thiourea to 2-aminothiazolo[4,5-c]porphycene (ATAZPo; Fig. 1 and Schemes S2 and S3, ESI‡), with a concomitant red-shift of its fluorescence spectrum by more than 70 nm. Formation of thiourea and its subsequent cyclisation proceeded almost quantitatively and with high selectivity at room temperature (Table 1), which is a pre-requisite for “click” labelling reactions.8 An additional benefit of the unexpected evolution of the porphycene-thiourea system is that it prevents the hydrolysis of the conjugates by urease enzymes.9
| Substrate | Compound | Solvent | Reaction time/h | Yield (%) |
|---|---|---|---|---|
| Ammonia | ATAZPo1 | THF | 3 | 97 |
| n-Butylamine | ATAZPo2 | CH2Cl2 | 3 | 95 |
| Diethylamine | ATAZPo3 | CH2Cl2 | 5 | 95 |
| Piperazine | ATAZPo4 | CH2Cl2 | 3 | 90 |
| Morpholine | ATAZPo5 | CH2Cl2 | 3 | 90 |
| Aniline | ATAZPo6 | CH2Cl2 | 24 | 80 |
| O-(2-aminoethyl)-O′-(2-azidoethyl)nonaethyleneglycol | ATAZPo7 | CH2Cl2 | 3 | 85 |
The formed ATAZPos have several relevant chemical, optical and photophysical properties that make them excellent candidates for theranostic applications (Table 2). Firstly, their absorption spectrum is shifted by more than 70 nm relative to that of the precursor 9-ITPPo, which allows their selective excitation even in the presence of any unbound precursor (Fig. S34, ESI‡). Secondly, their fluorescence shifts to the near-IR (Fig. 2), where tissue is most transparent and shows less autofluorescence.10a Thirdly, the fluorescence quantum yields are higher than those of the most common near-IR fluorophores, e.g., 0.01 for the IR800 dye10b and 0.04 for indocyanine green.10c Finally, ATAZPos produce singlet oxygen with high yields, which allows using them also for therapeutic purposes (Fig. S35, ESI‡).11
| Compound | λ m | ε | λ F | Φ F | τ s | τ T | Φ Δ |
|---|---|---|---|---|---|---|---|
| a Wavelength of the lowest-energy absorption band (nm). b Molar absorption coefficient at λm (M−1 cm−1). c Wavelength of fluorescence (nm). d Fluorescence quantum yield. e Singlet state lifetime in air (ns). f Triplet state lifetime under argon (μs). g Singlet oxygen quantum yield. | |||||||
| 9-ITPPo | 667 | 2.1 × 104 | 693 | 0.07 | 0.8 | 35 | 0.49 |
| ATAZPo1 | 745 | 9.3 × 103 | 750 | 0.03 | 0.5 | 30 | 0.30 |
| ATAZPo2 | 743 | 2.0 × 104 | 756 | 0.08 | 0.6 | 20 | 0.27 |
| ATAZPo3 | 763 | 1.4 × 104 | 775 | 0.05 | 0.7 | 50 | 0.24 |
| ATAZPo4 | 744 | 1.8 × 104 | 764 | 0.05 | 0.9 | 51 | 0.24 |
| ATAZPo5 | 740 | 1.2 × 104 | 758 | 0.06 | 0.9 | 32 | 0.30 |
| ATAZPo6 | 750 | 1.2 × 104 | 759 | 0.04 | 0.6 | 35 | 0.20 |
| ATAZPo7 | 748 | 1.7 × 104 | 768 | 0.07 | 0.9 | 23 | 0.31 |
 | ||
| Fig. 2 Solutions of 9-ITPPo and ATAZPo under white light (left) and under UV (365 nm) radiation (middle and right). While 9-ITPPo shows visible red fluorescence, ATAZPo emits mainly in the near-IR (λobs > 780 nm). | ||
Encouraged by the above results we set out to explore the potential of 9-ITPPo for conjugation to biomolecules and nanoparticles. Bovine serum albumin (BSA) and amino-functionalised gold nanoclusters (AuNCs) were selected for proof-of-concept purposes. In a typical experiment, a stock solution of 9-ITPPo was prepared and mixed with BSA in carbonate buffer (pH 9.2) or with AuNCs in ethanol. The reaction vessels were protected from light and stirred at room temperature. Formation of covalent adducts was unequivocally demonstrated by the changes in absorption and fluorescence spectra (Fig. 3 and Fig. S30, ESI‡) and the conjugates retained their ability to photosensitise the production of singlet oxygen.
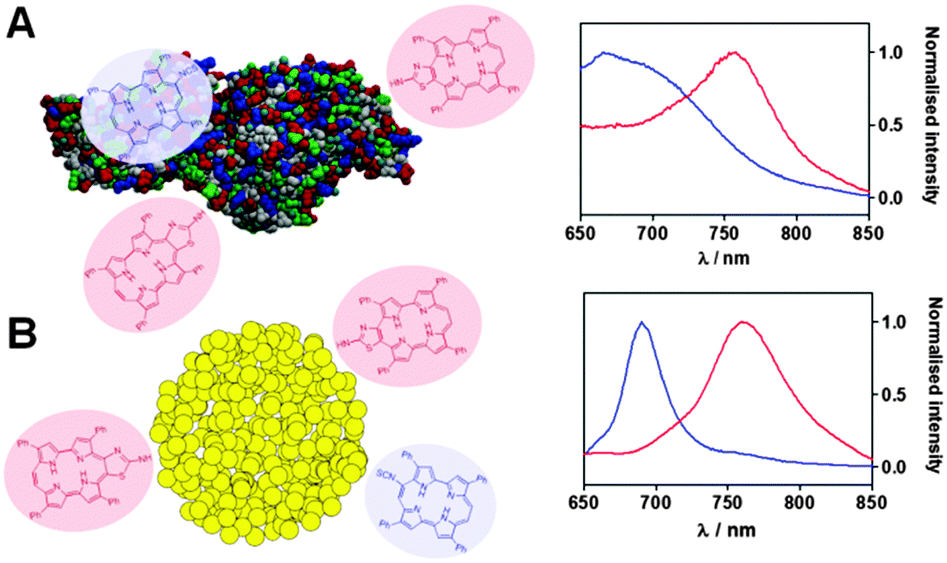 | ||
| Fig. 3 Fluorescence spectra of unbound 9-ITPPo (blue) and its covalent adduct ATAZPo (red) with BSA (A) and AuNCs (B). | ||
Insight into the formation of the thiazole ring was obtained from the study of the reaction kinetics. The time evolution of the absorbance of a mixture of 9-ITPPo and n-butylamine was measured in air-equilibrated toluene (Fig. 4). The isothiocyanate group disappeared within minutes with the concomitant formation of thiourea as the only product, as deduced from the observation of isosbestic points in the absorption spectra and the 1H-NMR spectrum (Fig. S33, ESI‡). This intermediate evolved over a period of several hours to the final product ATAZPo. The time-resolved spectra were analysed using the Specfit/32 software from Spectrum Software Associates and could be successfully fitted by a simple two-step consecutive pseudo-first order kinetic model (9-ITPPo → 9-TUPo → ATAZPo) with rate constants 0.32 s−1 and 6.5 × 10−4 s−1 for the first and second steps, respectively, at room temperature. Flushing the solutions with argon had no measurable effect on the rate constants.
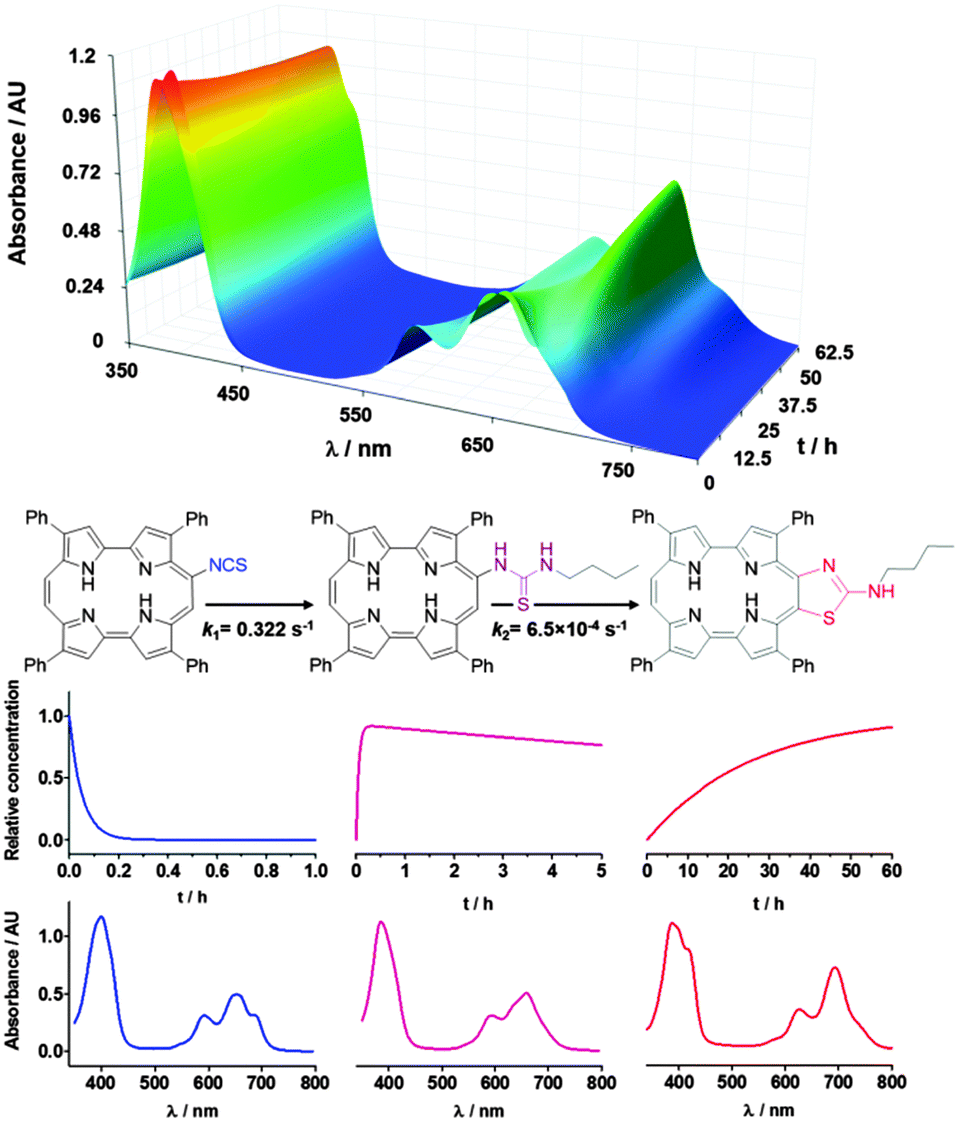 | ||
| Fig. 4 Top: time evolution of the absorbance of a mixture of 9-ITPPo (15 μM) and n-butylamine (3 mM) in toluene. Bottom: structures, concentration evolution and spectral profiles of the reactant, the intermediate and the product (from left to right) achieved through singular value decomposition of the spectral evolution in air. | ||
The Specfit software also yielded the spectra of the three species involved. An excellent match was obtained between the recovered and experimental spectra of the 9-ITPPo reactant, the 9-TUPo intermediate, and the ATAZPo product (Fig. 4).
The spontaneous cyclization of 9-TUPo was unexpected on the basis of the known literature for aromatic thioureas. Although thioureas are known to cyclize to the corresponding thiazoles, this is observed only when they either contain a halogen leaving group,12a,b or are activated with palladium12c or copper,12d or in the presence of added oxidants such as bromine or the tribromide anion.12e–h None of these factors is present in the porphycenes studied. Moreover, the thioureas of the closely-related porphyrins and phthalocyanines do not spontaneously cyclise to the corresponding thiazole derivatives.13 Thus, there is something unique in the electronic structure of porphycene that makes the cyclization reaction highly specific for this porphyrin isomer.
Additional insight was obtained by computational modelling studies. Using density functional methodology, we evaluated the molecular orbitals' energy and shape for a series of aromatic thioureas (Fig. 5). The results revealed that their HOMO is localized on the highly nucleophilic sulphur atom of these compounds, while the LUMO is mostly on the aromatic ring. This suggests that cyclisation of the thioureas has a substantial intramolecular sulphur-to-ring charge-transfer character.
 | ||
| Fig. 5 HOMO–LUMO energy gap (ΔE = ELUMO − EHOMO) of different aromatic thioureas, namely, (A) N-methyl-N′-phenylthiourea, (B) N-methyl-N′-(2-phthalocyanine)-thiourea, (C) N-methyl-N′-(2-porphyrin)-thiourea, and (D) N-methyl-N′-(9-porphycene)-thiourea. | ||
In the framework of the frontier molecular orbital theory, the reaction rate can thus be expected to be proportional to the energy gap between the HOMO of the thiourea nucleophile and the LUMO of the electrophile.14 As would probably be expected, the HOMO–LUMO gap for all porphyrinoids is lower than for the phenyl-thiourea, consistent with the larger size of their aromatic ring, which allows a better electron delocalization. The striking finding was that the HOMO–LUMO gap in 9-TUPo was roughly half that in the related porphyrin and phthalocyanine thioureas. This unique feature of porphycenes sets them apart from the more symmetrical porphyrinoids such as porphyrins and phthalocyanines and stems from their very particular electronic structure.15 Thus, porphycenes are ideal substrates for the formation of thiazole-fused derivatives, which could be exploited for a variety of purposes, particularly for labelling of biomolecules and nanosystems aimed at imaging and therapeutic applications.
As a further proof for the above mechanism, and in order to explore the scope of the new porphycene chemistry, 9-benzoamide porphycene (9-AmPo) was prepared from 9-amino-2,7,12,17-tetraphenylporphycene (9-ATPPo)5e and treated with Lawesson's reagent, which is known to promote the formation of the thioamide functionality.16 Once again, instead of the expected 9-benzothioamide porphycene, we isolated the corresponding thiazolo[4,5-c]porphycene (Scheme S5, Fig. S36 and Table S1, ESI‡). Thus, cyclisation is not restricted to thioureas but can also be realised with thioamides.
In summary, we report a novel specific reaction of porphycenes that allows the formation of thiazole-fused porphycene conjugates with unique absorption, fluorescence and photosensitizing properties. Thus, porphycene-ITCs are novel molecular entities with high potential for imaging and therapeutic applications. Work is currently in progress to explore such applications and shall be reported in due course.
This work has been supported by the Spanish Ministry of Economy and Competitiveness (grants No. CTQ2010-20870-C03-01 and CTQ2013-48767-C3-1-R). O. P. thanks the SUR del DEC de la Generalitat de Catalunya for his predoctoral fellowship (grant No. 2014FI_B 00777).
Notes and references
- (a) T. Lammers, S. Aime, W. E. Hennink, G. Storm and F. Kiessling, Acc. Chem. Res., 2011, 44, 1029–1038 CrossRef CAS PubMed; (b) R. Bardhan, S. Lal, A. Joshi and N. Halas, Acc. Chem. Res., 2011, 44, 936–946 CrossRef CAS PubMed; (c) J. Xie, S. Lee and X. Chen, Adv. Drug Delivery Rev., 2010, 62, 1064–1079 CrossRef CAS PubMed.
- (a) P. Huang, J. Lin, X. Wang, Z. Wang, C. Zhang, M. He, K. Wang, F. Chen, Z. Li, G. Shen, D. Cui and X. Chen, Adv. Mater., 2012, 24, 5104–5110 CrossRef CAS PubMed; (b) A. Nadler and C. Schultz, Angew. Chem., Int. Ed., 2013, 52, 2408–2410 CrossRef CAS PubMed; (c) R. Hudson, M. Carcenac, K. Smith, L. Madden, O. J. Clarke, A. Pèlegrin, J. Greenman and R. W. Boyle, Br. J. Cancer, 2005, 92, 1442–1449 CrossRef CAS PubMed; (d) S. Ohkuma and B. Poole, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 3327–3331 CrossRef CAS; (e) S. S. Ghosh, P. M. Kao, A. W. McCue and H. L. Chappelle, Bioconjugate Chem., 1990, 1, 71–76 CrossRef CAS; (f) M. Mitsunaga, M. Ogawa, N. Kosaka, L. T. Rosenblum, P. L. Choyke and H. Kobayashi, Nat. Med., 2011, 17, 1685–1691 CrossRef CAS PubMed; (g) M. Kobayashi and Y. Chiba, Anal. Biochem., 1994, 219, 189–194 CrossRef CAS PubMed; (h) Y. Hori and K. Kikuchi, Curr. Opin. Chem. Biol., 2013, 17, 644–650 CrossRef CAS PubMed; (i) S. Mizukami, Y. Hori and K. Kikuchi, Acc. Chem. Res., 2014, 47, 247–256 CrossRef CAS PubMed; (j) Q. Yan, S. L. Schwartz, S. Maji, F. Huang, C. Szent-Gyorgyi, D. S. Lidke, K. A. Lidke and M. P. Bruchez, ChemPhysChem, 2014, 15, 687–695 CrossRef CAS PubMed.
- (a) B. K. Wetzl, S. M. Yarmoluk, D. B. Craig and O. S. Wolfbeis, Angew. Chem., Int. Ed., 2004, 43, 5400–5402 CrossRef CAS PubMed; (b) B. K. Hoefelschweiger, A. Duerkop and O. S. Wolfbeis, Anal. Biochem., 2005, 344, 122–129 CrossRef CAS PubMed.
- E. Rosàs, P. Santomá, M. Duran-Frigola, B. Hernandez, M. C. Llinàs, R. Ruiz-González, S. Nonell, D. Sánchez-García, E. R. Edelman and M. Balcells, Langmuir, 2013, 29, 9734–9743 CrossRef PubMed.
- (a) E. Vogel, M. Kocher, H. Schmickler and J. Lex, Angew. Chem., Int. Ed., 1986, 25, 257–259 CrossRef; (b) P. F. Aramendia, R. W. Redmond, S. Nonell, W. Schuster, S. E. Braslavsky, K. Schaffner and E. Vogel, Photochem. Photobiol., 1986, 44, 555–559 CrossRef CAS PubMed; (c) S. E. Braslavsky, M. Müller, D. Mártire, S. Pörting, S. G. Bertolotti, S. Chakravorti, G. Koç-Weier, K. Bernd and K. Schaffner, J. Photochem. Photobiol., B, 1997, 40, 191–198 CrossRef CAS; (d) M. Duran-Frigola, R. Tejedor-Estrada, D. Sánchez-García and S. Nonell, Phys. Chem. Chem. Phys., 2011, 13, 10326–10332 RSC; (e) O. Planas, R. Tejedor-Estrada and S. Nonell, J. Porphyrins Phthalocyanines, 2012, 16, 633–640 CrossRef CAS; (f) H. Shimakoshi, K. Sasaki, Y. Iseki and Y. Hisaeda, J. Porphyrins Phthalocyanines, 2012, 16, 530–536 CrossRef CAS; (g) W. Brenner, J. Malig, R. D. Costa, D. M. Guldi and N. Jux, Adv. Mater., 2013, 25, 2314–2318 CrossRef CAS PubMed; (h) T. Kumagai, F. Hanke, S. Gawinkowski, J. Sharp, K. Kotsis, J. Waluk, M. Persson and L. Grill, Nat. Chem., 2014, 6, 41–46 CrossRef CAS PubMed; (i) T. Okabe, D. Kuzuhara, M. Suzuki, N. Aratani and H. Yamada, Org. Lett., 2014, 16, 3508–3511 CrossRef CAS PubMed.
- (a) C. Richert, J. M. Wessels, M. Muller, M. Kisters, T. Benninghaus and A. E. Goetz, J. Med. Chem., 1994, 37, 2797–2807 CrossRef CAS; (b) J. C. Stockert, M. Cañete, A. Juarranz, A. Villanueva, R. W. Horobin, J. I. Borrell, J. Teixido and S. Nonell, Curr. Med. Chem., 2007, 14, 997–1026 CrossRef CAS; (c) X. Ragàs, D. Sánchez-García, R. Ruiz-González, T. Dai, M. Agut, M. R. Hamblin and S. Nonell, J. Med. Chem., 2010, 53, 7796–7803 CrossRef PubMed; (d) M. García-Díaz, D. Sánchez-García, J. Soriano, M. L. Sagristà, M. Mora, A. Villanueva, J. C. Stockert, M. Cañete and S. Nonell, Med. Chem. Commun., 2011, 2, 616–619 RSC; (e) R. Ruiz-González, P. Acedo, D. Sánchez-García, S. Nonell, M. Cañete, J. C. Stockert and A. Villanueva, Eur. J. Med. Chem., 2013, 63, 401–414 CrossRef PubMed.
- (a) G. Grunwaldt, S. Haebel and C. Spitz, J. Photochem. Photobiol., B, 2002, 67, 177–186 CrossRef CAS; (b) N. Klonis and W. H. Sawyer, Photochem. Photobiol., 2003, 77, 502–509 CrossRef CAS.
- (a) M. O. Senge and J. C. Brandt, Photochem. Photobiol., 2011, 87, 1240–1296 CrossRef CAS PubMed; (b) P. Thirumurugan, D. Matosiuk and K. Jozwiak, Chem. Rev., 2013, 113, 4905–4979 CrossRef CAS PubMed.
- C. Lopreore and L. D. Byers, Arch. Biochem. Biophys., 1998, 349, 299–303 CrossRef CAS PubMed.
- (a) A. M. Smith, M. C. Mancini and S. Nie, Nat. Nanotechnol., 2009, 4, 710–711 CrossRef CAS PubMed; (b) X. Peng, H. Chen, D. R. Draney, W. Volcheck, A. Schutz-Geschwender and D. M. Olive, Anal. Biochem., 2009, 388, 220–228 CrossRef CAS PubMed; (c) R. Philip, A. Penzkofer and W. Bäumler, J. Photochem. Photobiol., A, 1996, 96, 137–148 CrossRef CAS.
- (a) S. S. Kelkar and T. M. Reineke, Bioconjugate Chem., 2011, 22, 1879–1903 CrossRef CAS PubMed; (b) T. Lammers, S. Aime, W. E. Hennink, G. Storm and F. Kiessling, Acc. Chem. Res., 2011, 44, 1029–1038 CrossRef CAS PubMed.
- (a) R. Wang, W. Yang, L. Yue, W. Pan and H. Zeng, Synlett, 2012, 1643–1648 CrossRef CAS PubMed; (b) S. K. Rout, S. Guin, J. Nath and B. K. Patel, Green Chem., 2012, 14, 2491–2498 RSC; (c) H.-Y. Thu, W.-Y. Yu and C.-M. Che, J. Am. Chem. Soc., 2006, 128, 9048–9049 CrossRef CAS PubMed; (d) Y.-J. Guo, R.-Y. Tang, P. Zhong and J.-H. Li, Tetrahedron Lett., 2010, 51, 649–652 CrossRef CAS PubMed; (e) A. D. Jordan, C. Luo and A. B. Reitz, J. Org. Chem., 2003, 68, 8693–8696 CrossRef CAS PubMed; (f) Z.-G. Le, J.-P. Xu, H.-Y. Rao and M. Ying, J. Heterocycl. Chem., 2006, 43, 1123–1124 CrossRef CAS; (g) R. B. Sparks, P. Polam, W. Zhu, M. L. Crawley, A. Takvorian, E. McLaughlin, M. Wei, P. J. Ala, L. Gonneville, N. Taylor, Y. Li, R. Wynn, T. C. Burn, P. C. C. Liu and A. P. Combs, Bioorg. Med. Chem. Lett., 2007, 17, 736–740 CrossRef CAS PubMed; (h) K. Inamoto, C. Hasegawa, J. Kawasaki, K. Hiroya and T. Doi, Adv. Synth. Catal., 2010, 352, 2643–2655 CrossRef CAS.
- (a) J. M. Sutton, O. J. Clarke, N. Fernandez and R. W. Boyle, Bioconjugate Chem., 2002, 13, 249–263 CrossRef CAS PubMed; (b) R. P. Hammer, C. V. Owens, S. Hwang, C. M. Sayes and S. A. Soper, Bioconjugate Chem., 2002, 13, 1244–1252 CrossRef CAS PubMed; (c) W. Duan, K. Smith, H. Savoie, J. Greenman and R. W. Boyle, Org. Biomol. Chem., 2005, 3, 2384–2386 RSC.
- (a) E. V. Anslyn and D. A. Dougherty, Modern Physical Organic Chemistry, University Science Books, Sausalito, 2006, p. 562 Search PubMed; (b) L.-G. Zhuo, W. Liao and Z.-X. Yu, Asian J. Org. Chem., 2012, 1, 336–345 CrossRef CAS.
- J. Waluk, M. Muller, P. Swiderek, M. Kocher, E. Vogel, G. Hohlneicher and J. Michl, J. Am. Chem. Soc., 1991, 113, 5511–5527 CrossRef CAS.
- T. Ozturk, E. Ertas and O. Mert, Chem. Rev., 2007, 107, 5210–5278 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Professor Juan C. “Tito” Scaiano on the occasion of his 70th birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental techniques and procedures, characterisation data, and additional results. See DOI: 10.1039/c4cc09070e |
| This journal is © The Royal Society of Chemistry 2015 |