
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Caught! Crystal trapping of a side-on peroxo bound to Cr(IV)†
David P.
de Sousa
 a,
Jennifer O.
Bigelow
b,
Jonas
Sundberg
a,
Lawrence
Que
Jr.
b and
Christine J.
McKenzie
*a
a,
Jennifer O.
Bigelow
b,
Jonas
Sundberg
a,
Lawrence
Que
Jr.
b and
Christine J.
McKenzie
*a
aDepartment of Physics, Chemistry and Pharmacy, University of Southern Denmark, Campusvej 55, 5230 Odense M, Denmark. E-mail: mckenzie@sdu.dk; Fax: +45 6615 8760; Tel: +45 6550 2518
bDepartment of Chemistry and Center for Metals in Biocatalysis, University of Minnesota, Minneapolis, Minnesota 55455, USA
First published on 7th January 2015
Abstract
A Cr(IV)η2–peroxo complex crystallizes from 33% aqueous H2O2. The complex is a likely intermediate in catalytic disproportionation of H2O2 proposed to occur through a single metal site mechanism in solution – and solid state.
The study of metal–dioxygen, peroxo and oxo adducts has attracted considerable attention as such metal-activated oxygen species have emerged as the common precursors and active oxidants in a wide range of biological and non-biological oxidation systems.1–5 The majority of first row transition metals are represented as co-factors in enzymes and proteins. However, in spite of its rich redox chemistry, reasonable availability and oxophilicity, chromium is by large absent in the natural world. Instead, it is generally considered a cell toxin capable of causing damage to DNA and RNA, through both oxidative mechanisms and adduct formation.6,7 Several molecular Cr(V)–oxo complexes have been reported in the literature over the last 40 years,8–13 whereas only a handful Cr–dioxygen complexes have been identified. In all cases these compounds are derived from the reaction of Cr(II) precursors with O2. An ion proposed to be a Cr(III)–superoxo complex [CrO2(bipy)2]2+ (bipy = 2,2′-bipyridine) was obtained by reacting coordinatively unsaturated [Cr(bipy)2]2+ with O2 in the gas phase.14,15 Subsequently Theopold et al.16 structurally characterized a “side-on” Cr(III)–superoxo adduct generated from the reaction of [CrII(TptBu,Me)NC6H5]+ (TptBu,Me = hydrotris(3-tert-butyl-5-methylpyrazolyl)borate) with O2 in diethylether. Recently Nam et al. have reported both an “end-on” Cr(III)–superoxo11 and a “side-on” Cr(IV)–peroxo17 adduct supported by macrocyclic tetramethylcyclams of different ring sizes.
Molecular Cr–dioxygen species are relevant with respect to the biotoxicity of chromium, and are useful as structural mimics of more elusive metal–dioxygen adducts of the later transition metals. We have previously used amino-acid derived pyridine ligands, such as tpena− (N,N,N′-tris(2-pyridylmethyl)ethylenediamine-N′-acetate), to generate reactive iron(IV)–oxo,18,19 manganese(IV)–oxo complexes and putative O2 adducts,20,21 and a metal–oxidant (iodosylbenzene) adduct.22 The instability of such high-valent iron and manganese complexes makes their structural characterization especially difficult. The isolation of more stable structural analogues based on earlier first row transition metals is a potentially fruitful strategy for gleaning structural information on such intermediates.18 Here we describe the structural trapping of a catalytically competent Cr(IV)–peroxo adduct formed from the reaction of a Cr(III) precursor with H2O2.
[CrIII(tpena)]2+ (1) is prepared in aqueous solution from the reaction of chromium(III)-nitrate with the sodium salt of the ligand, followed by crystallization as a diperchlorate salt. The X-ray crystal structure shows that tpena− coordinates through all six donor atoms. The Cr(III) atom displays a particularly irregular octahedral geometry, Fig. S1 (ESI†), most notable being an extremely obtuse N1–Cr–N5 angle of 115.40(4)°. When 1 is dissolved, UV-Vis spectra (pH 4–8) show that aqueous solutions contain an equilibrium between 1, its “pseudo hydrate”, [CrIII(tpenaH)OH]2+ (2) and the blue congener base [CrIII(tpena)OH]+ (3), Scheme 1. Complex 3 is the base peak in ESI-MS spectra and this speciation is supported by cyclic voltammetry, vide infra. Addition of base drives the equilibrium towards 3. When 100 eq. of H2O2 are added to solutions of 1, a colour change from red to violet occurs over 30 min accompanied by the evolution of dioxygen23 consistent with modest catalase activity. The solutions are EPR silent.
 | ||
| Scheme 1 Interrelationship between tpena− complexes of Cr(II), Cr(III) and Cr(IV), including a mechanistic proposal for the catalytic decomposition of H2O2. | ||
This observation, together with UV-Vis spectroscopy (λ, 539 nm; ε, 150 M−1 cm−1), Fig. 1, and electrospray ionization (ESI) mass spectra – which show the dominant presence of a Cr(tpena)–dioxygen adduct [CrO2(tpena)]+ at m/z 474.1 (calcd 474.1), suggests either of the isomeric species: a triplet Cr(III)–superoxo system consisting of a Cr(III) center anti-ferromagnetically coupled to a O2˙− radical, or a triplet Cr(IV)–peroxo system. A Cr(III)–hydroperoxo adduct was ruled out, since this formulation would give rise to a simple 3/2 system presumably with a spectrum similar to that obtained for 1, Fig. S5 (ESI†). The presence of a band at 878 cm−1 in the resonance Raman spectrum of the purple solutions, Fig. 2(a) and Fig. S8(b) (ESI†), (full spectrum) identifies the species unambiguously as a peroxo, rather than a superoxo complex.24 Consistently the IR spectrum of an EPR silent solid precipitated with diethyl ether–dioxane shows a strong sharp absorption at 871 cm−1 not seen in IR spectrum of 1, Fig. 2(b). The differences in the ligand vibrations at 1256, 1373 and 1669 cm−1 suggest that a pyridine arm of the ligand has become uncoordinated and protonated as seen in the salts of [VIVO(tpenaH)]2+ and [FeIII2O(tpenaH)2]4+,18,22 and the formulation [CrIVO2(tpenaH)]2+ (4) could be proposed.
 | ||
| Fig. 1 Aqueous UV-Vis spectra of 4 mM 1 (red line), 3.7 mM 4 (violet line, generated in situ from 1 and 200 eq. H2O2) and 3.6 mM 3 (blue line, generated in situ from 1 and 50 eq. NaOH). | ||
 | ||
| Fig. 2 (a) Frozen solution rRaman spectra of 9 mM 1 (black) and 4 (red) recorded at 77 K in CH3CN (λex = 514.5 nm, power = 65 mW). Asterisks denote solvent signals. (b) Solid state IR spectra of 1(ClO4)2 (black) and 4(ClO4)2 (red). | ||
Single crystals of 4(ClO4)2(H2O2)3(H2O) were obtained only on a couple of occasions by placing the largest crystals of 1(ClO4)2(C4H8O2)0.5 available into aqueous 33% H2O2. Over the course of a day at 4 °C the starting material was replaced by plates of the purple peroxo complex. Bubbles of O2 were observed to slowly emerge from both the solution and the surface of the crystals. The X-ray crystal structure, Fig. 3(a), shows that the peroxide is coordinated to a seven-coordinate Cr-centre in a side-on fashion, and charge balance dictates that the cation is a Cr(IV) species. As anticipated one methylpyridyl arm is uncoordinated and protonated. The distance between the O atoms of the O2 moiety is 1.383(8) Å. The average Cr–L distances for 1 and 4 respectively are 2.040(1) Å and 2.014(6) Å supporting a higher oxidation for the chromium ion in 4. The +4 oxidation state is confirmed by a Bond Valence Sum (BVS) analysis that yields a BVS = 4.001.25 A hydrogen-bonding motif is present in the crystal, with the cations linked together by the protonated dangling pyridyl arm of one molecule and the non-coordinated carbonyl oxygen of a neighbour (PyH+⋯−OOC, 1.904 Å), forming herringbone chains. The hydrogen-bonded chains stack regularly to form sheets parallel to the b-axis, forming the basis for a network of pores running along the c-axis. The pores are occupied by three co-crystallized H2O2 molecules per cation. The perchlorate ions H-bond predominantly to the substrate guest H2O2. The unusually large number of H2O2 molecules in the crystal lattice, and their disorder, suggest that these might be rather mobile guests. Thus we conclude that a “crystal trapping” of the reactive 4 is made possible because it is surrounded by substrate that is taken up in the solid state, selectively, from solution. A calculation of the pore dimensions after in silico removal of the H2O2 molecules reveals about 16% pseudo void space. These voids lie predominantly between internal lamellar surfaces with the coordinated peroxo ligands lining the surface of these sheets, Fig. 3(b). With such a spatial arrangement, it seems reasonable to assume that guest H2O2 molecules can creep between the sheets, while product O2 is able to leave the crystals the same way by diffusion.
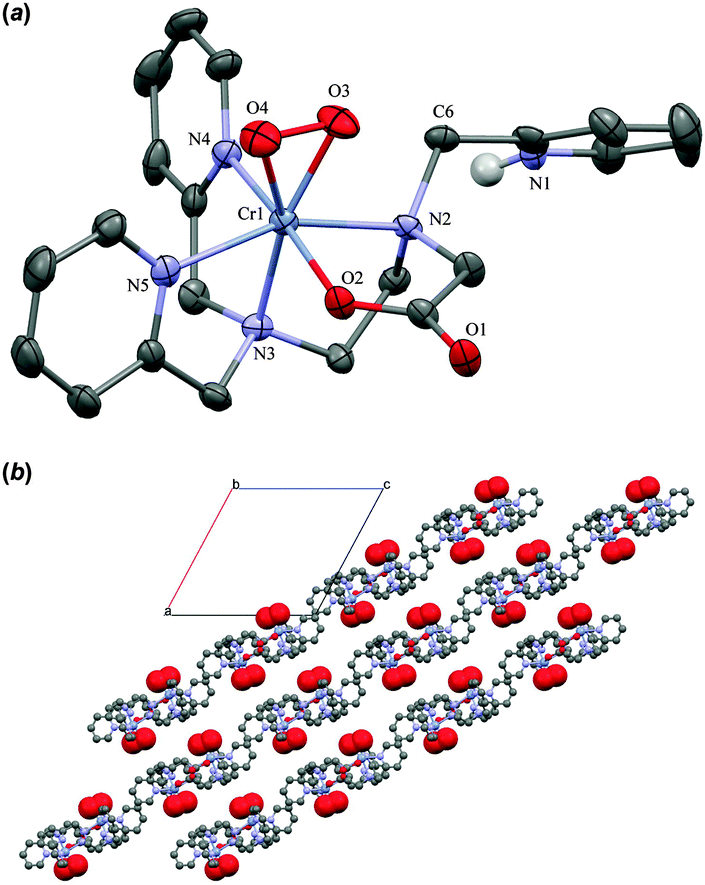 | ||
| Fig. 3 (a) ORTEP plot of the cation in 4(ClO4)2(H2O2)3(H2O) showing 50% probability ellipsoids. (b) Ball-and-stick illustration of the crystal lattice defined by the chains of H-bonded 4 with the peroxide ligand as space filled spheres after in silico removal of H2O2, H2O and ClO4−. Viewed down the b-axis. | ||
Deprotonated 4 (m/z 474.1, 4-H+) is present in ESI mass spectra obtained on both working solutions containing excess H2O2, and in the spectra of isolated 4(ClO4)2(H2O2)4 redissolved in acetonitrile. These spectra furnish some hints about the mechanism of the catalytic H2O2 disproportionation, and the decomposition of 4 in the absence of substrate H2O2, respectively. The dominant ion in both spectra is [CrII(tpena)]+ (5; m/z 442.1, calcd 442.1). While 5 is accessible electrochemically (vide infra), it was present, only occasionally, as a minor ion in the spectra of 1 (complex 3 dominates the spectra of 1, Fig. S2, ESI†). These observations suggest that 5 is not predominantly formed via an ionization induced one-electron reduction of 1. Collision Induced Dissociation (CID) experiments on the 474.1 ion (4-H+) result in the loss of the mass equivalent of two oxygen atoms to directly generate 5. That no stepwise O atom loss occurs, strongly indicates that the O–O bond remains intact during this process14 and that this is the dominant route for generation of the observed Cr(II) species 5. Ions assigned to [CrIVO(tpena)]+ (m/z 458.1, calcd 458.1) and [CrVO(tpena)]2+ (m/z 229.1, calcd 229.1), and its ion pair, [CrVO(tpena)]ClO4+ (m/z 557.1 calcd 557.1) and other high-valent O containing species appear only in the spectra of isolated 4(ClO4)2. These species might result from the inter- and intra-molecular O atom transfer from 4 to an unknown substrate or the ligand.26 It is noteworthy that a greater number of ligand decomposition products are observed in Fig. 4(b). On the basis of the electronic and geometrical structure of 4, the gas phase experiments through which various Cr(II), Cr(IV)–peroxo, Cr(IV)–oxo, and Cr(V)–oxo species are observed, we propose in Scheme 1, a catalytic acid–base type cycle for the relatively slow disproportionation of H2O2. The cycle is similar to mechanisms proposed for heme catalases.27 Indeed the possible involvement of a second coordination sphere basic pyridine to aid proton transfers is reminiscent of the role of the proximal histidine in peroxidases. The species labelled by a number have been detected, and a structural analogue for 6, [VIVO(tpenaH)]+ is known.18 The direct observation of the O2 release process step (vi) by CID suggests that the pro-catalyst 1 is not directly involved in the catalytic cycle, but requires reductive activation, by direct (i) + (ii) or indirect (iii) involvement by H2O2, before it can enter the cycle. Time-resolved UV-Vis spectroscopy indicates that the direct (i) + (ii) mechanism is the dominant activation pathway, since the time trace for the appearance of the λmax at 553 nm due to the chromophore of 4 can be modelled satisfactorily with a 2-step mechanism (see ESI,† Fig. S7). The cyclic voltammogram of 1(ClO4)2 reveals the presence of 1 and 3 in neutral aqueous solutions, with reversible Cr2+/3+ redox couples at E1/2 = −885 mV and −1099 mV (vs. Ag/Ag+), Fig. S3 (ESI†), supporting the plausibility of the involvement of 5 in the catalytic cycle. The Cr(IV)–oxo species, 6, formed by a O atom transfer from H2O2 to 5 (iv) is expected to be a strong base which can deprotonate H2O2 and assist the substitution step (v),28 perhaps with involvement of the dangling pyridine.
 | ||
| Fig. 4 (a) ESI mass spectrum of solution of 4 generated in situ from the reaction of 1 with 200 eq. H2O2 in H2O. (b) ESI mass spectrum of 4(ClO4)2 dissolved in CH3CN. Important assignments not in the text: m/z 490.1 [CrVI(O)(O2)(tpena)]+ (or [CrIV(O2)(tpenaO)]+), 366.1 ([CrIII(tpenaO-CHC5H4N)]+), 350.1 ([CrIII(tpena-CH2C5H4N)]+). | ||
In summary, we have isolated a rare example of a side-on Cr(IV)–peroxo adduct. This species is a catalytically competent species in solutions, and within crystals, for catalytic H2O2 dismutation. Its preparation involves the reaction of an unusually labile Cr(III) precursor and H2O2. This is an interesting contrast to the syntheses of the handful of other known Cr–O2 (superoxo, peroxo) adducts which typically involves the reaction of Cr(II) complexes with O2 – precisely the reverse of step (vi) in Scheme 1.
This work was supported by the Danish Council for Independent Research | Natural Sciences (grant 12-124985 to CMcK), and the US National Science Foundation (grants CHE-1058248 and CHE-1361773 to LQ). We thank Dr Mads S. Vad for recording EPR spectra and Simon Svane for performing CID experiments. The COST CM1003 action is acknowledged for travel funding.
Notes and references
- S. J. Lange and L. Que Jr., Curr. Opin. Chem. Biol., 1998, 2, 159 CrossRef CAS.
- P. C. A. Bruijnincx, G. van Koten and R. Gebbink, Chem. Soc. Rev., 2008, 37, 2716 RSC.
- W. A. Van der Donk, C. Krebs and J. M. Bollinger, Curr. Opin. Struct. Biol., 2010, 20, 673 CrossRef CAS PubMed.
- M. Yagi and M. Kaneko, Chem. Rev., 2001, 101, 21 CrossRef CAS PubMed.
- R. Huber, P. Hof, R. O. Duarte, J. J. Moura, I. Moura, M. Y. Liu, J. LeGall, R. Hille, M. Archer and M. J. Romão, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 8846 CrossRef CAS.
- A. Levina and P. A. Lay, Chem. Res. Toxicol., 2008, 21, 563 CrossRef CAS PubMed.
- A. Zhitkovich, Chem. Res. Toxicol., 2005, 18, 3 CrossRef CAS PubMed.
- K. Srinivasan and J. K. Kochi, Inorg. Chem., 1985, 24, 4671 CrossRef CAS.
- K. H. Nill, F. Wasgestian and A. Pfeil, Inorg. Chem., 1979, 18, 564 CrossRef CAS.
- T. J. Collins, C. Slebodnick and E. S. Uffelman, Inorg. Chem., 1990, 29, 3433 CrossRef CAS.
- J. Cho, J. Woo, J. E. Han, M. Kubo, T. Ogura and W. Nam, Chem. Sci., 2011, 2, 2057 RSC.
- M. E. O'Reilly, T. J. Del Castillo, J. M. Falkowski, V. Ramachandran, M. Pati, M. C. Correia, K. A. Abboud, N. S. Dalal, D. E. Richardson and A. S. Veige, J. Am. Chem. Soc., 2011, 133, 13661–13673 CrossRef PubMed.
- S. Liu, K. Mase, C. Bougher, S. D. Hicks, M. M. Abu-Omar and S. Fukuzumi, Inorg. Chem., 2014, 53, 7780–7788 CrossRef CAS PubMed.
- H. Molina-Svendsen, G. Bojesen and C. J. McKenzie, Inorg. Chem., 1998, 37, 1981 CrossRef CAS.
- P. R. Howe, J. E. McGrady and C. J. McKenzie, Inorg. Chem., 2002, 41, 2026 CrossRef CAS PubMed.
- K. Qin, C. D Incarvito, A. L. Rheingold and K. H. Theopold, Angew. Chem., Int. Ed., 2002, 41, 2333 CrossRef CAS.
- A. Yokoyama, J. E. Han, J. Cho, M. Kubo, T. Ogura, M. A. Siegler, K. D. Karlin and W. Nam, J. Am. Chem. Soc., 2012, 134, 15269 CrossRef CAS PubMed.
- M. S. Vad, A. Lennartson, A. Nielsen, J. Harmer, J. E. McGrady, C. Frandsen, S. Morup and C. J. McKenzie, Chem. Commun., 2012, 48, 10880 RSC.
- W. A. Donald, C. J. McKenzie and R. A O'Hair, Angew. Chem., Int. Ed., 2011, 50, 8379 CrossRef CAS PubMed.
- C. Baffert, M.-N. Collomb, A. Deronzier, S. Kjærgaard-Knudsen, J.-M. Latour, K. H. Lund, C. J. McKenzie, M. Mortensen, L. P. Nielsen and N. Thorup, Dalton Trans., 2003, 1765 RSC.
- A. K. Poulsen, A. Rompel and C. J. McKenzie, Angew. Chem., Int. Ed., 2005, 44, 6916 CrossRef CAS PubMed.
- A. Lennartson and C. J. McKenzie, Angew. Chem., Int. Ed., 2012, 51, 6767 CrossRef CAS PubMed.
- The gas was identified using gas chromatography. TCD detector.
- For a compilation of stretching vibrations for Cr–O2 complexes, see Table S1 in the ESI†.
- For details and validity of the BVS analysis, see the ESI†.
- A. Nielsen, F. B. Larsen, A. D. Bond and C. J. McKenzie, Angew. Chem., Int. Ed., 2006, 45, 1602 CrossRef CAS PubMed.
- M. Alfonso-Prieto, X. Biarnés, P. Vidossich and C. Rovira, J. Am. Chem. Soc., 2009, 131, 11751 CrossRef CAS PubMed.
- U. G. Nielsen, A. Hazell, J. Skibsted, H. J. Jakobsen and C. J. McKenzie, CrystEngComm, 2010, 12, 2826 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details of syntheses, crystal structure determination, spectroscopic techniques and supporting CV, EPR, ESI-MS, GC-TCD, rR and time-resolved UV-Vis spectra. CCDC 1031521 and 1031522. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc08785b |
| This journal is © The Royal Society of Chemistry 2015 |