
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mixed diboration of alkenes in a metal-free context†
Nuria
Miralles
,
Jessica
Cid
,
Ana B.
Cuenca
,
Jorge J.
Carbó
* and
Elena
Fernández
*
Department Química Física i Inorgànica, University Rovira i Virgili, C/Marcel·lí Domingo s/n, Tarragona, Spain. E-mail: mariaelena.fernandez@urv.cat; j.carbo@urv.cat
First published on 8th December 2014
Abstract
Experimental and theoretical rationalization on regioselective mixed diboration of alkenes, with the unsymmetrical diboron reagent Bpin–Bdan, providing the protecting Bdan moiety in the internal position.
Catalytic diboration reactions have been deeply studied from two standard perspectives: (1) the use of symmetrical diborons such as B2pin2 and B2cat2 (pin = pinacolate and cat = catecholate) and (2) the need of transition metal complexes to activate the diborons and transfer the two boryl units to unsaturated substrates promoting the 1,2-addition.1–3 Even nanoporous materials have been used to strategically activate the symmetrical diboron reagents to enhance the chemoselectivity of the reaction, avoiding secondary reactions involved in boryl addition.4 But two recent stalwart linchpins have changed the concept of diboration reactions.
First, the diboron reagent can be activated by simple alkoxides,5 to enhance the nucleophilic character of the trivalent boryl moiety and force the olefin to act as an electrophile (Scheme 1).6 This pull–push effect of diborons7 has gained potential application not only in enantioselective and diastereoselective diborations,8 but also in generating unusual stereoselectivity in the elegant trans 1,2-diboration assisted by a propargylic alcohol unit.9
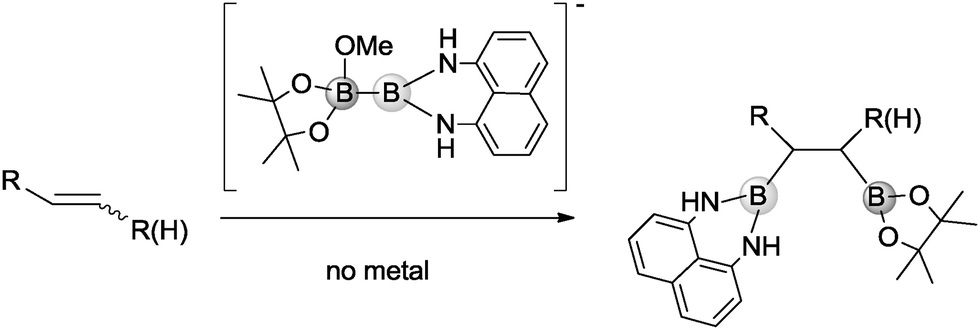 | ||
| Scheme 1 Mixed organocatalytic diboration of alkenes with the MeO− → Bpin–Bdan adduct. | ||
The second interesting input is the use of unsymmetrical diborons to difunctionalise targeted compounds. However, despite the fact that several efforts have been devoted to perform mixed diboration reactions, only Bpin–Bdan (dan = 1,8-diaminonaphthalene) has been successfully activated by Ir and Pt complexes and applied to the diboration of alkynes, giving the 1-alkene 1,2-diboronate derivative as the major regioisomer with the protecting Bdan unit at the terminal position.10 Interestingly, organoboranes with C-Bdan moieties are particularly easy to handle since the dan moiety acts as a masking group on B.11 Alternative unsymmetrical diboron compounds of the type pinB–B[(NR)2C6H4] (R = Me, Bn, SiMe3) have been recently isolated, but their application in catalysis has not yet been developed.12
Combining both challenges, we became interested in activating unsymmetrical diborons, such as Bpin–Bdan, with an alkoxide to generate the corresponding Lewis acid–base adduct and conduct an easy mixed diboration reaction, in the absence of metals or additives. Our goal was challenging, as the control of the diborated regioisomer in the organocatalytic approach could be a matter of concern. Nevertheless previous studies have shown that the MeO− → Bpin–Bdan adduct is formed preferentially because the π-donation from the lone pair of nitrogen to the empty orbital of boron protects the Bdan moiety from alkoxide attack.13 These conditions allowed the selective delivery of Bdan as a nucleophile. In this scenario, our objective in the present work is to activate the Bpin–Bdan reagent with methoxide and control the regioselective addition of Bdan and Bpin moieties to alkenes (Scheme 1).
We first attempted to find the optimal conditions for the diboration of allylbenzene (1) with Bpin–Bdan. When the reaction was carried out in THF as a solvent, at 70 °C, and 2 eq. of MeOH/30 mol% of Cs2CO3, (in order to mimic the reaction conditions used with B2pin2),5 we found that no diborated product was formed. Introducing MeOH as the solvent seemed to favour the reaction outcome, and moderate conversion from substrate 1 was achieved (Table 1, entry 1). The 1H NMR spectra of the crude reaction show two different groups of signals for the diborated product in a ratio of 4/1. The 1H, NOESY 1D experiment demonstrated that the major diborated regioisomer contained the Bdan moiety in the internal position. We next performed the mixed diboration reaction with vinylcyclohexane and in the presence of 30 mol% of Cs2CO3; conversion was high (81%) but even more interestingly, the regioselectivity was almost quantitative for the diborated product with Bdan in the internal position (Table 1, entry 2). A similar trend was observed with 50 mol% of Cs2CO3, with a slight increase in the global conversion (88%). Subsequently, terminal olefins of variable alkyl chain lengths were subjected to the same organocatalytic diboration reaction with Bpin–Bdan, and despite the amount of base used, conversions were very high and regioselectivity was favoured towards the regioisomer with the Bdan moiety in the internal position, by an average ratio of 9/1 (Table 1, entries 3–6). Isolated yields of the major regioisomer were in general low since the mixed diborated product partially decomposed along the purification procedure (flash chromatography). However, product 6a could be successfully isolated in up to 70% IY, (Table 1, entry 6), as we noted that the longer the aliphatic chain, the higher is the stability of the diborated product. For comparison, when NaOMe was used as base (30 mol%), the diboration of 6 was significantly diminished (Table 1, entry 6).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Cs2CO3 (mol%) | Conv.b (%) | a/b | [IY (%)]ca |
| a Reaction conditions: substrate (0.5 mmol), Bpin–Bdan (1.1 eq.), MeOH (2 mL), 70 °C, 12 h. b Conversion and regioselectivity calculated by GC/MS from an average of two essays. c Isolated yield of regioisomer a after purification by flash chromatography (see the ESI for details). d 30 mol% NaOMe. | |||||
| 1 |
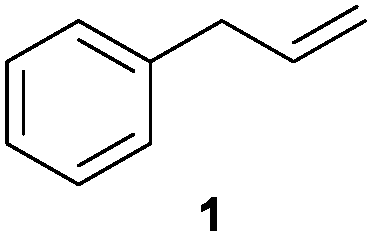
|
30 | 56 | 80/20 | 30 |
| 2 |
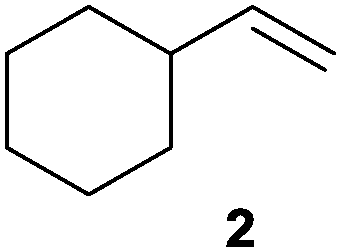
|
30 | 81 | 97/3 | 38 |
| 50 | 88 | 94/6 | 40 | ||
| 3 |
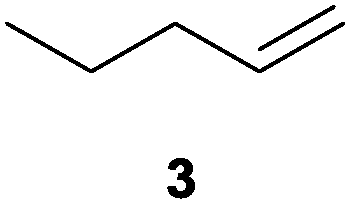
|
30 | 48 | 92/8 | 20 |
| 4 |
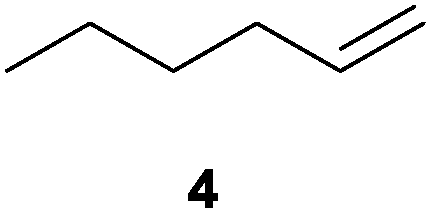
|
30 | 42 | 88/12 | 17 |
| 5 |
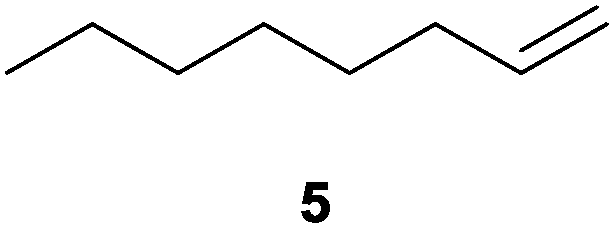
|
30 | 65 | 92/18 | 25 |
| 6 |
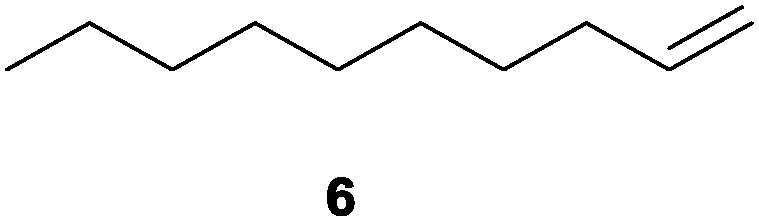
|
30 | 75 | 91/9 | 70 |
| 50 | 85 | 80/20 | 62 | ||
| 30d | 11 | 90/10 | — | ||
This new methodology to obtain mixed diborated products in the absence of transition metal complexes has a substrate scope limitation, because when vinylarenes were exposed to the diboration reaction with Bpin–Bdan under the same conditions, only the hydroborated product with Bdan in the terminal position was observed at the end of the reaction (Scheme 2).
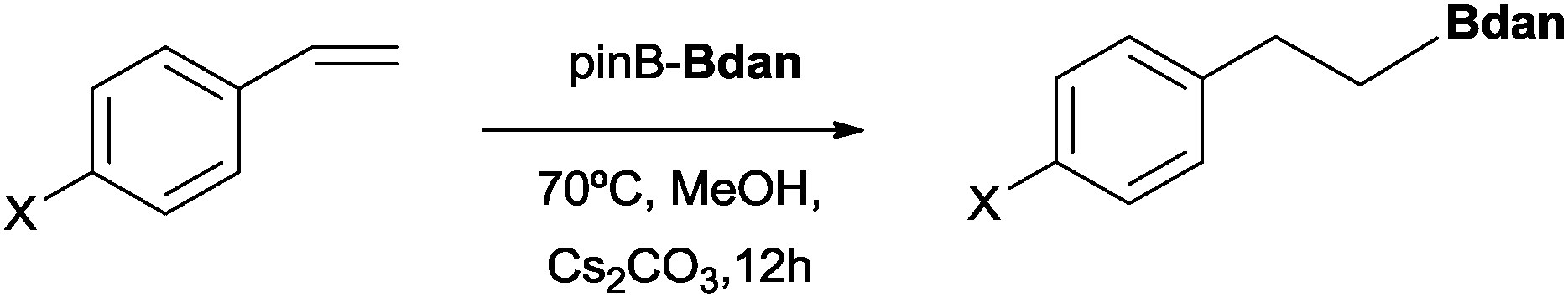 | ||
| Scheme 2 Reactivity of vinylarenes with the MeO− → Bpin–Bdan adduct, (X = Me, Cl, H). | ||
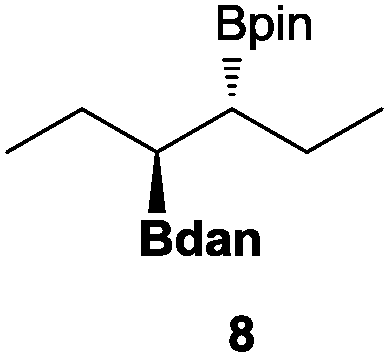
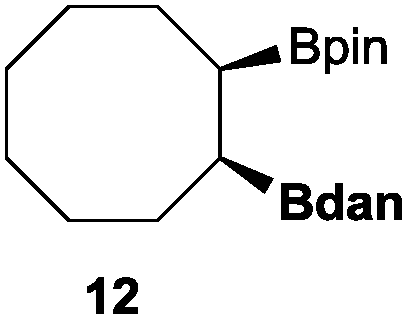
In the second set of experiments, internal olefins were subjected to the organocatalytic mixed diboration reaction and cis-3-hexene (7) was moderately converted into the desired product 8, despite the amount of base used (Table 2, entry 1). The 1,2-addition of Bpin and Bdan moieties to cyclic olefins took place in a syn fashion, (Table 1, entries 2–4), and despite the moderate conversion of the internal olefins to the diborated product, compound 10 could be isolated in up to 42% IY (Table 2, entry 2).
| Entry | Substrate | Cs2CO3 (mol%) | Conv.b (%) | Product | [IY (%)]c |
|---|---|---|---|---|---|
| a Reaction conditions: substrate (0.5 mmol), Bpin–Bdan (1.1 eq.), MeOH (2 mL), 70 °C, 12 h. b Conversion and calculated by GC/MS from an average of two essays. c Isolated yield of regioisomer a after purification by flash chromatography (see the ESI for details). | |||||
| 1 |

|
30 | 55 |
|
17 |
| 50 | 62 | 24 | |||
| 2 |
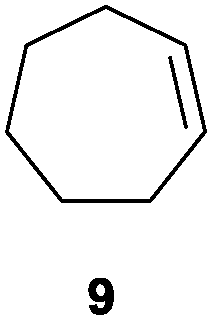
|
50 | 49 |
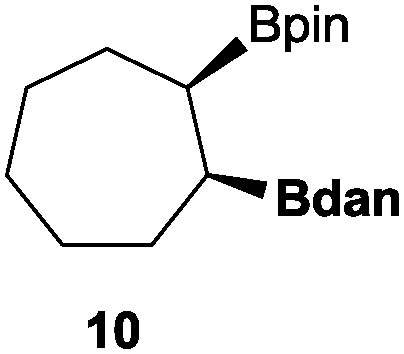
|
42 |
| 3 |

|
50 | 67 |
|
32 |
| 4 |
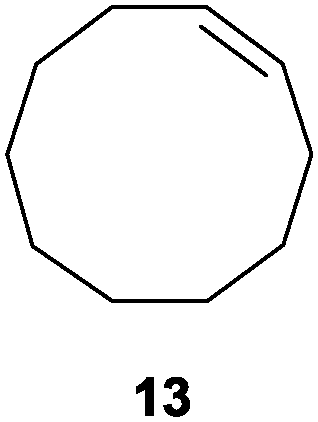
|
50 | 59 |
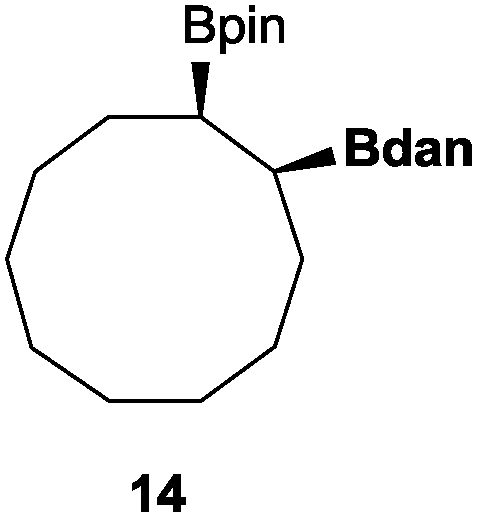
|
21 |
In order to have more insight into the mechanism of the metal-free mixed diboration reaction, we conducted DFT calculations14 with the aim to understand the intrinsic control of the regioselectivity. Bo and co-workers have computationally characterised the mechanism of the organocatalytic diboration reaction of non-activated olefins with the B2pin2 reagent.5 They proposed the nucleophilic attack of the boron reagent towards the substrate through an interaction between the strongly polarised B–B σ-bond of the activated diboron reagent and the antibonding π* orbital of the olefin.5 We used this proposal as a starting point to analyse the origin of the regioselectivity in diboration. Fig. 1 depicts the energy profile yielding the pathways of the two possible regioisomers.
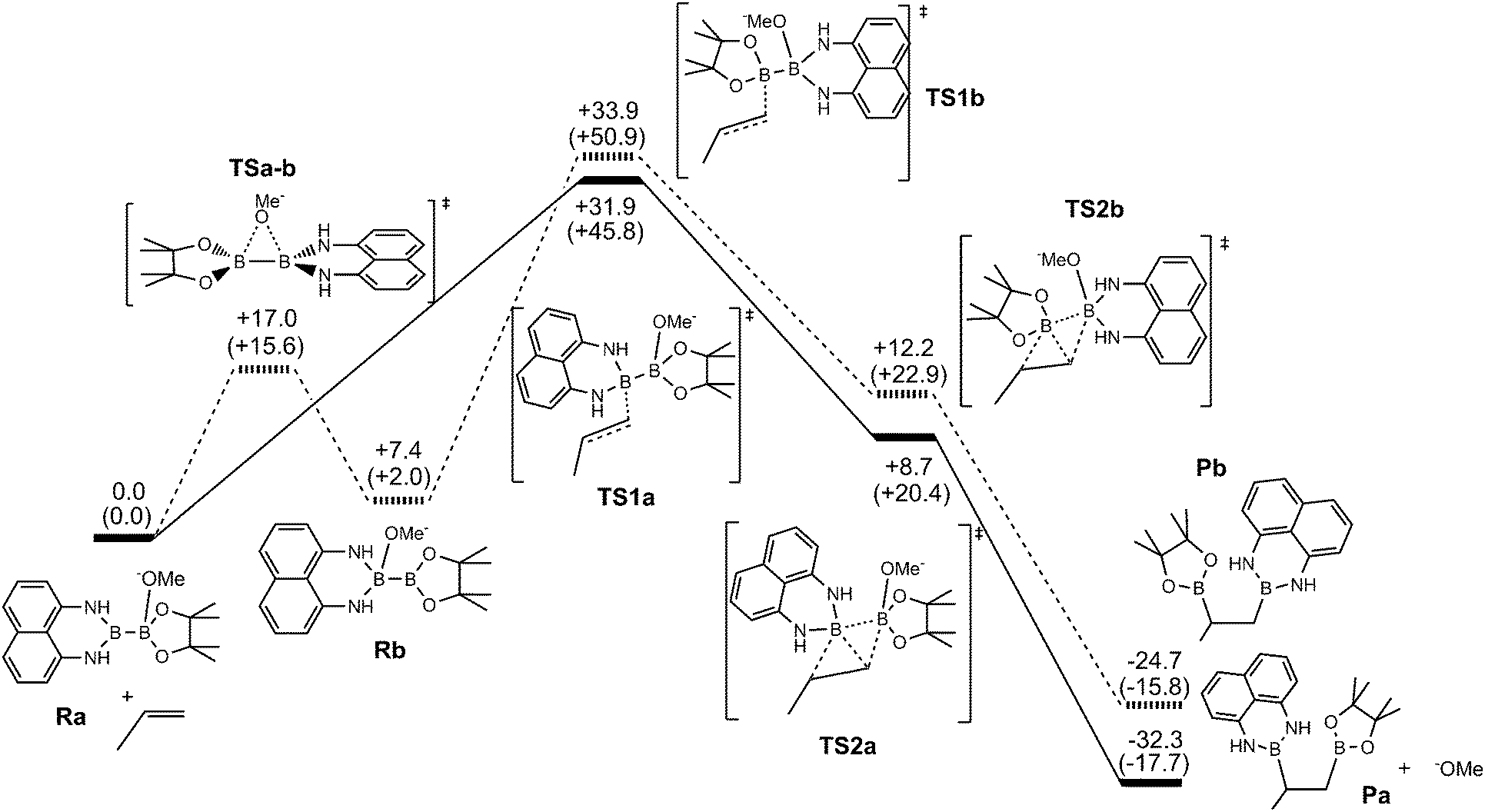 | ||
| Fig. 1 Calculated energy profile for the organocatalytic diboration of propene with the Bpin–Bdan diboron reagent in the presence of MeO−. Solid lines correspond to the attack of the Bdan moiety and dashed lines correspond to the attack of the Bpin moiety. Electronic energies and Gibbs free energies in MeOH (in parentheses) in kcal mol−1. | ||
Initially, the methoxide ions generated from MeOH in the presence of the base can form two different Lewis acid–base adducts with the unsymmetrical Bpin–Bdan: MeO− → Bpin–Bdan (Ra) and MeO− → Bdan–Bpin (Rb). Recently, we have shown that the Bpin moiety is a stronger Lewis acid than Bdan, and consequently, the former adduct Ra is 7.4 kcal mol−1 lower in energy than Rb.13 Here, Gibbs free energies including solvent effects via a continuum model show the same trends (Fig. 1).14 The energy barrier for the interconversion between the two adducts is modest (17.0 and 9.6 kcal mol−1 for Ra → Rb and Rb → Ra, respectively), and more importantly, lower than the energy required to transfer the boron moieties to alkenes (see below).
Diboration occurs sequentially through two connected transition states (TS1 and TS2).5 In TS1, the nucleophilic sp2 boryl unit interacts with the terminal carbon of the alkene.15 Then, the quaternised boron atom becomes electrophilic and capable of interacting with the negatively charged olefin-boryl fragment, as described in TS2. Nucleophilic boryl attack on the terminal carbon to reach TS1 is the most energy demanding step and the one that determines the regioselectivity. As expected,13 the computed energy barrier for the attack of the Bdan group in Ra (+31.9 kcal mol−1) is higher than that for the Bpin group in Rb, (+26.5 kcal mol−1). However, owing to the relatively low energy barrier for interconversion of Ra and Rb (see Fig. 1), the product distribution should be determined by the relative energy between the transition states of both paths, TS1a and TS1b. Thus overall, the attack on the alkene through the Bdan moiety (TS1a) is lower in energy than through Bpin (TS1b) by 2 kcal mol−1. The transition state TS1a is connected with another lower energy-lying transition state, TS2a, in which the Bdan moiety shifts to the internal olefinic carbon and the Bpin(OMe) moiety binds to the terminal carbon leading to the diborated product, Pa, and a methoxide molecule (see Fig. 1). These results are in good agreement with experimentally observed quantitative selectivity for Bdan addition in the internal position, albeit with non-negligible formation of the opposite regioisomer. Moreover, they explain the observed regioselectivity that is opposite to that found for diboration of alkynes by Ir and Pt complexes.10 In our case, the alkoxide Lewis base does not only activate the boron–boron bond, but also inverts its polarity driving diboron addition to a specific regioisomer.
Finally, we performed additional calculations to gain some insight into the substrate dependence of the reaction outcome: diboration vs. hydroboration. The mechanistic proposal by Bo and co-workers can also explain the hydroboration side reaction.5 According to this mechanism, when the system reaches transition state TS1, the reaction path bifurcates connecting with transition state TS2 or with intermediate I1 that yields hydroboration product via protonation and releases Bpin–OMe (see Scheme 3). For styrene, the computed energy of TS1a and I1a is lowered substantially compared to propene (Scheme 3). The phenyl substituent of the alkene stabilizes the negative charge generated at the internal alkene carbon, as well as the negative charge of intermediate I1a. Thus, the formation of hydroborated products is favoured for vinylarenes, which is in full agreement with the experimental findings.
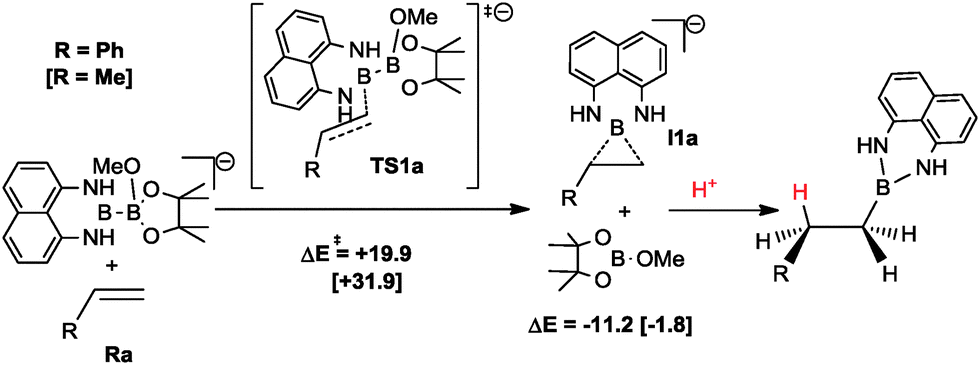 | ||
| Scheme 3 Proposed mechanism for hydroboration of propene and styrene leading to Bdan transfer. Calculated electronic energies for styrene (and in brackets for propene) in kcal mol−1. | ||
In summary, we have found that mixed diboration reactions with the Bpin–Bdan reagent, can be carried out in a metal-free context, with high control of regioselectivity, locating the Bdan unit in the internal position. Ten new diborated products have been isolated, most of them in a pure form but some as a mixture of the two regioisomers. The B nucleophile in the Bdan moiety is generated from the easily accessible, chemically resistant diboron reagent Bpin–Bdan and methoxide. The possibility to subsequently create an electrophilic Bpin unit on the activated diboron reagent makes the mixed diboration possible. Calculations on the energy profile for the organocatalytic diboration of propene with the Bpin–Bdan diboron reagent in the presence of alkoxide, support the experimental observation and rationalise the reaction outcome.
Notes and references
- The original Pt mediated diboration of alkynes: T. Ishiyama, N. Matsuda, N. Miyaura and A. Suzuki, J. Am. Chem. Soc., 1993, 115, 11018 CrossRef CAS ; the original Rh mediated diboration of alkenes: T. R. Baker, P. Nguyen, T. B. Marder and S. A. Westcott, Angew. Chem., Int. Ed. Engl., 1995, 34, 1336 CrossRef ; the original Pt mediated diboration of alkenes: T. Ishiyama, M. Yamamoto and N. Miyaura, Chem. Commun., 1997, 689 RSC.
- Reviews: (a) J. Takaya and N. Iwasawa, ACS Catal., 2012, 2, 1993 CrossRef CAS; (b) M. Suginome and T. Ohmura, In Boronic Acids, Wiley-VCH, New York, 2nd edn, 2011, vol. 1, 171 Search PubMed; (c) H. E. Burks and J. P. Morken, Chem. Commun., 2007, 4717 RSC; (d) J. Ramírez, V. Lillo, A. M. Segarra and E. Fernandez, C. R. Chim., 2007, 10, 138 CrossRef PubMed; (e) T. Ishiyama and N. Miyaura, Chem. Rec., 2004, 3, 271 CrossRef CAS PubMed; (f) T. B. Marder and N. C. Norman, Top. Catal., 1998, 63 CrossRef CAS.
- (a) T. Ishiyama, N. Matsuda, M. Murata, F. Ozawa, A. Suzuki and N. Miyaura, Organometallics, 1996, 15, 713 CrossRef CAS; (b) T. Ishiyama, T. Kitano and N. Miyaura, Tetrahedron Lett., 1998, 39, 2357 CrossRef CAS; (c) T. Ishiyama, S. Momota and N. Miyaura, Synlett, 1999, 1790 CrossRef CAS PubMed; (d) F. Y. Yang and C.-H. Cheng, J. Am. Chem. Soc., 2001, 123, 761 CrossRef CAS; (e) J. B. Morgan, S. P. Miller and J. P. Morken, J. Am. Chem. Soc., 2003, 125, 8702 CrossRef CAS PubMed; (f) N. F. Pelz, A. R. Woodward, H. E. Burks, H. E. Sieber and J. D. Morken, J. Am. Chem. Soc., 2004, 126, 16328 CrossRef CAS PubMed; (g) J. D. Sieber and J. P. Morken, J. Am. Chem. Soc., 2006, 128, 74 CrossRef CAS PubMed; (h) H. Y. Cho and J. P. Morken, J. Am. Chem. Soc., 2008, 130, 16140 CrossRef CAS PubMed; (i) T. Ishiyama and N. Miyaura, J. Organomet. Chem., 2000, 611, 392 CrossRef CAS; (j) G. Lesley, P. Nguyen, N. J. Taylor, T. B. Marder, A. J. Scott, W. Clegg and N. C. Norman, Organometallics, 1996, 15, 5137 CrossRef CAS; (k) R. L. Thomas, F. E. S. Souza and T. B. Marder, J. Chem. Soc., Dalton Trans., 2001, 1650 RSC; (l) S. Trudeau, J. B. Morgan, M. Shrestha and J. P. Morken, J. Org. Chem., 2005, 70, 9538 CrossRef CAS PubMed; (m) L. T. Kliman, S. N. Mlynarski and J. P. Morken, J. Am. Chem. Soc., 2009, 131, 13210 CrossRef CAS PubMed; (n) J. R. Coombs, F. Haeffner, L. T. Kliman and J. P. Morken, J. Am. Chem. Soc., 2013, 135, 11222 CrossRef CAS PubMed; (o) N. Iwadate and M. Suginome, J. Am. Chem. Soc., 2010, 132, 2548 CrossRef CAS PubMed; (p) V. Lillo, M. R. Fructos, J. Ramírez, A. A. C. Braga, F. Maseras, M. M. Díaz-Requejo, P. J. Pérez and E. Fernández, Chem. – Eur. J., 2007, 13, 2614–2621 CrossRef CAS PubMed; (q) H. Yoshida, S. Kawashima, Y. Takemoto, K. Okada, J. Ohshita and K. Takaki, Angew. Chem., Int. Ed., 2012, 51, 235 CrossRef CAS PubMed; (r) C. J. Adams, R. A. Baber, A. S. Batsanov, G. Bramham, J. P. H. Charmant, M. F. Haddow, J. A. K. Howard, W. H. Lam, Z. Lin, T. B. Marder, N. C. Norman and A. G. Orpen, Dalton Trans., 2006, 1370 RSC.
- (a) J. Ramírez, M. Sanaú and E. Fernández, Angew. Chem., Int. Ed., 2008, 47, 5194 CrossRef PubMed; (b) Q. Chen, J. Zhao, Y. Ishikawa, N. Asao, Y. Yamamoto and T. Jin, Org. Lett., 2013, 15, 5766 CrossRef CAS PubMed.
- A. Bonet, C. Pubill-Ulldemolins, C. Bo, H. Gulyás and E. Fernández, Angew. Chem., Int. Ed., 2011, 50, 7158 CrossRef CAS PubMed.
- (a) J. Cid, H. Gulyás, J. J. Carbó and E. Fernández, Chem. Soc. Rev., 2012, 41, 3558 RSC; (b) J. Cid, J. J. Carbó and E. Fernández, Chem. – Eur. J., 2012, 18, 12794 CrossRef CAS PubMed.
- (a) K. Lee, A. R. Zhugralin and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131, 7253 CrossRef CAS PubMed; (b) A. Bonet, H. Gulyás and E. Fernández, Angew. Chem., Int. Ed., 2010, 49, 5130 CrossRef CAS PubMed; (c) C. Pubill-Ulldemolins, A. Bonet, C. Bo, H. Gulyás and E. Fernández, Chem. – Eur. J., 2012, 18, 1121 CrossRef CAS PubMed; (d) C. Solé and E. Fernández, Angew. Chem., Int. Ed., 2013, 52, 11351 CrossRef PubMed; (e) H. Ito, Y. Horita and E. Yamamoto, Chem. Commun., 2012, 48, 8006 RSC; (f) I. Ibrahem, P. Breistein and A. Córdova, Chem. – Eur. J., 2012, 18, 5175 CrossRef CAS PubMed; (g) H. Wu, S. Radomkit, J. M. O'Brien and A. H. Hoveyda, J. Am. Chem. Soc., 2012, 134, 8277 CrossRef CAS PubMed; (h) C. Kleeberg, A. G. Crawford, A. S. Batsanov, P. Hodgkinson, D. C. Apperley, M. S. Cheung, Z. Y. Lin and T. B. Marder, J. Org. Chem., 2012, 77, 785 CrossRef CAS PubMed; (i) C. Sole, H. Gulyás and E. Fernández, Chem. Commun., 2012, 48, 3769 RSC.
- (a) A. Bonet, C. Sole, H. Gulyás and E. Fernández, Org. Biomol. Chem., 2012, 10, 9677 RSC; (b) Th. P. Blaisdell, Th. C. Caya, L. Zhang, A. Sanz-Marco and J. P. Morken, J. Am. Chem. Soc., 2014, 136, 9256 CrossRef PubMed.
- Y. Nagashima, K. Hirano, R. Takita and M. Uchiyama, J. Am. Chem. Soc., 2014, 136, 8532 CrossRef CAS PubMed.
- N. Iwadate and M. Suginome, J. Am. Chem. Soc., 2010, 132, 2548 CrossRef CAS PubMed.
- (a) H. Noguchi, K. Hojo and M. Suginome, J. Am. Chem. Soc., 2007, 129, 758 CrossRef CAS PubMed; (b) N. Iwadate and M. Suginome, J. Organomet. Chem., 2009, 694, 1713 CrossRef CAS PubMed; (c) H. Noguchi, T. Shioda, Ch.-M. Chou and M. Suginome, Org. Lett., 2008, 10, 377 CrossRef CAS PubMed; (d) L. Iannazzo, K. P. C. Vollhardt, M. Malacria, C. Aubert and V. Gandon, Eur. J. Org. Chem., 2011, 3283 CrossRef CAS; (e) X. Feng, H. Jeon and J. Yun, Angew. Chem., Int. Ed., 2013, 52, 3989 CrossRef CAS PubMed; (f) H. Yoshida, Y. Takemoto and K. Takaki, Chem. Commun., 2014, 50, 8299 RSC.
- C. Borner and Ch. Kleeberg, Eur. J. Inorg. Chem., 2014, 2486 CrossRef CAS.
- J. Cid, J. J. Carbó and E. Fernández, Chem. – Eur. J., 2014, 20, 3616 CrossRef CAS PubMed.
- Calculations were performed using Gaussian09 (B3LYP functional) and the 6-31g(d,p) basis set. In parenthesis we also provide the free energy corrections including the solvent effect of methanol (ε = 32.613) that introduced into the optimised vacuum geometries by using the IEFPCM continuum model. Main discussion used electronic energies because ΔG values overestimate the entropic cost for bimolecular processes. See the ESI† for details.
- We have also considered the attack to the internal carbon of the alkene but the corresponding TSs are 2 and 5 kcal mol−1 higher than TS1a and TS1b. See the ESI† for details.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cc08743g |
| This journal is © The Royal Society of Chemistry 2015 |