
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProtecting group free enantiospecific total syntheses of structurally diverse natural products of the tetrahydrocannabinoid family†
Dattatraya H.
Dethe
*,
Rohan D.
Erande‡
,
Samarpita
Mahapatra‡
,
Saikat
Das
and
Vijay Kumar
B.
Department of Chemistry, Indian Institute of Technology Kanpur, Kanpur-208016, India. E-mail: ddethe@iitk.ac.in; Fax: +91-512-2597436; Tel: +91-512-2596537
First published on 5th January 2015
Abstract
A simple, highly diastereoselective, Lewis acid catalyzed Friedel–Crafts coupling of a cyclic allylic alcohol with resorcinol derivatives has been developed. The method was applied for the enantiospecific total syntheses of structurally diverse natural products such as machaeriol-D, Δ8-THC, Δ9-THC, epi-perrottetinene and their analogues. Synthesis of both natural products and their enantiomers has been achieved with high atom economy, in a protecting group free manner and in less than 6 steps, the longest linear sequence, in a very good overall yield starting from R-(+) and S-(−)-limonene.
The ever-growing field of total synthesis of natural products continues to be the source of inspiration for many synthetic chemists worldwide.1–3 Natural product synthesis also plays an important role in developing many areas of modern day biology. Over the century, total synthesis has now reached a stage where, given sufficient time and effort, synthetic chemists are able to construct almost any known natural product in a small quantity. However, the gram scale synthesis of complex natural products for further biological studies, using a minimum number of synthetic transformations, labour and material expenses presents significant challenges to organic chemists. Total synthesis in the 21st century should be an ideal synthesis4 starting with readily available, inexpensive starting materials in a simple, protecting group free, safe, environmental friendly and cost effective manner, which proceeds quickly and in quantitative yield. In this context, we have developed a simple, short, protecting group free, atom economical and universal strategy for the synthesis of structurally diverse natural products and natural product analogues of different classes. We are able to solve several different and difficult problems in complex molecule synthesis by using the simple and well studied reaction, the Friedel–Crafts reaction. Machaeriols are a structurally diverse and biologically potent group of tetrahydrocannabinoids containing linearly fused 6,6,6-tricyclic ring systems. Machaeridiol A–C, 1–3, and machaeriol A–D, 4–7, were isolated by Muhammad et al. in 2003, from the stem bark of Machaerium multiflorum spruce.5 The first member of this family, Δ9-trans-tetrahydrocannabinol 8 (Δ9-THC) was isolated from Cannabis sativa var. indica in 1964.6 Subsequently, several additional cannabinoids, such as Δ8-trans-THC 9,7 conicol 10,8 perrottetinene 11,9 bisabosqual-A 1210 have been isolated and structurally characterized (Fig. 1). Since their isolation Δ9-THC, Δ8-THC and related tetrahydrocannabinols have been among the most highly sought synthetic targets.11 Recently, the research groups of She and Pan have reported an elegant approach for the first enantioselective total synthesis of (+)-machaeriol-D 7 using SN2′ reaction as a key step with the longest linear sequence of 18 steps.12 Herein we report the atom economic, protecting group free six step total synthesis of both the enantiomers of machaeriol-D 7 facilitated by a newly developed methodology for one pot C–C and C–O bond formation and a strategic effort to avoid the use of a protecting group and expensive reagents.
 | ||
| Fig. 1 Selected naturally occurring hexahydro-6H-benzo[c]-chromenes. | ||
It was envisioned that machaeriol-D 7 could be synthesized from compound 13 by allylic oxidation and further diastereoselective double bond reduction. Compound 13 in turn could be prepared via the coupling of allylic alcohol 14 and the electron rich aromatic moiety 15 by concomitant formation of C–C and C–O bonds. So our strategy was based on the well studied Friedel–Crafts reaction but in a modified way which is unprecedented in the literature (Scheme 1).
 | ||
| Scheme 1 Retrosynthetic analysis for machaeriol-D. | ||
To begin with, compound 15 was prepared using the known two step protocol by the Suzuki coupling of compounds 16 and 17 followed by demethylation (Scheme 2). Allylic alcohol 14 was obtained from limonene by allylic oxidation followed by reduction of the ketone thus formed (see ESI†). After having key coupling partners in hand, various acids were screened such as p-TSA, TFA, BF3·OEt2, AlCl3, InCl3 for the coupling reaction. Among these BF3·OEt2 was found to be the best catalyst for the cyclization reaction. Compound 15 and allylic alcohol 14 were merged without protecting groups using 10 mol% BF3·OEt2 to furnish compound 18 as a single diastereomer in 90% isolated yield in just 5 min at room temperature. Excellent diastereoselectivity was observed possibly due to the adjacent bulkier isopropenyl group.13 Interestingly when 50 mol% of BF3·OEt2 was used and the reaction mixture was allowed to stir for 2 h, we also observed formation of a pyran ring along with isomerization of the double bond to generate the core of machaeriol-D 19. It is presumed that double bond isomerisation might be due to the thermodynamic stability of compound 19 over 13. m-CPBA mediated epoxidation of the double bond in compound 19 generated the epoxide 20 in 74% yield as a single diastereoisomer. At this stage we could not assign the stereochemistry of epoxide. Regioselective opening of the epoxide using combination of NaBH3CN and BF3·OEt2 generated compound 21, whose 1H and 13C data did not match with machaeriol-D 7. This made us realize that epoxidation has occurred from the α-face. Interestingly, BF3·OEt2 catalyzed semipinacol rearrangement14 of the epoxide generated the ketone 22 in 82% yield as a single diastereoisomer. The structure and stereochemistry of ketone 22 was established by single crystal X-ray analysis.15 Reduction of ketone 22 using NaBH4 at 0 °C furnished the natural product (+)-machaeriol-D 7 in 96% yield (Scheme 3).
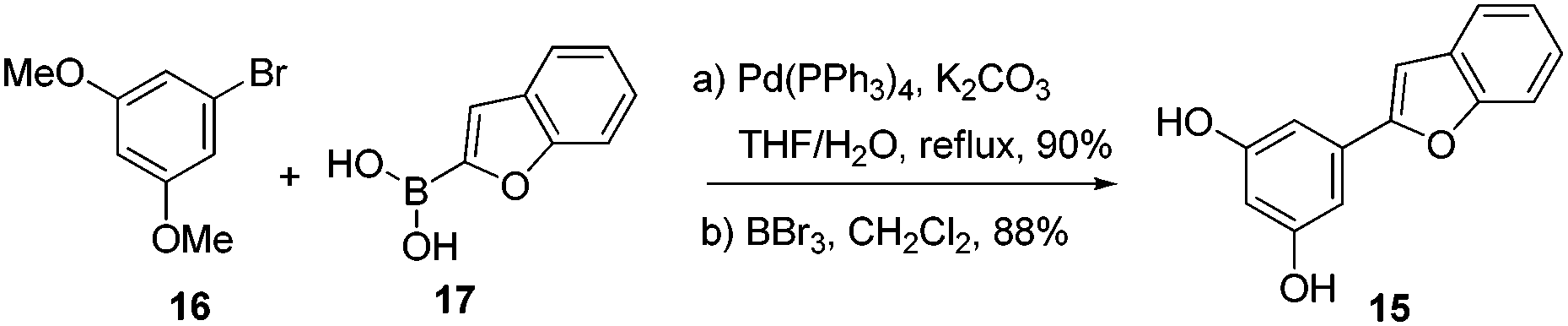 | ||
| Scheme 2 Synthesis of precursor 15. | ||
 | ||
| Scheme 3 Total synthesis of (+) and (−)-machaeriol-D and epi-machaeriol-D. | ||
When NaBH4 reduction was carried out at room temperature, a minor amount (6%) of another diastereomer was observed, whose spectral data were identical with compound 21 obtained from epoxide 20 by reductive epoxide opening. This further confirms the stereochemistry of epoxide 20 and epi-machaeriol 21. So in six simple steps from S-(−)-limonene, (+)-machaeriol-D is now accessible in large quantities via direct coupling of alcohol 14 and resorcinol derivative 15. Similarly (−)-machaeriol-D 7 was synthesized starting from R-(+)-limonene. On the way, we have also accomplished the one pot total synthesis of (+) and (−)-Δ9-THC 8, (+) and (−)-Δ8-THC 9, epi-perrottetinene 23 and their analogues as shown in Scheme 4.
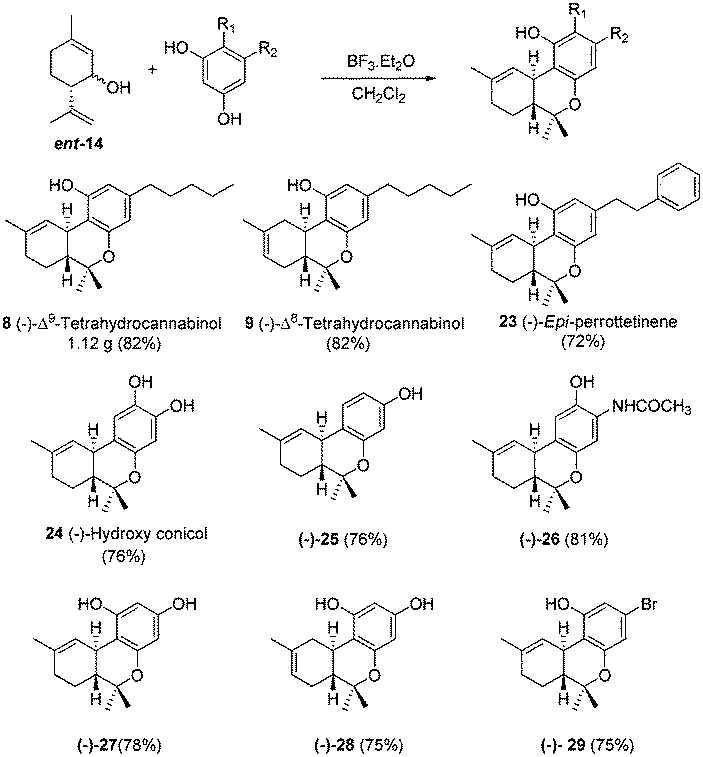 | ||
| Scheme 4 Synthesis of analogues of tetrahydrocannabinols. | ||
Independent coupling of alcohol 14 and ent-14 with olivetol furnished both the enantiomers of Δ8- and Δ9-THC. Furthermore, this reaction was robust and was conducted on a gram-scale synthesis of Δ9-THC yielding 1.12 g of it. Coupling of alcohol 14 with various resorcinol derivatives generated half a dozen congeners of tetrahydrocannabinols 23–29. Although to date many syntheses of Δ9-THC have been reported in the literature, to best of our knowledge, only one synthesis each of Δ8-THC 9,16 conicol 10,17 perrottetinene 1118 and epi-perrottetinene 2318 have been reported in the literature.
Using a simple strategy we have achieved enantiospecific total syntheses of structurally diverse natural products isolated from different sources and having a wide range of biological activities. The synthesis of both natural products and their enantiomers has been achieved in a highly atom economical, protecting group free manner and in less than 6 steps, the longest linear sequence, starting from R-(+) and S-(−)-limonene. Finally it is worth mentioning that using a good strategy, even the Friedel–Crafts reaction can help solve total synthesis problems that have either not yet been solved or have required many steps through other routes.
We thank Prof. Viresh H. Rawal, University of Chicago, for his suggestions and comments to improve the manuscript substantially. We thank Mr Dipankar Sahoo and Prosenjit Daw for crystal structures. R. D. E., S. D. and V. K. thank CSIR, New Delhi, and S. M. thanks UGC, New Delhi, for the award of research fellowships. Financial support from IIT Kanpur and DST, New Delhi (SB/S1/OC-01/2014) is gratefully acknowledged.
Notes and references
- K. C. Nicolaou and E. J. Sorensen, Classics in Total Synthesis, VCH, New York, 1996 Search PubMed.
- K. C. Nicolaou and S. A. Snyder, Classics in Total Synthesis II, Wiley-VCH, Weinheim, 2003 Search PubMed.
- K. C. Nicolaou and J. S. Chen, Classics in Total Synthesis III, Wiley-VCH, New York, 2011 Search PubMed.
- T. Gaich and P. S. Baran, J. Org. Chem., 2010, 75, 4657 CrossRef CAS PubMed.
- I. Muhammad, X. C. Li, M. R. Jacob, B. L. Tekwani, D. C. Dunbar and D. Ferreira, J. Nat. Prod., 2003, 66, 804 CrossRef CAS PubMed.
- Y. Gaoni and R. Mechoulam, J. Am. Chem. Soc., 1964, 86, 1646 CrossRef CAS.
- E. C. Taylor, K. Lenard and Y. Shvo, J. Am. Chem. Soc., 1966, 88, 367 CrossRef CAS.
- L. Garrido, E. Zubia, M. J. Ortega and J. Salva, J. Nat. Prod., 2002, 65, 1328 CrossRef CAS PubMed.
- M. Toyota, T. Shimamura, H. Ishii, M. Renner, J. Braggins and Y. Asakawa, Chem. Pharm. Bull., 2002, 50, 1390 CrossRef CAS.
- K. Minagawa, S. Kouzuki, K. Nomura, Y. Kawamura, H. Tani, Y. Terui, H. Nakai and T. Kamigauchi, J. Antibiot., 2001, 54, 896 CrossRef CAS.
- (a) W. E. Childers and H. W. Pinnick, J. Org. Chem., 1984, 49, 5276 CrossRef CAS; (b) L. Crombie, W. M. L. Crombie, S. V. Jamieson and C. J. Palmer, J. Chem. Soc., Perkin Trans. 1, 1988, 1243 RSC; (c) D. A. Evans, E. A. Shaughnessy and D. M. Barnes, Tetrahedron Lett., 1997, 38, 3193 CrossRef CAS; (d) L. Crombie, W. M. L. Crombie, C. J. Palmer and S. V. Jamieson, Tetrahedron Lett., 1983, 24, 3129 CrossRef CAS; (e) R. W. Rickards and H. Ronnen-berg, J. Org. Chem., 1984, 49, 572 CrossRef CAS; (f) S. H. Baek, M. Srebnik and R. Mechoulam, Tetrahedron Lett., 1985, 26, 1083 CrossRef CAS; (g) V. Vaillancourt and K. F. Albizati, J. Org. Chem., 1992, 57, 3627 CrossRef CAS; (h) A. D. William and Y. Kobayashi, Org. Lett., 2001, 3, 2017 CrossRef CAS PubMed; (i) A. D. William and Y. Kobayashi, J. Org. Chem., 2002, 67, 8771 CrossRef CAS PubMed; (j) L. E. Pearson, N. Kanizaj, A. C. Willis, M. N. Paddon-Row and M. S. Sherburn, Chem. – Eur. J., 2010, 16, 8280 CrossRef PubMed; (k) E. Ballerini, L. Minuti and O. Piermatti, J. Org. Chem., 2010, 75, 4251 CrossRef CAS PubMed; (l) Q. Huang, B. Ma, X. Li, X. Pan and X. She, Synthesis, 2010, 1766 CAS; (m) L. Minuti and E. Ballerini, J. Org. Chem., 2011, 76, 5392 CrossRef CAS PubMed; (n) D. A. Evans, E. A. Shaughnessy and D. M. Barnes, Tetrahedron Lett., 1997, 38, 3193 CrossRef CAS; (o) D. A. Evans, D. M. Barnes, J. S. Johnson, T. Lectka, P. V. Matt, S. J. Miller, J. A. Murry, R. D. Norcross, E. A. Shaughnessy and K. R. Campos, J. Am. Chem. Soc., 1999, 121, 7582 CrossRef CAS; (p) B. M. Trost and K. Dogra, Org. Lett., 2007, 9, 861 CrossRef CAS PubMed.
- Q. Wang, Q. Huang, B. Chen, J. Lu, H. Wang, X. She and X. Pan, Angew. Chem., Int. Ed., 2006, 45, 3651 CrossRef CAS PubMed.
- A. Srikrishna and D. H. Dethe, Tetrahedron Lett., 2003, 44, 7817 CrossRef CAS PubMed.
- Z. L. Song, C. A. Fan and Y. Q. Tu, Chem. Rev., 2011, 111, 7523 CrossRef CAS PubMed.
- CCDC 1017678 (+)-22.
- L. J. Cheng, J. H. Xie, Y. Chen, L. X. Wang and Q. L. Zhou, Org. Lett., 2013, 15, 764 CrossRef CAS PubMed.
- B. C. Hong, P. Kotame, C. W. Tsai and J. H. Liao, Org. Lett., 2010, 12, 776 CrossRef CAS PubMed.
- Y. Song, S. Hwang, P. Gong, D. Kim and S. Kim, Org. Lett., 2008, 10, 269 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1017678 (+22). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc08562k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |