
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chiral Brønsted acid-catalyzed Friedel–Crafts alkylation of electron-rich arenes with in situ-generated ortho-quinone methides: highly enantioselective synthesis of diarylindolylmethanes and triarylmethanes†
Satyajit
Saha
,
Santosh Kumar
Alamsetti
and
Christoph
Schneider
*
Institut für Organische Chemie, Universität Leipzig, Johannisallee 29, D-4103 Leipzig, Germany. E-mail: schneider@chemie.uni-leipzig.de
First published on 3rd December 2014
Abstract
We disclose herein a highly enantioselective protocol for the Brønsted acid-catalyzed addition of indoles and phenols to in situ-generated ortho-quinone methides which deliver broadly substituted diarylindolylmethanes and triarylmethanes, respectively, in a one-pot reaction under very mild conditions. A chiral phosphoric acid catalyst has been developed for this process serving to convert the starting ortho-hydroxybenzhydryl alcohols into the reactive ortho-quinone methides and to control the enantioselectivity of the carbon–carbon bond-forming event via hydrogen-bonding.
Triarylmethanes have gained substantial attention from the synthetic community because of their importance in medicinal chemistry, materials science and as dye precursors.1 Several of them are known to be potential drug candidates for the treatment of cancer, bacterial infections, and diabetes2 and are also core structures in natural products such as for example in cassigarol B.3 Similarly, heteroaryl-substituted analogues of this product class have been shown to be powerful pharmaceuticals and bioactive molecules like letrozole, vorozole, and paraphenyl-substituted diindolylmethanes.4 Accordingly, novel synthetic methods to access enantiomerically highly enriched triarylmethanes and related compounds continue to be highly desirable.
Although a variety of racemic routes have been developed5 only a few enantioselective syntheses are currently available based upon reports from the groups of Jarvo,6 Watson,7 and Crudden8 who employed asymmetric cross-coupling technology to construct the target triarylmethanes in the optically highly enriched form. Apart from that You,9 Zhang,10 and Han11 developed Brønsted acid-catalyzed, enantioselective syntheses of special aryldiindolylmethanes starting from both aryl(3-indolyl)methanols and aryl(2-indolyl)methanols.
Recently, we have disclosed the phosphoric acid-catalyzed, highly enantioselective conjugate addition of 1,3-dicarbonyl compounds to in situ generated ortho-quinone methides (o-QM).12 This strategy was applied to a one-pot and straightforward synthesis of optically highly enriched 4-aryl-4H-chromenes and related heterocycles through a subsequent cyclodehydration reaction (Scheme 1, pathway a). o-QM constitute highly reactive synthetic intermediates participating easily in conjugate additions, hetero Diels–Alder reactions, and 6π-electrocyclizations.13 It was only recently that a range of catalytic, enantioselective processes have been successfully developed for o-QM chemistry including palladium-, cinchona alkaloid-, BINOL- and NHC-catalyzed reactions.14
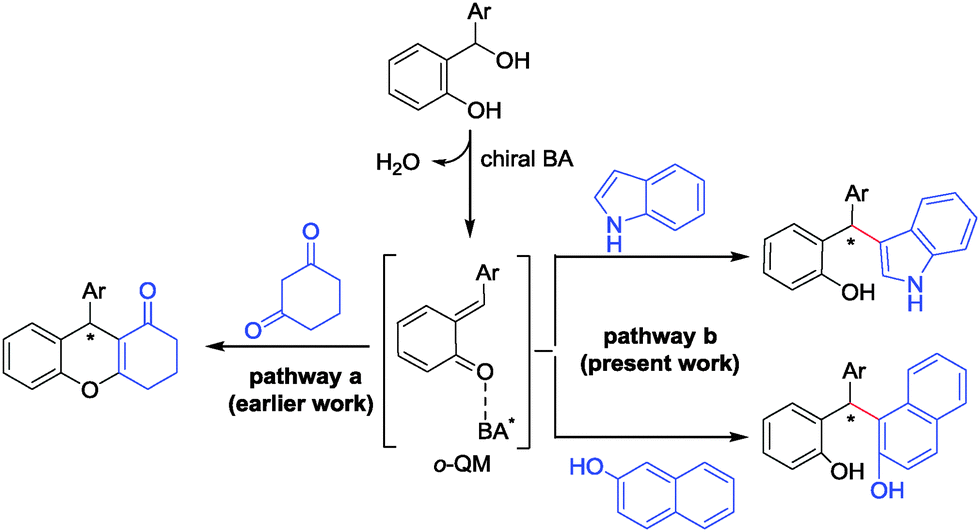 | ||
| Scheme 1 Brønsted acid-catalyzed reaction of o-QM with 1,3-diketones (pathway a) and with indoles and 2-naphthols (pathway b). | ||
In continuation of our interest in enantioselective reactions of hydrogen-bonded o-QM we now report that both indoles and naphthols are highly suitable nucleophiles for this purpose and deliver broadly substituted diarylindolylmethanes and triarylmethanes with excellent yields and enantioselectivities (Scheme 1, pathway b). Bach and coworkers pursued this strategy previously and obtained some addition products with moderate enantioselectivity.15 Very recently Sun et al. have shown that tertiary benzylic alcohols can form triarylmethanes that carry exclusively quaternary chiral centers upon indole addition.16
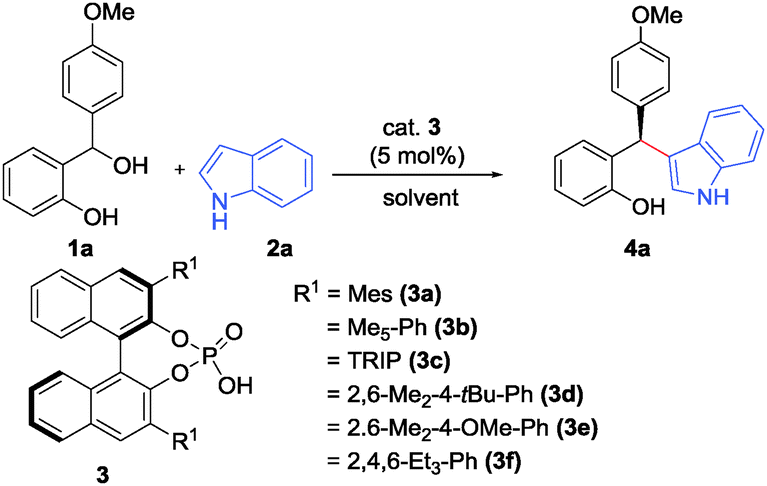
We initiated our studies by investigating the reaction of ortho-hydroxybenzhydryl alcohol 1a (1 equiv.) with indole (2a) (1.2 equiv.) in CH2Cl2 in the presence of various chiral phosphoric acids 3a–g (5 mol%) (Table 1). The (F–C) adduct 4a was obtained in good yields in almost all cases within 2.5 h at room temperature. Systematic screening of the catalysts further revealed that sterically demanding 3,3′-aryl substituents in the BINOL-backbone of the Brønsted acid catalyst gave improved enantioselectivities. An optimal selectivity was eventually obtained with phosphoric acid 3d (Ar = 2,6-Me2-4-tBuC6H2) which delivered the (F–C) addition product 4a with 94% yield and 91![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 er (Table 1, entry 4). Solvents like toluene and CH3CN were found to be inferior as compared to CH2Cl2, reducing the selectivity to 79:21 er and 81:19 er, respectively, albeit in excellent yields (entries 9 and 10). The amount of indole was further reduced to 1.0 equiv. without compromising the yield or selectivity of the reaction. Under these conditions the (F–C) adduct was isolated in 92% overall yield and with 92:8 er (entry 11). It is important to note that the free NH-moiety within the indole component is crucial for high enantioselectivity presumably via additional hydrogen-bonding to the phosphoric acid catalyst.17
:9 er (Table 1, entry 4). Solvents like toluene and CH3CN were found to be inferior as compared to CH2Cl2, reducing the selectivity to 79:21 er and 81:19 er, respectively, albeit in excellent yields (entries 9 and 10). The amount of indole was further reduced to 1.0 equiv. without compromising the yield or selectivity of the reaction. Under these conditions the (F–C) adduct was isolated in 92% overall yield and with 92:8 er (entry 11). It is important to note that the free NH-moiety within the indole component is crucial for high enantioselectivity presumably via additional hydrogen-bonding to the phosphoric acid catalyst.17
|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. | Solvent | Time (h) | Yieldb (%) | Erc |
| a All reactions were carried out with 0.23 mmol (1 equiv.) of 1a, 0.27 mmol (1.2 equiv.) of 2a and 5 mol% of catalyst 3a–3f in 3 mL of CH2Cl2 at rt. b Isolated yield of the purified product. c Er determined through chiral HPLC-analysis (see the ESI). d Reaction was carried out at 0 °C. e 1.0 equiv. of indole (2a) was used as the substrate. | |||||
| 1 | 3a | CH2Cl2 | 1.5 | 96 | 80:20 |
| 2 | 3b | CH2Cl2 | 1.5 | 97 | 89:11 |
| 3 | 3c | CH2Cl2 | 2 | 94 | 80:20 |
| 4 | 3d | CH2Cl2 | 1.5 | 94 |
91:9 |
| 5 | 3e | CH2Cl2 | 1.5 | 94 | 72:28 |
| 6 | 3f | CH2Cl2 | 1.5 | 96 | 89:11 |
| 7 | 3h | CH2Cl2 | 1.5 | 97 | 65:35 |
| 8d | 3d | CH2Cl2 | 4 | 95 | 86:14 |
| 9 | 3d | CH3CN | 2.5 | 96 | 79:21 |
| 10 | 3d | Toluene | 1.5 | 94 | 81:19 |
| 11 | 3d | CH2Cl2 | 1.5 | 92 |
92:8 |
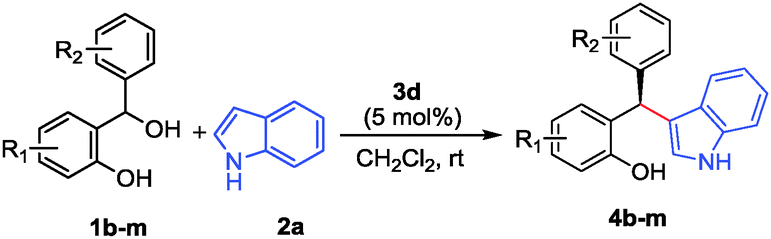
To evaluate the scope of this process various ortho-hydroxy benzhydrols 1b–m were subsequently reacted with indole (2a) under the above-optimized reaction conditions and the results are summarized in Table 2. In almost all cases studied, the reaction proceeded smoothly and was typically completed within 2–12 h at rt. The products 4b–m were obtained in excellent yields and with good to excellent enantioselectivities (entries 1–12). A range of substituents was readily accommodated at various positions both within the o-QM fragment as well as the β-aryl substituent delivering the products with excellent results as well.
|
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | Product | Yieldb (%) | Erc |
| a All reactions were carried out with 0.42 mmol (1 equiv.) of 1b–m, 0.42 mmol (1.0 equiv.) of indole (2a) and 5 mol% of catalyst 3d in 4 mL of CH2Cl2 at rt for 2–12 h. b Isolated yield of the purified product. c Er determined through chiral HPLC-analysis (see the ESI). | |||||
| 1 | 4-OMe | 2-Me | 4b | 87 | 96:4 |
| 2 | 4-OMe | 2-Ph | 4c | 85 | 98:2 |
| 3 | 4-OMe | 2-OMe | 4d | 83 | 95:5 |
| 4 | 4-OMe | 2-iPr | 4e | 85 | 96:4 |
| 5 | 4-OMe | 2,3-Me2 | 4f | 91 | 95:5 |
| 6 | 4-OMe | 4-Me | 4g | 86 | 92:8 |
| 7 | 4-Me | 2-Et | 4h | 92 | 95:5 |
| 8 | 4-Me | 2-Me | 4i | 82 | 94:6 |
| 9 | 4-tBu | 4-Me | 4j | 87 | 92:8 |
| 10 | 4-tBu | 2-OMe | 4k | 82 | 93:7 |
| 11 | 4-Br | 2-Et | 4l | 88 | 97:3 |
| 12 | 4-Br | 2-OMe | 4m | 84 | 99:1 |
A crystal structure of diarylindolylmethane 4c (entry 2) obtained by slow evaporation of CH2Cl2 revealed its absolute configuration which was assigned to all other products as well (Fig. 1).18
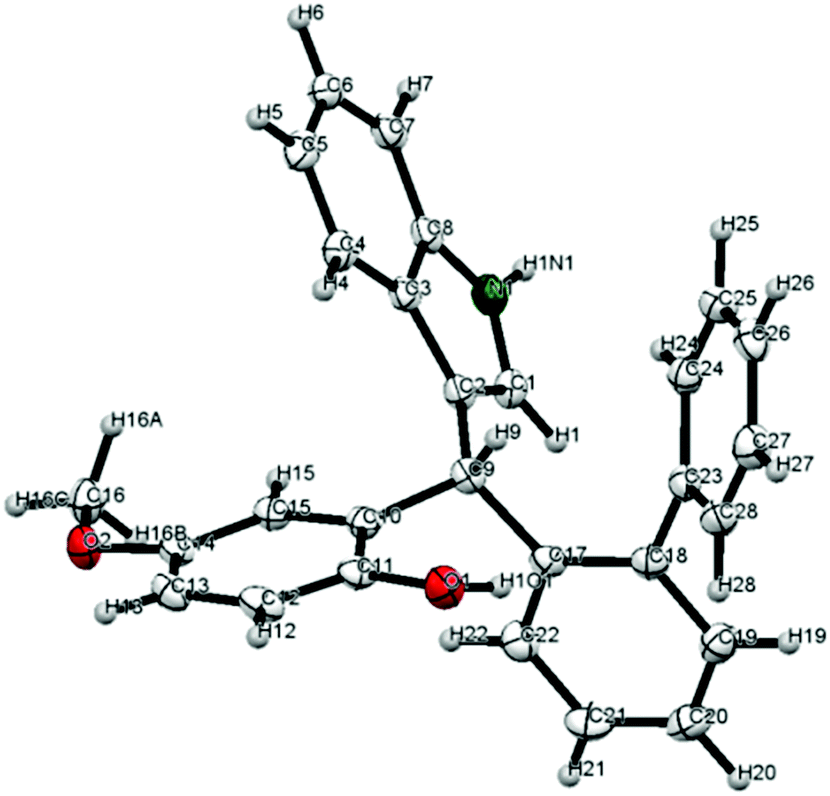 | ||
| Fig. 1 X-ray crystal structure of 4c. | ||
In addition, the influence of various functional groups and substituents in the indole component was investigated in this study (Table 3). These results clearly reveal an excellent functional group tolerance of this process and a broad set of heteroaryl-substituted triarylmethanes was obtained in good yields and enantioselectivities irrespective of the electronic properties of the substituents on the indole ring.
|
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | Yieldb (%) | Erc |
| a All reactions were carried out with 0.42 mmol (1 equiv.) of 1, 0.42 mmol (1.0 equiv.) of indoles 2b–j and 5 mol% of catalyst 3d in 4 mL of CH2Cl2 at rt for 2–12 h. b Isolated yield of the purified product. c Er determined through chiral HPLC-analysis (see the ESI). | |||||
| 1 | 4-OMe | 2,3-di-Me | 5-Me | 88 (5a) | 94:6 |
| 2 | 4-OMe | 2-Et | 5-Me | 86 (5b) | 95:5 |
| 3 | 4-OMe | 2-Ph | 5-Me | 85 (5c) | 92:8 |
| 4 | 4-tBu | 2-OMe | 5-Br | 84 (6a) | 94:6 |
| 5 | 4-OMe | 2-OMe | 5-Br | 90 (6b) | 95:5 |
| 6 | 4-OMe | 2-Et | 4-Br | 87 (7a) | 96:4 |
| 7 | 4-OMe | 2-Br | 4-Br | 82 (7b) | 92:8 |
| 8 | 4-OMe | 2-OMe | 6-Cl | 84 (8a) | 94:6 |
| 9 | 4-OMe | 2-Et | 6-Cl | 86 (8b) | 97:3 |
| 10 | 4-OMe | 2-Ph | 4-CHO | 82 (9a) | 97:3 |
| 11 | 4-OMe | 2-OMe | 4-CHO | 89 (9b) | 93:7 |
| 12 | 4-OMe | 2,3-(Me)2 | 4,5-(–OC2H4O–) | 90 (10a) | 96:4 |
| 13 | 4-Br | 2-Et | 4,5-(–OC2H4O–) | 89 (10b) | 94:6 |
| 14 | 4-OMe | 2-Ph | 5-CO2Me | 82 (11a) | 98:2 |
| 15 | 4-Br | 2-Et | 5-CO2Me | 81 (11b) | 93:7 |
| 16 | 4-Br | 2-Et | 4-OH | 70 (12) | 95:5 |
| 17 | 4-OMe | 2-Et | 4-CN | 88 (13a) | 96:4 |
| 18 | 4-OMe | 2-Br | 4-CN | 86 (13b) | 96:4 |
| 19 | 4-OMe | 2-iPr | 5-CO2H | 83 (14) | 86:14 |
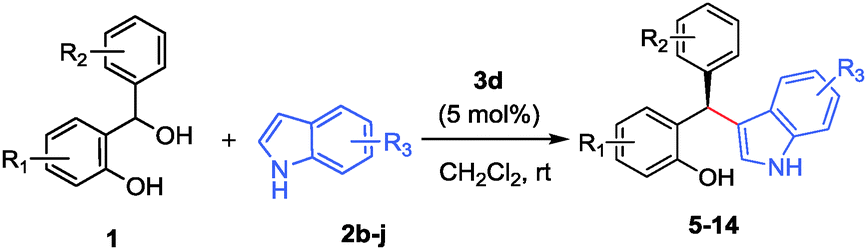
Challenging substituents like phenol, formyl, acid, cyano, ester, ether, and halide groups did not interfere with this process and furnished the desired (F–C) addition products in good to excellent enantioselectivities and high yields. The ability to withstand all these functional groups without compromising the yield or selectivity is testimony to the scope of this methodology.
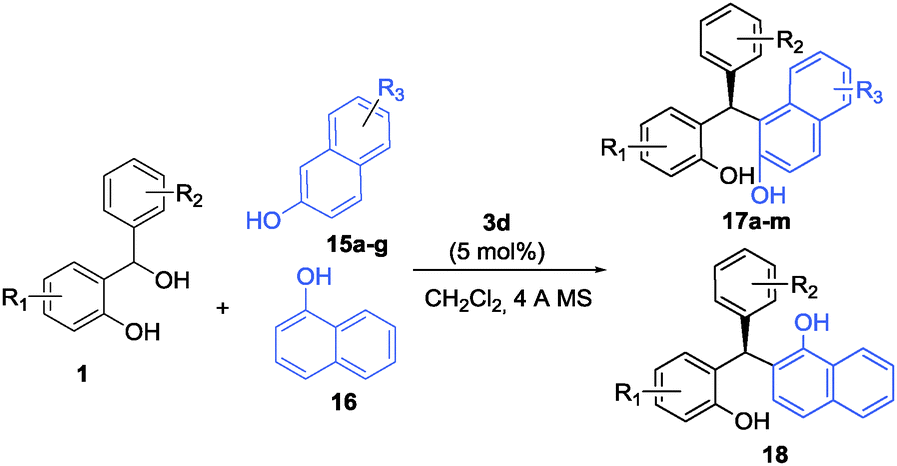
The excellent results obtained for indoles inspired us to pursue this strategy further and extend it to electron-rich naphthols which are also known to be excellent substrates to undergo (F–C) alkylation reactions. A range of Brønsted acid catalysts were screened for the (F–C) alkylation of 2-naphthol (15a) with ortho-hydroxybenzhydrol 1a (see the ESI†). It turned out that in the presence of 5 mol% of catalysts 3a, 3c, and 3d, respectively, the unsymmetrically substituted triarylmethanes were obtained in excellent yields in all cases studied. The highest enantioselectivity was obtained again with catalyst 3d when 4 Å MS was used as an additive in CH2Cl2 as the solvent at room temperature.
With these conditions established we examined a range of 2-naphthols in reactions of hydrogen-bonded o-QM (Table 4). A series of differently substituted ortho-hydroxybenzhydryl alcohols were tested as o-QM precursors (entries 1–7). In almost all cases studied the triarylmethanes 17a–g were isolated in excellent yields and with >96:4 er. Further substitution within the 2-naphthol ring with halogen, ester, ether, and aryl substituents was readily tolerated and delivered the products 17h–m in comparably high yields and enantioselectivities (entries 8–13). Quite interestingly, 1-naphthol (16) worked equally well as the substrate and furnished triarylmethane 18 in almost quantitative yield and 93:7 er (entry 14).
|
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | Yieldb (%) | Erc |
| a All reactions were carried out with 0.2 mmol (1 equiv.) of 1, 0.24 mmol (1.2 equiv.) of naphthols 15a–g and 16 using 5 mol% of catalyst 3d in 2 mL of CH2Cl2 at rt for 4–6 h in the presence of a catalytic amount of 4 Å MS. b Isolated yield of the purified product. c Er determined through chiral HPLC-analysis (see the ESI). d 1-Naphthol (16) was used as the substrate. | |||||
| 1 | H | 4-OMe | H | 95 (17a) | 99:1 |
| 2 | H | 3,4-(–OC2H4O–) | H | 95 (17b) | 97:3 |
| 3 | H | 3,4-di-Me | H | 95 (17c) | 96:4 |
| 4 | H | 2,5-di-Me-4-OMe | H | 90 (17d) | 96:4 |
| 5 | H | 4-F | H | 90 (17e) | 87:13 |
| 6 | 4-Br | 4-OMe | H | 94 (17f) | 96:4 |
| 7 | 4-tBu | 4-OMe | H | 94 (17g) | 96:4 |
| 8 | H | 4-OMe | 6-Br | 93 (17h) | 97:3 |
| 9 | H | 4-OMe | 7-Br | 94 (17i) | 97:3 |
| 10 | H | 4-OMe | 7-CO2Me | 90 (17j) | 99:1 |
| 11 | H | 4-OMe | 6-Ph | 96 (17k) | 97:3 |
| 12 | H | 4-OMe | 7-OMe | 92 (17l) | 95:5 |
| 13 | H | 4-OMe | 6-Naphth | 94 (17m) | 97:3 |
| 14d | H | 4-OMe | H | 97 (18) | 93:7 |
To further reveal the synthetic potential of this new process some of the diarylindolylmethanes were subsequently converted into highly versatile dihydrochromeno[2,3-b]indoles 19a–b through a one-pot bromination followed by cyclization and base-catalyzed elimination (Scheme 2).14h The products were obtained in good yields and retained their optical purity almost completely.
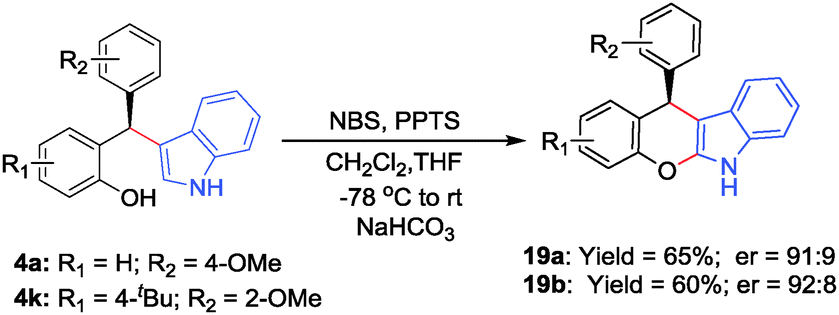 | ||
| Scheme 2 Conversion into dihydrochromeno[2,3-b]indoles 19a–b. | ||
On the basis of the crystal structure which we obtained for the (F–C)-product 4c, we propose a transition structure as shown in Fig. 2, which accommodates double hydrogen-bonding of the catalyst to both the o-QM and the nucleophile and intramolecular delivery of the nucleophile to the re-face of the o-QM because the opposite face is effectively shielded by the neighbouring 3′-Ar-group.19
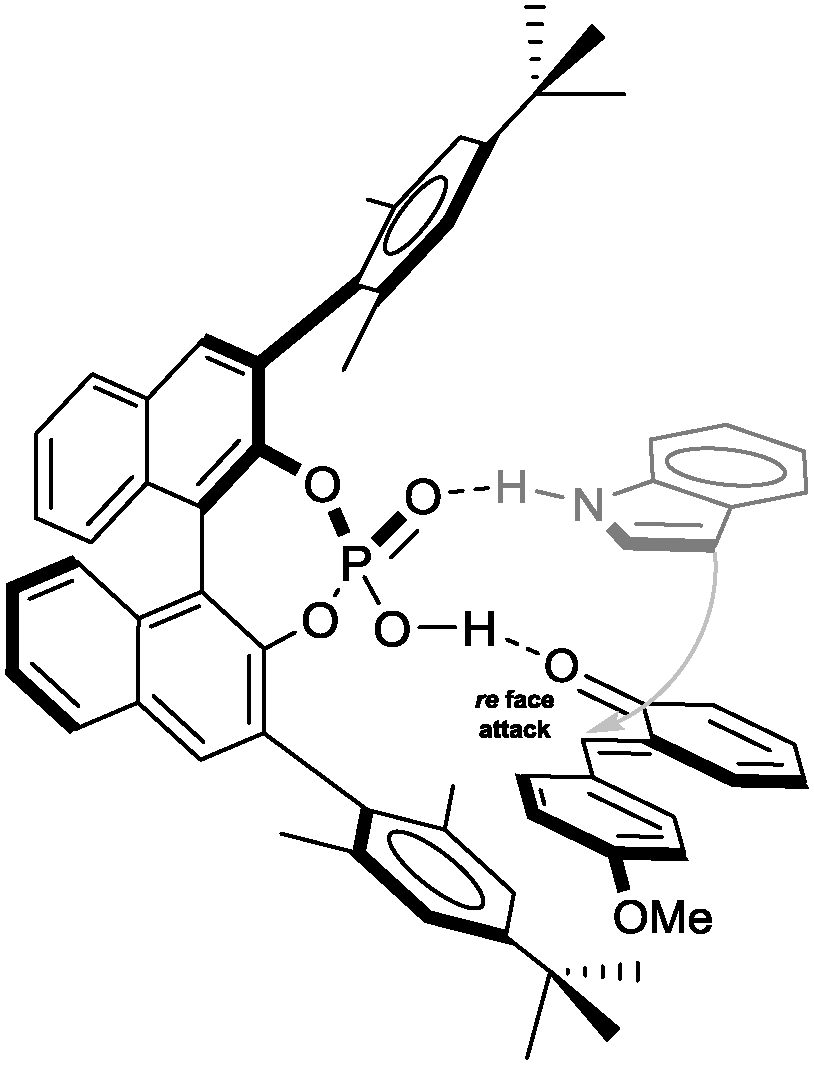 | ||
| Fig. 2 Plausible transition state structure. | ||
In summary, we have developed a highly efficient, Brønsted acid-catalyzed (F–C)-alkylation of electron-rich indoles and naphthols with in situ-generated o-QM which furnished a broad range of synthetically useful diarylindolylmethanes and triarylmethanes with excellent yields and enantioselectivities. The diarylindolylmethanes were subsequently converted into valuable dihydrochromeno[2,3-b]indoles through a base-catalyzed addition–elimination reaction with full retention of absolute configuration. This study further underlines the utility and power of phosphoric acid-catalyzed, enantioselective reactions of o-QM and significantly extends the scope of this strategy.
We thank Dr P. Lönnecke (University of Leipzig) for the crystal structure analysis. We gratefully acknowledge the donation of chemicals from Evonik and BASF.
Notes and references
- (a) D. F. Duxbury, Chem. Rev., 1993, 93, 381 CrossRef CAS; (b) R. Muthyala, A. R. Katritzky and X. Lan, Dyes Pigm., 1994, 25, 303 CrossRef CAS; (c) A. R. Katritzky, V. Gupta, C. Garot, C. V. Stevens and M. F. Gordeev, Heterocycles, 1994, 38, 345 CrossRef CAS; (d) M. S. Shchepinov and V. A. Korshun, Chem. Soc. Rev., 2003, 32, 170 RSC; (e) Y.-Q. Xu, J.-M. Lu, N.-J. Li, F. Yan, X. Xia and Q. Xu, Eur. Polym. J., 2008, 44, 2404 CrossRef CAS PubMed; (f) N. Herron, G. A. Johansson and N. S. Radu, US Pat. 2005/0187364, 2005 Search PubMed.
- (a) Shagufta, A. K. Srivastava, R. Sharma, R. Mishra, A. K. Balapure, P. S. R. Murthy and G. Panda, Bioorg. Med. Chem., 2006, 14, 1497 CrossRef CAS PubMed; (b) R. Palchaudhuri, V. Nesterenko and P. J. Hergenrother, J. Am. Chem. Soc., 2008, 130, 10274 CrossRef CAS PubMed; (c) M. K. Parai, G. Panda, V. Chaturvedi, Y. K. Manju and S. Sinha, Bioorg. Med. Chem. Lett., 2008, 18, 289 CrossRef CAS PubMed; (d) B. A. Ellsworth, W. R. Ewing and E. Jurica, US Pat. 2011/0082165A1, 2011 Search PubMed.
- (a) K. Baba, K. Maeda, Y. Tabata, M. Doi and M. Kozawa, Chem. Pharm. Bull., 1988, 36, 2977 CrossRef CAS; for a recent total synthesis of cassigarol B involving Friedel–Crafts methodology see: (b) S. A. Snyder, S. P. Breazzano, A. G. Ross, Y. Lin and A. L. Zografos, J. Am. Chem. Soc., 2009, 131, 1753 CrossRef CAS PubMed.
- For select examples of letrozole- and vorozole-type triarylmethanes see: (a) H. V. Bossche, G. Willemsens, I. Roels, D. Bellens, H. Moereels, M. C. Coene, L. L. Jeune, W. Lauwers and P. A. Janssen, Biochem. Pharmacol., 1990, 40, 1707 CrossRef; (b) M. Recanatini, A. Cavalli and P. Valenti, Med. Res. Rev., 2002, 22, 282 CrossRef CAS PubMed; (c) P. M. Wood, L. L. Woo, J. R. Labrosse, M. N. Trusselle, S. Abbate, G. Longhi, E. Castiglioni, F. Lebon, A. Purohit and M. K. Reed, J. Med. Chem., 2008, 51, 4226 CrossRef CAS PubMed; (d) K. Takahashi, G. Yamagishi, T. Hiramatsu, A. Hosoya, K. Onoe, H. Doi, H. Nagata, Y. Wada, H. Onoe, Y. Watanabe and T. Hosoya, Bioorg. Med. Chem., 2011, 19, 1464 CrossRef CAS PubMed; (e) P. M. Wood, L. W. Woo, M. P. Thomas, M. F. Mahon, A. Purohit and B. V. Potter, ChemMedChem, 2011, 6, 1423 CrossRef CAS PubMed; for select examples of para-phenyl-substituted diindolylmethanes see: (f) S. D. Cho, K. Yoon, S. Chintharlapalli, M. Abdelrahim, P. Lei, S. Hamilton, S. Khan, S. K. Ramaiah and S. Safe, Cancer Res., 2007, 67, 674 CrossRef CAS PubMed; (g) T. Inamoto, S. Papineni, S. Chintharlapalli, S. D. Cho, S. Safe and A. M. Kamat, Mol. Cancer Ther., 2008, 7, 3825 CrossRef CAS PubMed; (h) S. D. Cho, S. O. Lee, S. Chintharlapalli, M. Abdelrahim, S. Khan, K. Yoon, A. M. Kamat and S. Safe, Mol. Pharmacol., 2010, 77, 396 CrossRef CAS PubMed; (i) K. Yoon, S. O. Lee, S. D. Cho, K. Kim, S. Khan and S. Safe, Carcinogenesis, 2011, 32, 836 CrossRef CAS PubMed.
- Select recent examples: (a) V. Nair, K. G. Abhilash and N. Vidya, Org. Lett., 2005, 7, 5857 CrossRef CAS PubMed; (b) T. Niwa, H. Yorimitsu and K. Oshima, Org. Lett., 2007, 9, 2373 CrossRef CAS PubMed; (c) J. Y. Yu and R. Kuwano, Org. Lett., 2008, 10, 973 CrossRef CAS PubMed; (d) C. R. Liu, M. B. Li, C. F. Yang and S. K. Tian, Chem. Commun., 2008, 1249 RSC; (e) Y. G. I. McGrew, J. Temaismithi, P. J. Carroll and P. J. Walsh, Angew. Chem., Int. Ed., 2010, 49, 5541 CrossRef PubMed; (f) J. Zhang, A. Bellomo, A. D. Creamer, S. D. Dreher and P. J. Walsh, J. Am. Chem. Soc., 2012, 134, 13765 CrossRef CAS PubMed; (g) S. Tabuchi, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2014, 79, 5401 CrossRef CAS PubMed; (h) J. Zhang, A. Bellomo, A. D. Creamer, S. D. Dreher and P. J. Walsh, J. Am. Chem. Soc., 2014, 136, 6276 CrossRef CAS PubMed.
- (a) B. L. H. Taylor, M. R. Harris and E. R. Jarvo, Angew. Chem., Int. Ed., 2012, 51, 7790 CrossRef CAS PubMed; (b) M. R. Harris, L. E. H. M. A. Greene, C. E. Moore and E. R. Jarvo, J. Am. Chem. Soc., 2013, 135, 3303 CrossRef CAS PubMed.
- Q. Zhou, H. D. Srinivas, S. Dasgupta and M. P. Watson, J. Am. Chem. Soc., 2013, 135, 3307 CrossRef CAS PubMed.
- (a) S. C. Matthew, B. W. Glasspoole, P. Eisenberger and C. M. Crudden, J. Am. Chem. Soc., 2014, 136, 5828 CrossRef CAS PubMed; (b) M. Nambo and C. M. Crudden, Angew. Chem., Int. Ed., 2014, 53, 742 CrossRef CAS PubMed.
- F. L. Sun, X. J. Zheng, Q. Gu, Q. L. He and S. L. You, Eur. J. Org. Chem., 2010, 47 CrossRef CAS.
- M. H. Zhuo, Y. J. Jiang, Y. S. Fan, Y. Gao, S. Liu and S. Zhang, Org. Lett., 2014, 16, 1096 CrossRef CAS PubMed.
- S. Qi, C. Y. Liu, J. Y. Ding and F. S. Han, Chem. Commun., 2014, 50, 8605 RSC.
- (a) O. El-Sepelgy, S. Haseloff, S. K. Alamsetti and C. Schneider, Angew. Chem., Int. Ed., 2014, 53, 7923 CrossRef CAS PubMed; (b) After publication of our results another communication appeared: C. C. Hsiao, H. H. Liao and M. Rueping, Angew. Chem., Int. Ed., 2014, 53, 13258 CrossRef CAS PubMed.
- Reviews: (a) R. W. van de Water and T. R. R. Pettus, Tetrahedron, 2002, 58, 5367 CrossRef CAS; (b) T. P. Pathak and M. S. Sigman, J. Org. Chem., 2011, 76, 9210 CrossRef CAS PubMed; (c) N. J. Willis and C. D. Bray, Chem. – Eur. J., 2012, 18, 9160 CrossRef CAS PubMed.
- (a) Y. Zhang and M. S. Sigman, J. Am. Chem. Soc., 2007, 129, 3076 CrossRef CAS PubMed; (b) A. E. Mattson and K. A. Scheidt, J. Am. Chem. Soc., 2007, 129, 4508 CrossRef CAS PubMed; (c) E. Alden-Danforth, M. T. Scerba and T. Lectka, Org. Lett., 2008, 10, 4951 CrossRef CAS PubMed; (d) K. H. Jensen, T. P. Pathak, Y. Zhang and M. S. Sigman, J. Am. Chem. Soc., 2009, 131, 17074 CrossRef CAS PubMed; (e) T. P. Pathak, K. M. Gligorich, B. E. Welm and M. S. Sigman, J. Am. Chem. Soc., 2010, 132, 7870 CrossRef CAS PubMed; (f) K. H. Jensen, J. D. Webb and M. S. Sigman, J. Am. Chem. Soc., 2010, 132, 17471 CrossRef CAS PubMed; (g) M. Rueping, U. Uria, M. Y. Lin and I. Atodiresei, J. Am. Chem. Soc., 2011, 133, 3732 CrossRef CAS PubMed; (h) R. Jana, T. P. Pathak, K. H. Jensen and M. S. Sigman, Org. Lett., 2012, 14, 4074 CrossRef CAS PubMed; (i) Y. Luan and S. E. Schaus, J. Am. Chem. Soc., 2012, 134, 19965 CrossRef CAS PubMed; (j) H. Lv, W. Q. Jia, L. H. Sun and S. Ye, Angew. Chem., Int. Ed., 2013, 52, 8607 CrossRef CAS PubMed; (k) J. Izquierdo, A. Orue and K. A. Scheidt, J. Am. Chem. Soc., 2013, 135, 10634 CrossRef CAS PubMed; (l) B. S. Zeng, X. Yu, P. W. Siu and K. A. Scheidt, Chem. Sci., 2014, 5, 2277 RSC.
- D. Wilcke, E. Herdtweck and T. Bach, Synlett, 2011, 1235 CAS.
- W. Zhao, Z. Wang, B. Chu and J. Sun, Angew. Chem., Int. Ed., 2014, 53 DOI:10.1002/anie.201405252.
-
N-Boc indole gives rise to the corresponding diarylindolylmethane in 94% yield and with 56:44 er.
- CCDC 1028558 (4c).
- The intermediacy of ortho-quinone methides in this transformation is supported by an experiment in which a stable ortho-quinone methide prepared separately is treated with indole (2a) and phosphoric acid catalyst 3d under otherwise identical reaction conditions and furnishes the diarylindolylmethane in comparable yield and enantioselectivity.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization and copies of 1H, 13C NMR spectra and HPLC profiles for novel compounds. CCDC 1028558. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc08559k |
| This journal is © The Royal Society of Chemistry 2015 |