
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A mild TCEP-based para-azidobenzyl cleavage strategy to transform reversible cysteine thiol labelling reagents into irreversible conjugates†
Antoine
Maruani
,
Shamim
Alom
,
Pierre
Canavelli
,
Maximillian T. W.
Lee
,
Rachel E.
Morgan
,
Vijay
Chudasama
* and
Stephen
Caddick
Department of Chemistry, University College London, 20 Gordon Street, London, WC1H OAJ, UK. E-mail: v.chudasama@ucl.ac.uk; Tel: +44 (0)20 7679 2077
First published on 12th November 2014
Abstract
It has recently emerged that the succinimide linkage of a maleimide thiol addition product is fragile, which is a major issue in fields where thiol functionalisation needs to be robust. Herein we deliver a strategy that generates selective cysteine thiol labelling reagents, which are stable to hydrolysis and thiol exchange.
Advances in protein modification by chemical means have led to the development of a range of protein bioconjugation methodologies.1 These methodologies have been successfully applied to a number of fields such as the fluorescent tagging of proteins,2 and the development of therapeutic protein conjugates3,4 to treat indications such as HIV,5 cancer,6 and malaria.7 Chemically modified proteins are also utilised as diagnostics.8
The use of synthetic methodology to modify proteins has to overcome many major obstacles, the most significant of which is the need for high selectivity, i.e. modifying only one amino acid type by discriminating against the other natural amino acids in a protein.9 As free cysteines are extremely rare in proteins10 and the thiol side chain has the highest nucleophilicity of all proteinogenic groups at physiological conditions,11 it is a very popular target for the selective and site-specific modification of proteins.12 Moreover, with the possibility of facile cysteine introduction by site-directed mutagenesis, cysteine modification is a leading approach. The most popular strategy for labelling the thiol moiety of cysteine residues is by alkylation with maleimides to form thioether-succinimides.12,13 However, it has recently come to light that such an appendage is sub-optimal owing to issues of hydrolysis, and thiol exchange with reactive thiols in the blood (e.g. albumin).14 This has major implications for biologics that employ a maleimide motif to functionalise a protein thiol for in vivo applications. For example, in antibody-drug conjugates (ADCs), where an antibody delivers a toxic payload to cancerous tissue selectively, the use of maleimides to attach cytotoxic drugs to an antibody is not ideal as thiol exchange onto human serum albumin in the blood results in off-site toxicity.14 Although recent advances have been made in this area through the use of hydrolysed maleimides and succinimides,14,15 a strong drive to develop novel reagents for reliable, chemoselective, stable and irreversible thiol labelling remains, and particularly for the construction of ADCs.16
Recently, we have described a novel, reversible approach to cysteine bioconjugation through the use of bromomaleimides and bromopyridazinediones.17 To date, our approach has provided access to complex bioconjugates in high yields, without prior activation of reagents with reliable, reversible conjugation. Owing to the demand for hydrolytically stable and thiol irreversible bioconjugates that react in a chemoselective manner, we naturally sought to explore the use of reagents that would meet these criteria. During the course of developing bromopyridazinediones for reversible cysteine bioconjugation, we became intrigued by the prospect of pyridazinediones (PDs) as irreversible cysteine functionalisation reagents. Previously we have shown that if one of the nitrogen atoms on the PD core is unsubstituted the molecule does not react with thiols at physiological pH or higher.17a We postulate that this is a consequence of such a structure existing as its enol tautomer, which is likely to be significantly deprotonated under physiological pH (or higher), based on the reported pKa of 1-methyl-3,6-(1H,2H)-pyridazinedione being ∼5.7 in H2O and the calculated pKa of its thioether analogue,‡ 1-methyl-4-(methylthio)-3,6-(1H,2H)-pyridazinedione, being ∼5.9.18 Thiol reactivity will therefore be greatly reduced as the electrophilicity of the resulting PD-core moiety will be tuned down considerably. As such, we set about developing a strategy where we could generate a mono-alkylated-PD species post-bioconjugation to a cysteine thiol to afford a thiol stable construct (see Fig. 1).
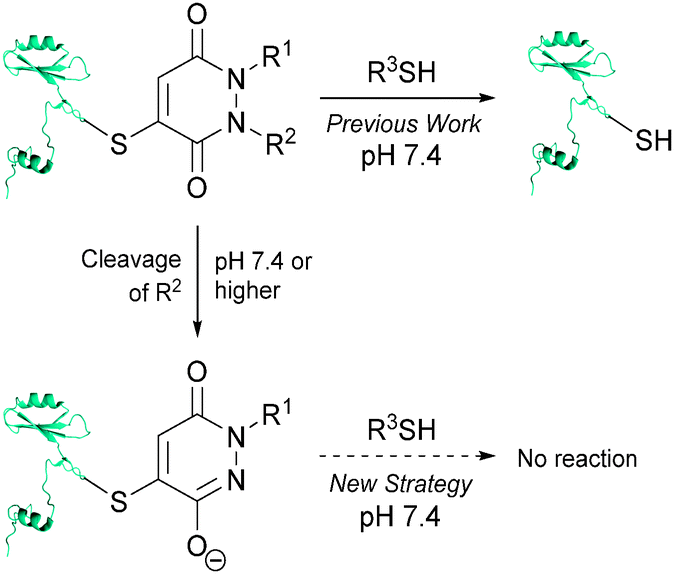 | ||
| Fig. 1 A novel strategy for developing thiol-stable pyridazinedione bioconjugates. | ||
Our study began with the reaction of model protein GFP-S147C 1 with pyridazinediones 2 and 3 to confirm our previous observations when using protein Grb2-L111C (see Fig. 2).17a These results were consistent with our previous work and confirmed that a mono-alkylated-PD is unreactive to thiol (or other nucleophilic functional groups on amino acid side-chains).
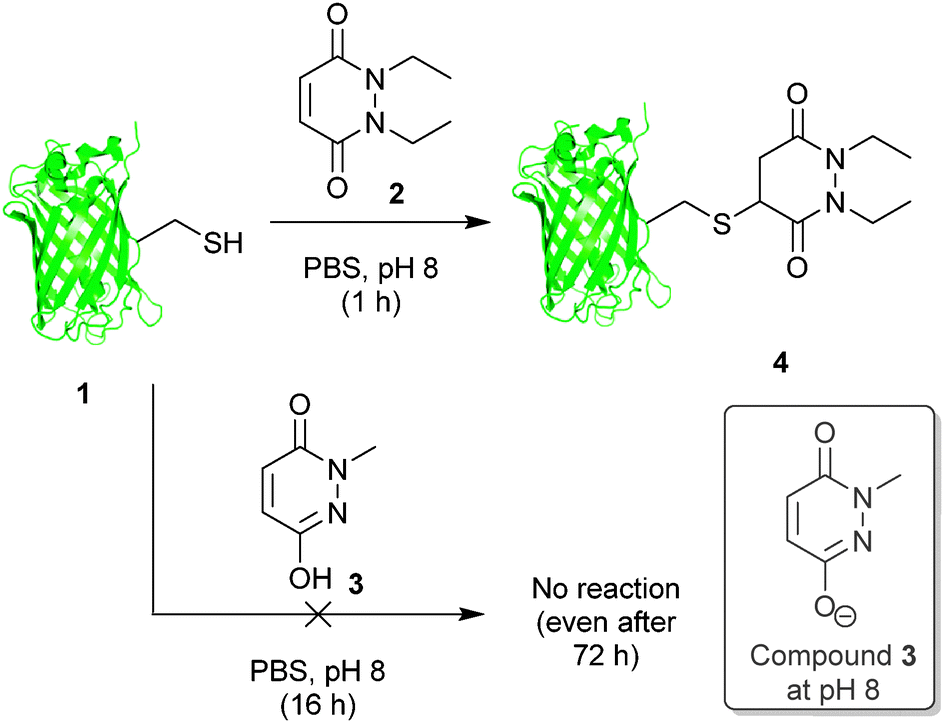 | ||
| Fig. 2 Incubation of GFPS147C 1 with pyridazinediones 2 and 3. | ||
These initial studies paved the way for us to appraise the use of a novel strategy for developing thiol-stable pyridazinedione bioconjugates (see Fig. 1). To do so, we needed to develop a selective method for cleavage of R2 from the PD core. There are many strategies that could be applied, however, at this juncture we took the opportunity to develop a novel, mild and simple method based on an azide trigger. Our desire to use an azide-based cleavable handle originates from the bioorthogonality of the azide functional group. Taking inspiration from the well-documented work on p-aminobenzyloxycarbonyl (PABC) linkers,19 we set about using a p-azidobenzyl cleavage strategy (see Fig. 3).
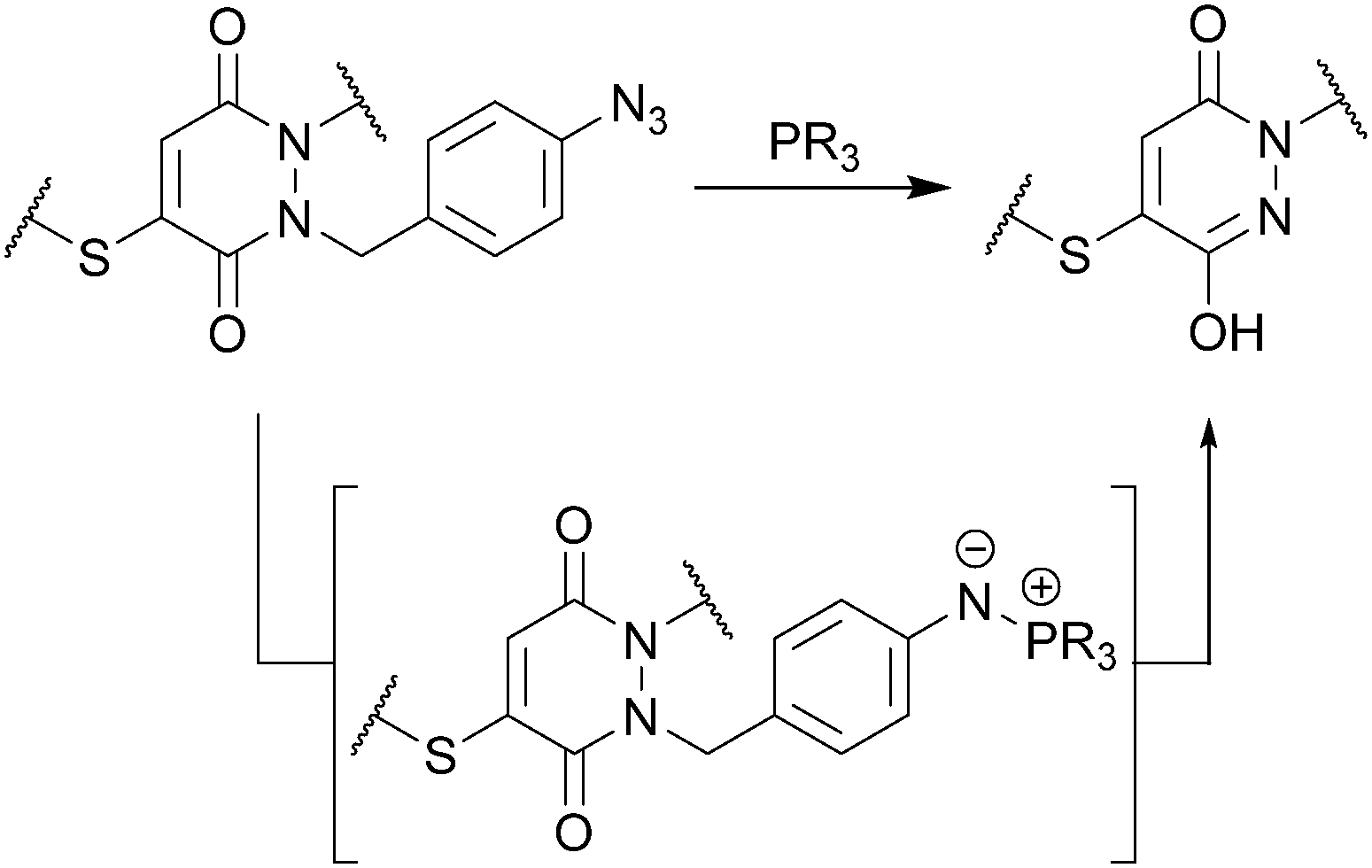 | ||
| Fig. 3 Use of a p-azidobenzyl cleavage strategy to generate a thiol-stable pyridazinedione bioconjugate. | ||
We initially evaluated our p-azidobenzyl cleavage strategy in a small molecule study through the use of cysteine derivative 6, formed by reaction of N-(tert-butoxycarbonyl)-L-cysteine methyl ester and mono-bromo PD 5 (see ESI† for details on synthesis). The use of an alkyne handle, which would conceptually be retained post p-azidobenzyl cleavage, would allow for the resulting construct to be readily functionalised by a Cu(I)-catalyzed Azide-Alkyne Cycloaddition (CuAAC). To our delight, treatment of derivative 6 with TCEP led to clean conversion to derivative 7, thus providing proof of concept for our novel cleavage strategy. Moreover, incubation of derivatives 6 and 7 with 15 equivalents of 1-hexanethiol in THF/PBS buffer (pH 7.4) only led to thiol exchange in the case of derivative 6. This provided encouragement for our hypothesis of a mono-alkylated-PD being thiol unreactive under physiological pH or higher (Fig. 4).
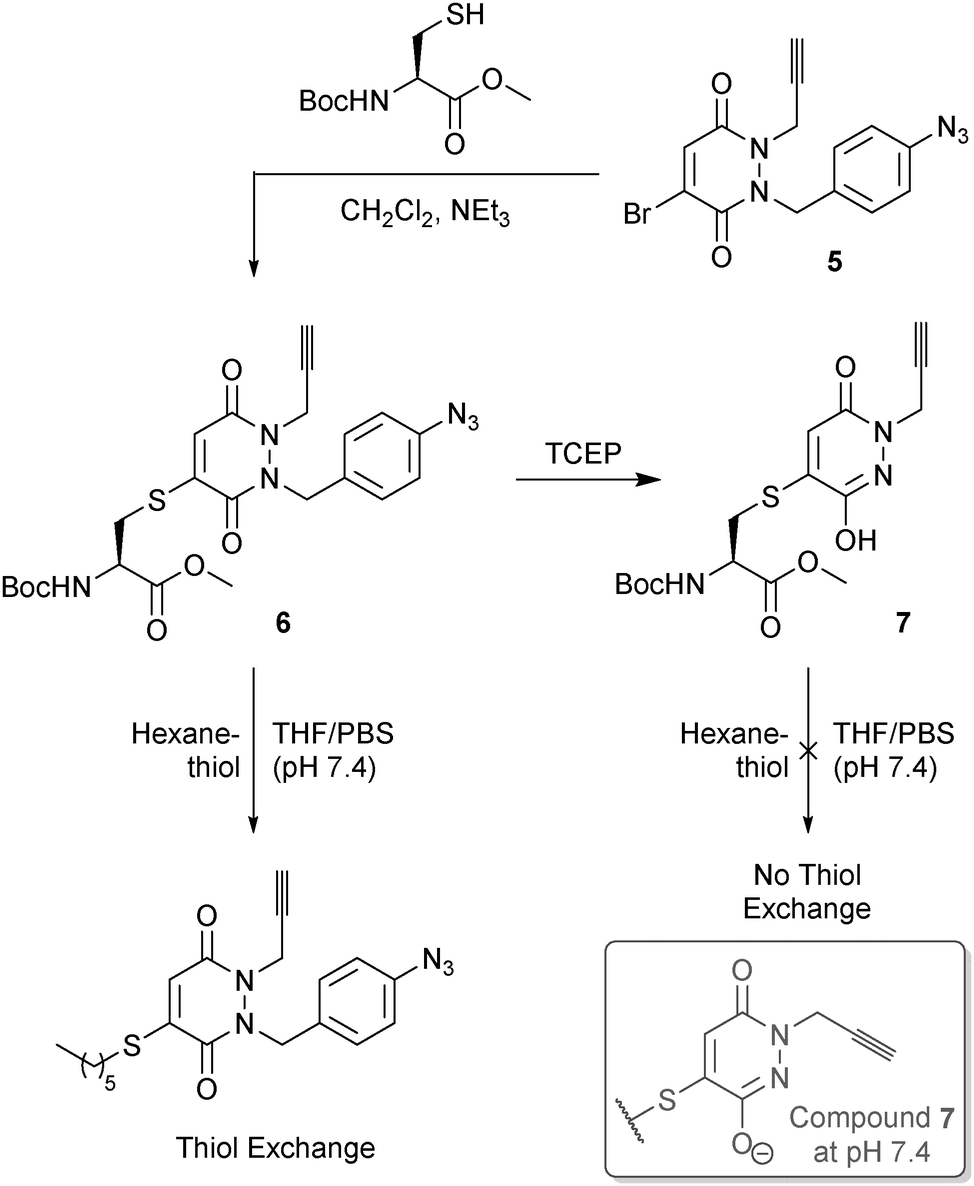 | ||
| Fig. 4 Use of TCEP in our p-azidobenzyl cleavage strategy to generate pyridazinedione derivative 7 from derivative 6, and the thiol stability of each construct. | ||
Following these encouraging results on a small molecule study, we appraised our strategies on a model protein with a single cysteine mutation, GFPS147C 1. Initially, GFPS147C 1 was incubated with mono-bromo-PD 5 in sodium phosphate buffer (pH 8.0) for 1 h at 37 °C. As expected, this proceeded with complete conversion and afforded GFP-derivative 8. We next applied our TCEP cleavage strategy, by incubation of this derivative with 10 equivalents of TCEP in phosphate buffer at pH 8.0. Satisfyingly, clean conversion to bioconjugate 9 was observed, which is consistent with our small molecule study. It is also noteworthy that no hydrolysis occurred under these conditions, which is consistent with our previous observations on the PD core being hydrolytically stable.17a Having established, using mass spectrometry, that our cleavage strategy is applicable on a protein, we next compared the thiol stability of 8 and 9 by incubation with glutathione (0.5 mM) for 72 h at pH 7.4 and 37 °C. Gratifyingly, GFP-derivative 9 was completely stable under the reaction conditions, whereas derivative 8 showed complete thiol exchange with glutathione. This therefore established proof of concept for both our strategies on a model protein scaffold. Moreover, this work also highlights the versatility of the PD platform with a facile shift from reversible to irreversible constructs achieved under mild conditions (Fig. 5).
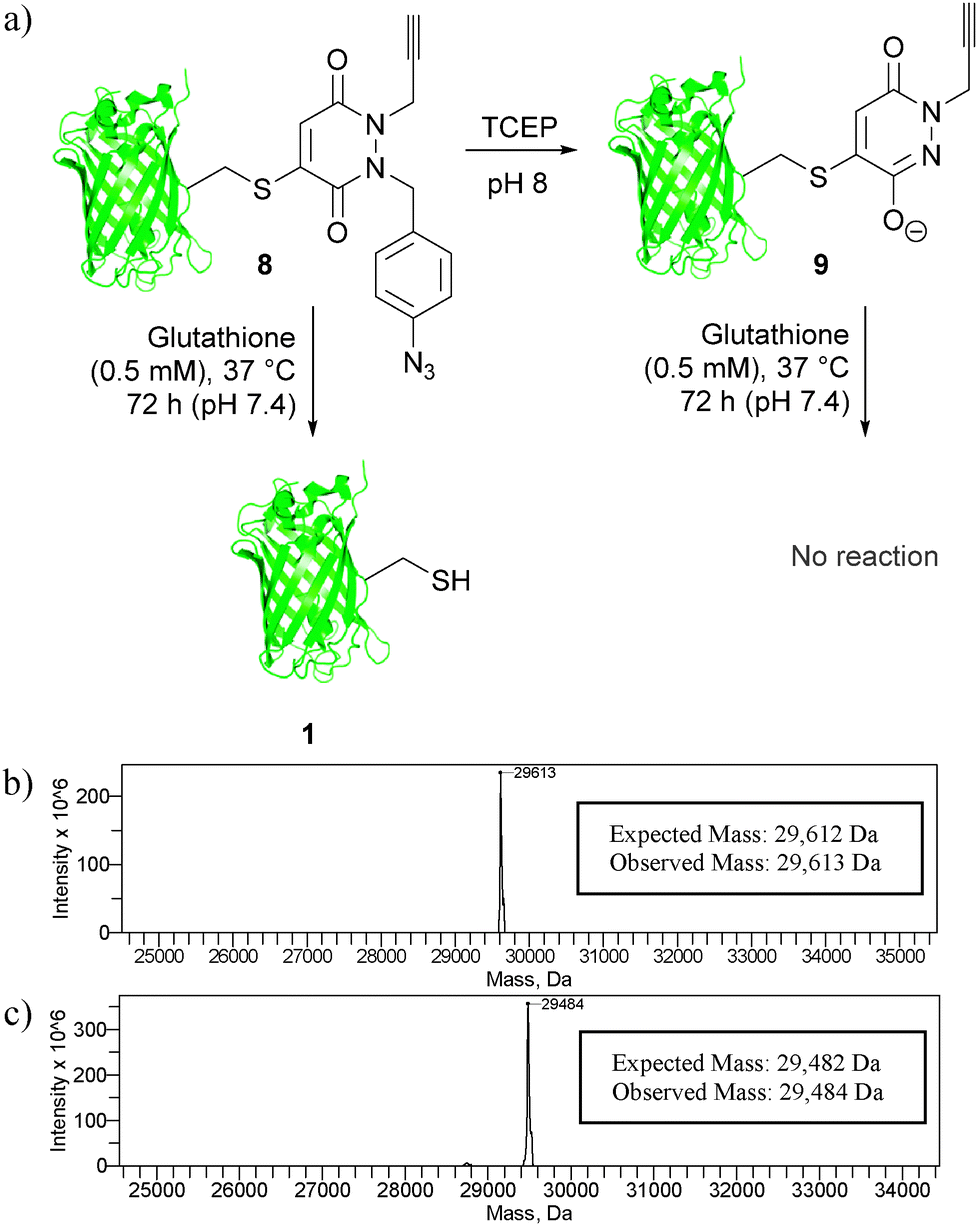 | ||
| Fig. 5 (a) Translation of our thiol cleavage and thiol stability strategies onto protein bioconjugates 8 and 9, and deconvoluted MS data for (b) bioconjugate 8 and (c) bioconjugate 9. | ||
Following our work on developing a novel p-azidobenzyl cleavage strategy and obtaining a thiol stabile construct, we set about functionalising protein scaffold 9 by the use of ‘click’ chemistry. If successful, this would result in a facile method for functionalising the thiol-stable bioconjugate. A number of ‘click’ conditions were trialled using benzyl azide as our model azide. The most promising conditions were the use of Cu(I)Br as copper source and THPTA as ligand. These conditions gave complete conversion of starting material alkyne 9 to triazole bioconjugate 10a. Moreover, these conditions also allowed for clean reaction of the alkyne derivative with a dansyl azide and a sulfo-cyanine5 azide to afford 10b and 10c, respectively (Fig. 6).
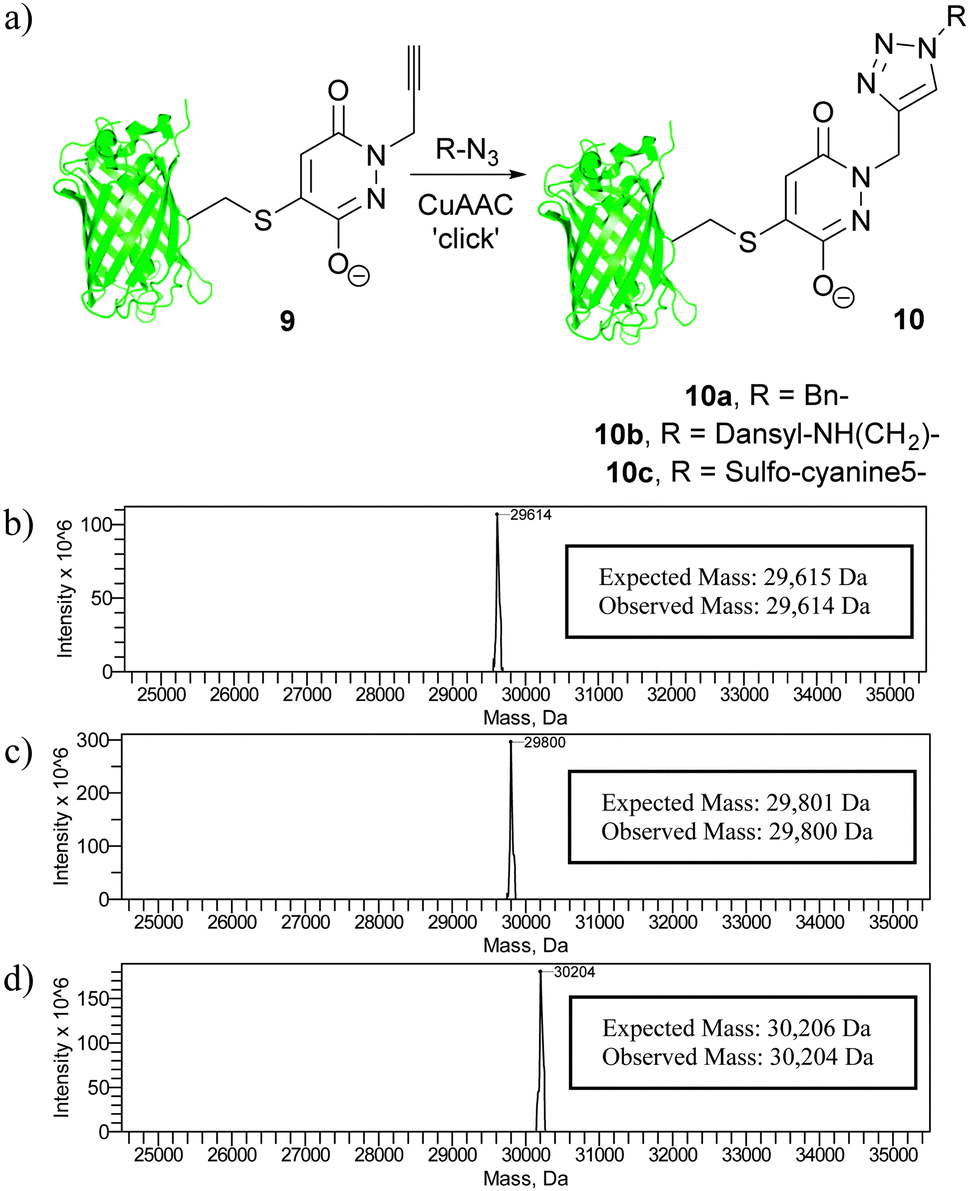 | ||
| Fig. 6 (a) The use of ‘click’ chemistry to functionalise bioconjugate 9, and deconvoluted MS data for (b) bioconjugate 10a, (c) bioconjugate 10b and (d) bioconjugate 10c. | ||
In conclusion, we have developed, via a novel p-azidobenzyl cleavage strategy, a route to thiol stable cysteine-bioconjugates that has a clear advantage over conventional maleimide chemistry. The strategy has been demonstrated on both a small molecule system and on a model protein, GFPS147C. Owing to the plethora of fields where thiol functionalisation needs to be robust and irreversible, e.g. in antibody-drug conjugates (ADCs), imaging and theranostics, we believe this work will find use in a variety of domains. We hope to deliver on the application of our platform in a range of contexts, including ADCs, in the near future.
The authors gratefully acknowledge the EPSRC, Ramsay Memorial Trust and UCL for support of our programme.
Notes and references
- C. D. Spicer and B. G. Davis, Nat. Commun., 2014, 5, 5740 CrossRef PubMed and references therein.
- S. Girouard, M. H. Houle, A. Grandbois, J. W. Keillor and S. W. Michnick, J. Am. Chem. Soc., 2005, 127, 559–566 CrossRef CAS PubMed.
- D. H. Dube and C. R. Bertozzi, Nat. Rev. Drug Discovery, 2005, 4, 477–488 CrossRef CAS PubMed.
- S. Jevševar, M. Kunstelj and V. G. Porekar, Biotechnol. J., 2010, 5, 113–128 CrossRef PubMed.
- R. D. Astronomo, H. K. Lee, C. N. Scanlan, R. Pantophlet, C. Y. Huang, I. A. Wilson, O. Blixt, R. A. Dwek, C. H. Wong and D. R. Burton, J. Virol., 2008, 82, 6359–6368 CrossRef CAS PubMed.
- L. M. Krug, G. Ragupathi, K. K. Ng, C. Hood, H. J. Jennings, Z. Guo, M. G. Kris, V. Miller, B. Pizzo, L. Tyson, V. Baez and P. O. Livingston, Clin. Cancer Res., 2004, 10, 916–923 CrossRef CAS.
- L. Schofield, M. C. Hewitt, K. Evans, M.-A. Siomos and P. H. Seeberger, Nature, 2002, 18, 785–789 CrossRef PubMed.
- J. J. Day, B. V. Marquez, H. E. Beck, T. A. Aweda, P. D. Gawande and C. F. Meares, Curr. Opin. Chem. Biol., 2010, 14, 803–809 CrossRef CAS PubMed.
- I. S. Carrico, Chem. Soc. Rev., 2008, 37, 1423–1431 RSC.
- R. Bhattacharyya, D. Pal and P. Chakrabarti, Protein Eng., Des. Sel., 2004, 17, 795–808 CrossRef CAS PubMed.
- R. L. Lundblad, Chemical Reagents for Protein Modification, CRC Press, Boca Raton, FL, 3rd edn, 2005 Search PubMed.
- J. M. Chalker, G. J. L. Bernardes, Y. A. Lin and B. G. Davis, Chem. – Asian J., 2009, 4, 630–640 CrossRef CAS PubMed and references therein.
- (a) D. J. Betting, K. Kafi, A. Abdollahi-Fard, S. A. Hurvitz and J. M. Timmerman, J. Immunol., 2008, 181, 4131–4140 CrossRef CAS; (b) Y. Zhang, V. S. Bhatt, G. Sun, P. G. Wang and A. F. Palmer, Bioconjugate Chem., 2008, 19, 2221–2230 CrossRef CAS PubMed; (c) I. Shin, H. J. Jung and M. R. Lee, Tetrahedron Lett., 2001, 42, 1325–1328 CrossRef CAS; (d) C. Linda, R. Gerdesa, Y. Hamnella, I. Schuppe-Koistinenc, H. B. von Löwenhielmd, A. Holmgrene and I. A. Cotgreavea, Arch. Biochem. Biophys., 2002, 406, 229–240 CrossRef.
- B.-Q. Shen, K. Xu, L. Liu and H. Raab, et al. , Nat. Biotechnol., 2012, 30, 184–189 CrossRef CAS PubMed.
- (a) R. P. Lyon, J. R. Setter, T. D. Bovee, S. O. Doronina, J. H. Hunter, M. E. Anderson, C. L. Balasubramanian, S. M. Duniho, C. I. Leiske, F. Li and P. D. Senter, Nat. Biotechnol., 2014, 32, 1059–1062 CrossRef CAS PubMed; (b) L. N. Tumey, M. Charati, T. He, E. Sousa, D. Ma, X. Han, T. Clark, J. Casavant, F. Loganzo, F. Barletta, J. Lucas and E. I. Graziani, Bioconjugate Chem., 2014, 25, 1871–1880 CrossRef CAS PubMed; (c) C. P. Ryan, M. E. B. Smith, F. F. Schumacher, D. Grohmann, D. Papaioannou, G. Waksman, F. Werner, J. R. Baker and S. Caddick, Chem. Commun., 2011, 47, 5452–5454 RSC.
- (a) P. M. S. D. Cal, G. J. L. Bernardes and P. M. P. Gois, Angew. Chem., Int. Ed., 2014, 53, 10585–10587 CrossRef CAS PubMed; (b) R. V. J. Chari, M. L. Miller and W. C. Widdison, Angew. Chem., Int. Ed., 2014, 53, 3796–3827 CrossRef CAS PubMed.
- (a) V. Chudasama, M. E. B. Smith, F. F. Schumacher, D. Papaioannou, G. Waksman, J. R. Baker and S. Caddick, Chem. Commun., 2011, 47, 8781–8783 RSC; (b) R. I. Nathani, V. Chudasama, C. P. Ryan, P. R. Moody, R. E. Morgan, R. J. Fitzmaurice, M. E. B. Smith, J. R. Baker and S. Caddick, Org. Biomol. Chem., 2013, 11, 2408–2411 RSC; (c) M. E. B. Smith, F. F. Schumacher, C. P. Ryan, L. M. Tedaldi, D. Papaioannou, G. Waksman, S. Caddick and J. R. Baker, J. Am. Chem. Soc., 2010, 132, 1960–1965 CrossRef CAS PubMed.
- (a) N. A. Burton, D. V. S. Green, I. H. Millier, P. J. Taylor, M. A. Vincent and S. Woodcock, J. Chem. Soc., Perkin Trans. 2, 1993, 331–335 RSC; (b) H. Feuer, G. B. Silverman, H. P. Angstadt and A. R. Fauke, J. Org. Chem., 1962, 27, 2081–2084 CrossRef CAS; (c) D. M. Miller and R. W. White, Can. J. Chem., 1956, 34, 1510–1512 CrossRef CAS; (d) S. Du Breuil, J. Org. Chem., 1961, 26, 3382–3386 CrossRef.
- P. L. Carl, P. K. Chakravarty and J. A. Katzenellenbogen, J. Med. Chem., 1981, 24, 479–480 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: LC-MS, ES-MS and deconvoluted spectra for all reactions with proteins described herein, and 1H and 13C NMR spectra for all small molecule constructs. See DOI: 10.1039/c4cc08515a |
| ‡ MarvinSketch and its calculator plugins were used for pKa prediction, MarvinSketch 14.11.3.0, ChemAxon (http://www.chemaxon.com). |
| This journal is © The Royal Society of Chemistry 2015 |