
Visible light induced cyclopropanation of dibromomalonates with alkenes via double-SET by photoredox catalysis†
Yanbin
Zhang‡
a,
Rong
Qian‡
b,
Xingliang
Zheng
c,
Yi
Zeng
c,
Jing
Sun
a,
Yiyong
Chen
a,
Aishun
Ding
a and
Hao
Guo
*ad
aDepartment of Chemistry, Fudan University, 220 Handan Road, Shanghai, 200433, P. R. China. E-mail: Hao_Guo@fudan.edu.cn; Fax: +86-21-55664361; Tel: +86-21-55664361
bShanghai Mass Spectrometry Center, Shanghai Institute of Ceramics, Chinese Academy of Sciences, Shanghai 200050, P. R. China
cInstitute of Chemistry & Biological Engineering, Changsha University of Science and Technology, Changsha 410114, P. R. China
dKey Laboratory of Organofluorine Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, P. R. China
First published on 10th November 2014
Abstract
We report herein a visible light induced generation of a carbanion via double-SET and its application in cyclopropanation of alkenes. This new synthetic approach to form cyclopropane derivatives was conducted under mild conditions, using sunlight in open air, showing the features such as environmental benignness and an easy to handle procedure.
Visible light photoredox catalysis (VLPC) has a rich history in organic chemistry.1 Research on using solar energy to achieve new photocatalysis strategies has attracted continuous interest of chemists. Thanks to the contributions from MacMillan,2 Yoon,3 Stephenson,4 and other researchers,5 VLPC has attracted much attention of chemists. A large number of outstanding studies in this field2–5 have proved that visible light is mild, environmentally benign, and infinitely available. Most importantly, visible light induced transformations are highly tolerant and selective, since undesired side reactions of UV-sensitive compounds can be minimized or even avoided. Upon irradiation with visible light, photoredox catalysts can easily reach a photoexcited state which is readily available for both oxidative and reductive quenching.6 This feature makes VLPC a powerful tool for the generation of a reactive radical intermediate via a single electron transfer (SET) process.7 However, to date, the application of VLPC is still quite limited to radical formation and subsequent transformations.2–5 Reports on carbocation or carbanion generation and following bond formations are rare.8 Thus, studies to extend the application of VLPC and develop non-radical-limited visible light photoreactions are highly desired.
It has been reported that benzyl bromide can be reduced into carbanion PhCH2−via double-SET under the catalysis of Ru(bpy)32+ (Scheme 1).8
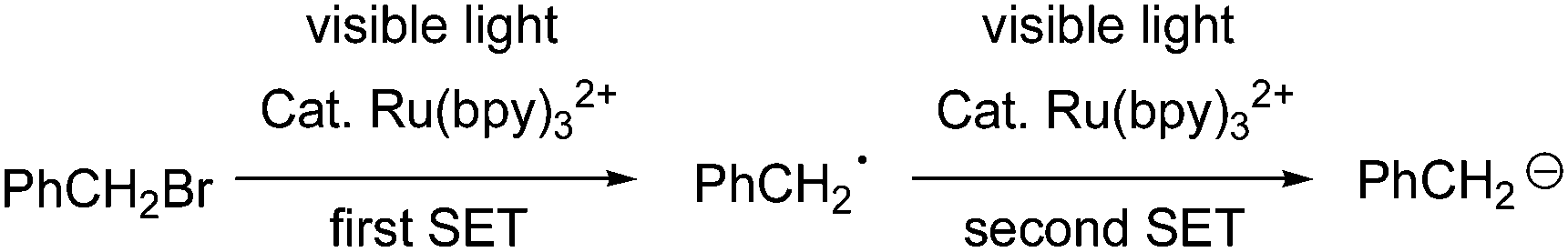 | ||
| Scheme 1 The reduction of PhCH2Br into carbanion PhCH2−via double-SET. | ||
It is also known that bromomalonate 1 can be converted into a carbon-centered radical via SET under the catalysis of Ru(bpy)32+ (Scheme 2).9 Considering that two strong electron-withdrawing groups were attached on the carbon center of this radical, it might be reduced into a malonate carbanion.10 We hypothesized that a second SET to the in situ formed radical intermediate might result in such a malonate carbanion (Scheme 2).
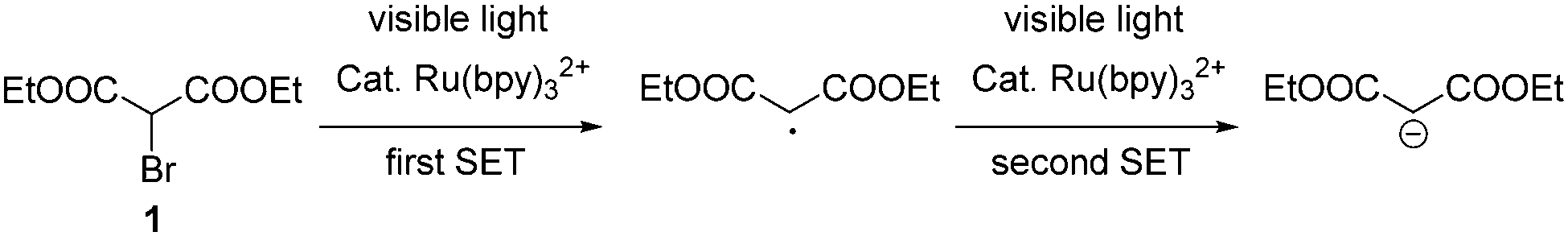 | ||
| Scheme 2 The formation of a carbanion via hypothesized double-SET. | ||
Based on the above idea, a cyclopropanation via intermolecular Michael addition and subsequent intramolecular nucleophilic substitution was designed.11 A prospective mechanism for this double-SET transformation is shown in Scheme 3. Under the visible light irradiation, Ru(bpy)32+ accepts a photon to reach the excited state Ru(bpy)32+*.6a This high energy species is able to grab a single electron from the amine to form the highly reducing Ru(bpy)3+.6b Then the first SET to dibromomalonate 2 leads to the electron-deficient bromomalonate radical 5 and regenerates Ru(bpy)32+.12 Next, a similar catalytic circle processes the second SET to the carbon-centered radical 5 and results in the desired bromomalonate carbanion 6. Its Michael addition11,13 to an electron-deficient alkene 3 will afford a new carbanion 7 which undergoes intramolecular nucleophilic substitution to produce a cyclopropane derivative 4 as the final product. The base might have two effects: donating electrons and trapping the in situ formed HBr.
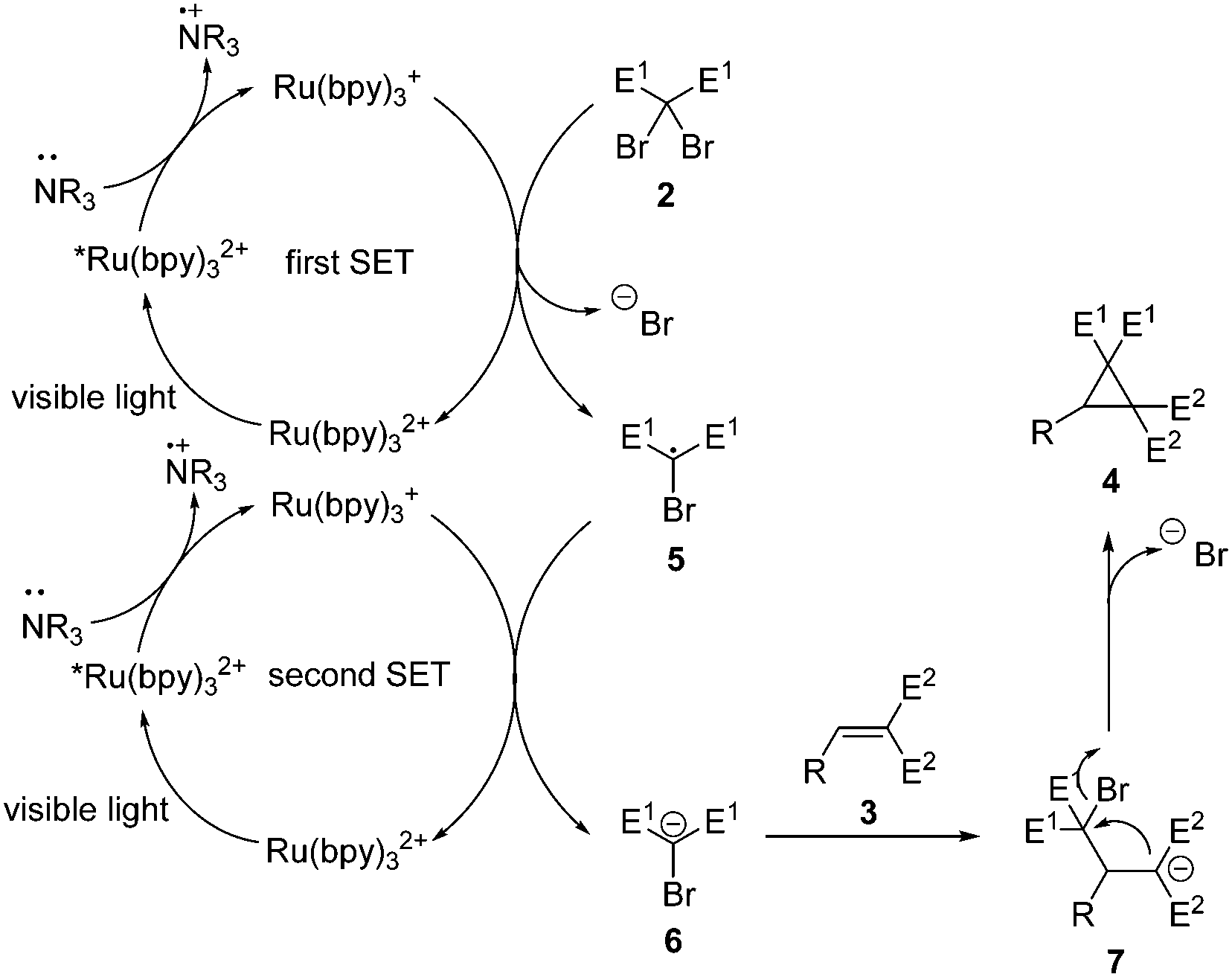 | ||
| Scheme 3 The prospective mechanism. | ||
Close attention should be paid to several key features in the above-mentioned dual-catalysis design plan. First of all, carbanion 6 should be easily available from the reduction of the corresponding radical 5 and reactive enough for Michael addition. Secondly, very strong electron-withdrawing groups should be attached at the α-position of the Michael addition acceptor, so that the in situ formed carbanion 7 will be highly nucleophilic for the following transformation. Thirdly, the steric hindrance at the α-position should be very little to enable the three-membered ring formation process.
With these considerations in mind, diethyl 2,2-dibromomalonate 2a and 2-benzylidenemalononitrile 3a were chosen as the model substrates to examine this new cyclopropanation protocol. To our delight, the reaction underwent smoothly in our initial attempt. Then a series of control reactions were examined. Without light, neither with nor without the catalyst, this reaction proceeded slowly to give very low yields of cyclopropane 4aa after full conversion of the starting material 2a (entries 1 and 2, Table 1). Under the irradiation with visible light and in the absence of the catalyst, a similar low yield of 4aa was achieved (entry 3, Table 1). When irradiated by visible light and in the presence of 1 mol% of the photocatalyst, the reaction speed dramatically increased, affording 4aa in 94% isolated yield (entry 4, Table 1). Notably, this reaction could be carried out in air, yielding 4aa in almost the same excellent yield (entry 5, Table 1). As the time is shortened to approximately 5 hours, it enables this transformation to be conducted under sunlight. Indeed, higher efficiency and better yield were observed (entry 6, Table 1). So condition A (2 equiv. of 3, 2 equiv. of Pri2NEt, 1 mol% Ru(bpy)3Cl2·6H2O, methanol, visible light, and rt) and condition B (2 equiv. of 3, 2 equiv. of Pri2NEt, 1 mol% Ru(bpy)3Cl2·6H2O, methanol, sunlight, and rt) were applied for the following studies.14
|
|
||||
|---|---|---|---|---|
| Entry | Light source | Catalyst [Ru] | Time (h) | Isolated yield (%) |
| a All reactions were carried out using 2a (0.2 mmol), 3a (0.4 mmol), and Pri2NEt (0.4 mmol) in deaerated methanol (10 mL) at rt. b The reaction was carried out under an argon atmosphere. c The reaction was carried out under an air atmosphere. | ||||
| 1b | Dark | — | 32 | 16 |
| 2b | Dark | 1 mol% | 30 | 21 |
| 3b | Visible light | — | 18 | 23 |
| 4b | Visible light | 1 mol% | 5 | 94 |
| 5c | Visible light | 1 mol% | 5 | 92 |
| 6c | Sunlight | 1 mol% | 4 | 97 |
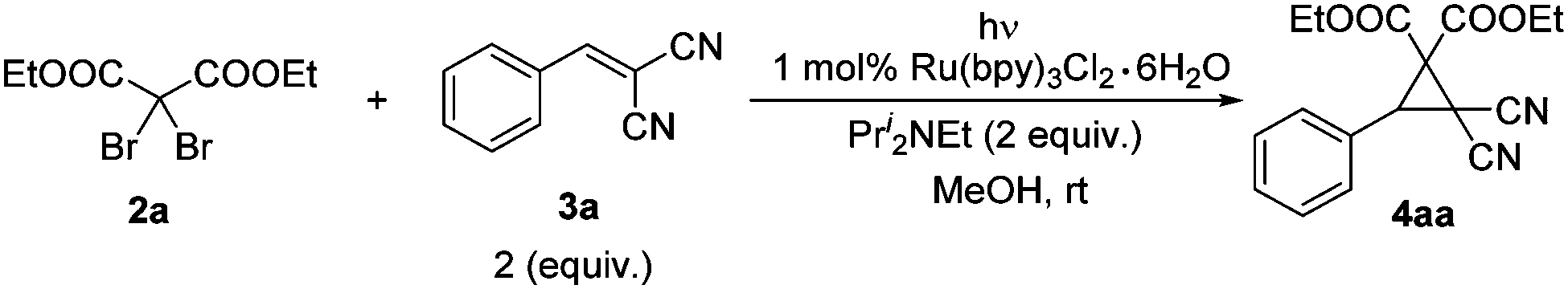
With the optimal conditions in hand, we investigated the scope of this photocyclopropanation with a series of dibromomalonate derivatives and alkenes. Reactions under conditions A and B gave similar results (Table 2). Firstly, the electronic effect of substituents on the phenyl ring of the alkenes was studied carefully. With strong electron-withdrawing groups, such as methoxycarbonyl (entry 1, Table 2), trifluoromethyl (entry 2, Table 2), and nitro (entries 3–5, Table 2), the corresponding cyclopropane derivatives were formed in good to excellent yields. Excellent yields were also obtained for weak electron-withdrawing groups substituted substrates, like fluorine (entry 6, Table 2) and chlorine (entry 7, Table 2), and weak electron-donating groups, like alkyl (entries 9–13, Table 2) and phenyl (entry 14, Table 2). For substrates bearing strong electron-donating groups, like alkoxy (entries 15–18, Table 2) and acetoxy groups (entry 19, Table 2), the yields decreased slightly. The electronic effect of the substrate strongly suggested a carbanion intermediated mechanism.11 Then other dibromides 2b and 2c were applied in the photocyclopropanation of 3a, which gave the desired products 4ba and 4ca in excellent isolated yields, respectively (entries 20 and 21, Table 2). Finally, electron-rich alkene 3t, aliphatic alkene 3u, and 2-methylenemalononitrile 3v were applied under condition A. No cyclopropanes were formed (Scheme S1 in ESI†). In all the above cases, no reduced 3 was formed.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | 2 (E) | 3 (Ar) | 4 | Condition A | Condition B | ||
| Time (h) | Yieldc (%) | Time (h) | Yieldc (%) | ||||
| a All reactions were carried out using 2 (0.2 mmol), 3 (0.4 mmol), and Pri2NEt (0.4 mmol) in deaerated methanol (10 mL) under a 23 W household lamp at rt under an air atmosphere. b All reactions were carried out using 2 (0.2 mmol), 3 (0.4 mmol), and Pri2NEt (0.4 mmol) in deaerated methanol (10 mL) under sunlight at rt under an air atmosphere. c Isolated yield. | |||||||
| 1 | 2a (COOEt) | 3b (4-COOMeC6H4) | 4ab | 4 | 98 | 4 | 98 |
| 2 | 2a (COOEt) | 3c (4-CF3C6H4) | 4ac | 4 | 96 | 2.5 | 98 |
| 3 | 2a (COOEt) | 3d (4-NO2C6H4) | 4ad | 2 | 92 | 2 | 91 |
| 4 | 2a (COOEt) | 3e (3-NO2C6H4) | 4ae | 2 | 84 | 2 | 81 |
| 5 | 2a (COOEt) | 3f (2-NO2C6H4) | 4af | 2 | 85 | 2 | 84 |
| 6 | 2a (COOEt) | 3g (4-FC6H4) | 4ag | 3 | 95 | 3 | 97 |
| 7 | 2a (COOEt) | 3h (4-ClC6H4) | 4ah | 3.5 | 95 | 3 | 95 |
| 8 | 2a (COOEt) | 3a (C6H5) | 4aa | 5 | 92 | 4 | 97 |
| 9 | 2a (COOEt) | 3i (4-MeC6H4) | 4ai | 2.5 | 95 | 2 | 96 |
| 10 | 2a (COOEt) | 3j (3-MeC6H4) | 4aj | 3 | 95 | 3 | 95 |
| 11 | 2a (COOEt) | 3k (2-MeC6H4) | 4ak | 2.5 | 96 | 3 | 98 |
| 12 | 2a (COOEt) | 3l (4-EtC6H4) | 4al | 3 | 94 | 3 | 95 |
| 13 | 2a (COOEt) | 3m (4-PriC6H4) | 4am | 2.5 | 96 | 2.5 | 96 |
| 14 | 2a (COOEt) | 3n (4-PhC6H4) | 4an | 4 | 89 | 4.5 | 90 |
| 15 | 2a (COOEt) | 3o (4-MeOC6H4) | 4ao | 5 | 70 | 4 | 85 |
| 16 | 2a (COOEt) | 3p (3-MeOC6H4) | 4ap | 3 | 85 | 3 | 85 |
| 17 | 2a (COOEt) | 3q (2-MeOC6H4) | 4aq | 3 | 80 | 3 | 79 |
| 18 | 2a (COOEt) | 3r (4-EtOC6H4) | 4ar | 4 | 81 | 4 | 81 |
| 19 | 2a (COOEt) | 3s (4-AcOC6H4) | 4as | 4 | 77 | 4 | 77 |
| 20 | 2b (COOMe) | 3a (C6H5) | 4ba | 3 | 96 | 3 | 95 |
| 21 | 2c (COOPri) | 3a (C6H5) | 4ca | 4 | 93 | 4 | 93 |
Mechanistically, a set of reactions were performed to gain more information. Firstly, radical scavenger TEMPO was added to this reaction system. The addition of 1 equiv. of TEMPO resulted in a slightly decreased yield. Further increase in the amount of TEMPO to 5 equiv. led to similar results. In both cases, no obvious changes in the reaction speed were observed. Next, 5 equiv. of BHT was used instead of TEMPO, similarly, no inhibition was observed (Scheme S2, ESI†). Considering that no obvious feature of a free radical was observed, since O2 in air, TEMPO, or BHT could not significantly affect this transformation, the radical species might not be involved in the rate-determining step.15,16 This might mean that the reduction of a carbon radical into a carbanion proceeds at an extremely rapid rate. Then a dark reaction after irradiation for 5 minutes was also carried out yielding only 29% of 4aa, which ruled out a radical chain mechanism, since the reaction gave a similar yield of 4aa under condition A without light (Scheme S3 in ESI†).
Next, alkenes 8 with different configurations were employed to investigate the stereochemistry of this cyclopropanation. As shown in Scheme 4, under the standard conditions, reactions of 2a with either Z-8 or E-8 gave the same trans-product 9 in nearly the same yield. Notably, no cis-9 was formed in either case. To check whether there was an isomerization of Z-8 into E-8 under these reaction conditions, the unconsumed reactants Z-8 and E-8 were recovered. Careful 1H NMR analysis proved that there was no change in their double bond configuration in the recovered starting material. These results suggest that a free bond rotation occurred during the cyclization procedure, which fits well with our designed reaction pathway via Michael addition and subsequent intramolecular nucleophilic attraction.
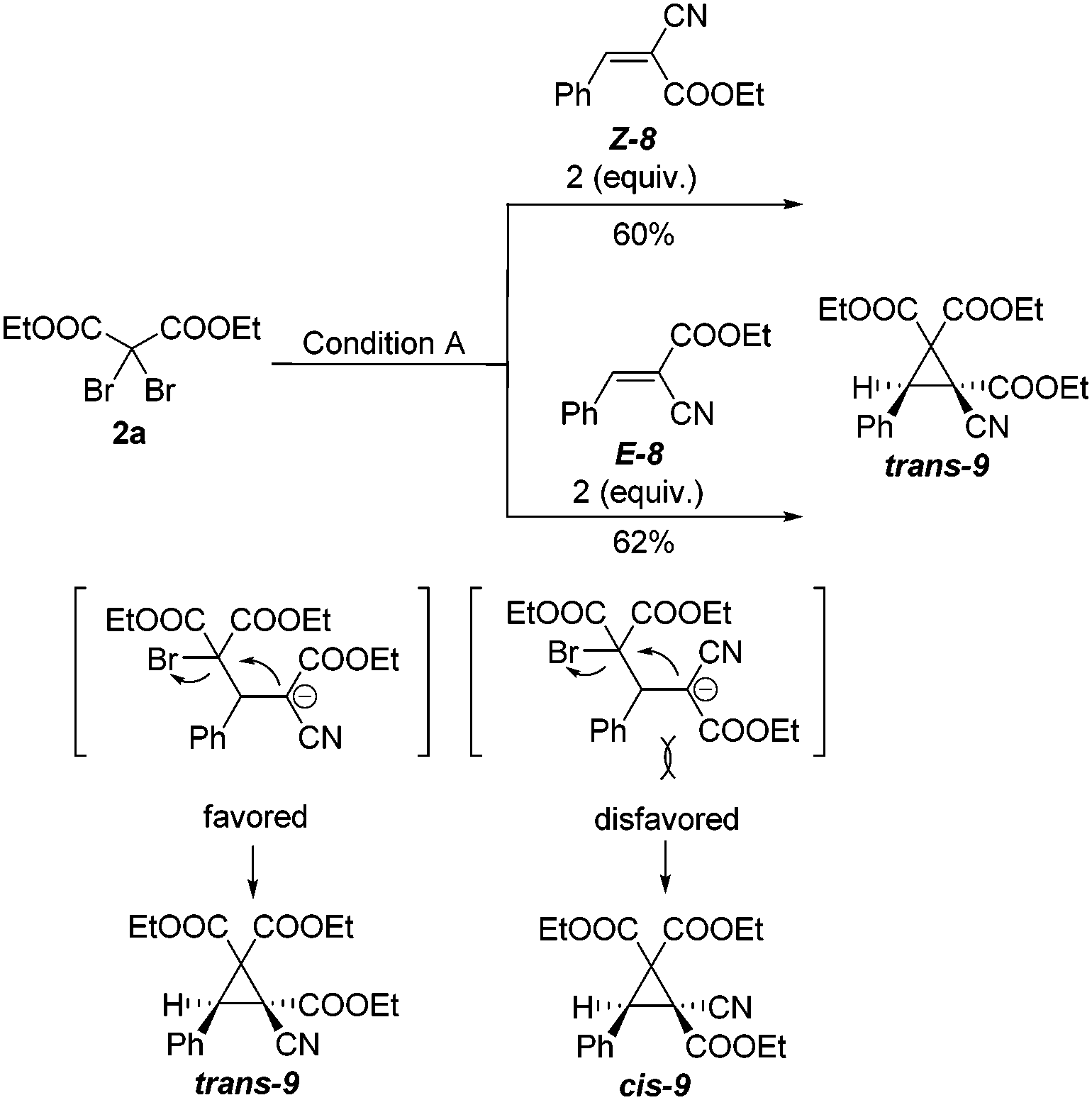 | ||
| Scheme 4 Cyclopropanation of two stereoisomeric alkenes Z-8 and E-8. | ||
Although the above results fitted well with our initially proposed mechanism as shown in Scheme 3, some other possibilities of reaction pathways should also be considered (for further discussion, see the ESI†).
In summary, we have developed a visible light induced generation of carbanions to achieve cyclopropanation of dibromomalonates with alkenes by VLPC, which provides a novel catalytic protocol for the synthesis of multisubstituted cyclopropane derivatives under mild conditions. The high yield of this transformation shows a broad range of synthetic application prospects. A carbanion pathway with double-SET has been proposed to explain the mechanism of this reaction. Further investigations on the applications of this system to new reactions for organic synthesis are in progress in our laboratory.
We greatly acknowledge the financial support from the Shanghai Rising-Star Program (14QA1400500), the National Basic Research Program of China (973 Program, 2012CB720300), the National Nature Science Foundation of China (21274023, 21102016, and 21102157), the Shanghai Scientific and Technological Innovation Project (13520711500), and Development and Innovation of Instrument Functions, CAS (No. Y27YQ1110G).
Notes and references
- (a) D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed; (b) T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527–532 CrossRef CAS PubMed; (c) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2010, 40, 102 RSC; (d) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (e) J. Xuan and W. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828–6838 CrossRef CAS PubMed; (f) Y. Zou, J. Chen and W. Xiao, Angew. Chem., Int. Ed., 2013, 52, 11701–11703 CrossRef CAS PubMed; (g) D. P. Hari and B. König, Angew. Chem., Int. Ed., 2013, 52, 4734–4743 CrossRef CAS PubMed.
- (a) A. McNally, C. K. Prier and D. W. C. MacMillan, Science, 2011, 334, 1114–1117 CrossRef CAS PubMed; (b) D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS PubMed; (c) M. T. Pirnot, D. A. Rankic, D. B. C. Martin and D. W. C. MacMillan, Science, 2013, 339, 1590–1593 CrossRef PubMed; (d) D. A. Nagib and D. W. C. MacMillan, Nature, 2011, 480, 224–228 CrossRef CAS PubMed.
- J. Du, K. L. Skubi, D. M. Schultz and T. P. Yoon, Science, 2014, 344, 392–396 CrossRef CAS PubMed.
- (a) C. Dai, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2011, 3, 140–145 CrossRef CAS PubMed; (b) J. D. Nguyen, E. M. D'Amato, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2012, 4, 854–859 CrossRef CAS PubMed.
- (a) D. Kalyani, K. B. McMurtrey, S. R. Neufeldt and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 18566–18569 CrossRef CAS PubMed; (b) Y. Miyake, K. Nakajima and Y. Nishibayashi, J. Am. Chem. Soc., 2012, 134, 3338–3341 CrossRef CAS PubMed; (c) X. Shu, M. Zhang, Y. He, H. Frei and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 5844–5847 CrossRef CAS PubMed; (d) Y. Ye and M. S. Sanford, J. Am. Chem. Soc., 2012, 134, 9033–9037 Search PubMed; (e) S. Cai, X. Zhao, X. Wang, Q. Liu, Z. Li and D. Z. Wang, Angew. Chem., Int. Ed., 2012, 51, 8050–8053 CrossRef CAS PubMed; (f) G. Deng, Z. Wang, J. Xia, P. Qian, R. Song, M. Hu, L. Gong and J. Li, Angew. Chem., Int. Ed., 2013, 52, 1535–1538 CrossRef CAS PubMed; (g) N. Iqbal, J. Jung, S. Park and E. J. Cho, Angew. Chem., Int. Ed., 2014, 53, 539–542 CrossRef CAS PubMed; (h) J. Liu, Q. Liu, H. Yi, C. Qin, R. Bai, X. Qi, Y. Lan and A. Lei, Angew. Chem., Int. Ed., 2014, 53, 502–506 CrossRef CAS PubMed.
- (a) A. Juris, V. Balzani, P. Belser and A. von Zelewsky, Helv. Chim. Acta, 1981, 64, 2175–2182 CrossRef CAS; (b) K. Kalyanasundaram, Coord. Chem. Rev., 1982, 46, 159–244 CrossRef CAS.
- C. Pac, M. Ihama, M. Yasuda, Y. Miyauchi and H. Sakurai, J. Am. Chem. Soc., 1981, 103, 6495–6497 CrossRef CAS.
- K. Hironaka, S. Fukuzumi and T. Tanaka, J. Chem. Soc., Perkin Trans. 2, 1984, 1705–1709 RSC.
- (a) J. D. Nguyen, J. W. Tucker, M. D. Konieczynska and C. R. J. Stephenson, J. Am. Chem. Soc., 2011, 133, 4160–4163 CrossRef CAS PubMed; (b) C. Wallentin, J. D. Nguyen, P. Finkbeiner and C. R. J. Stephenson, J. Am. Chem. Soc., 2012, 134, 8875–8884 CrossRef CAS PubMed.
- J. C. Le Menn, J. Sarrazin and A. Tallec, Electrochim. Acta, 1990, 35, 563–566 CrossRef CAS.
- (a) M. N. Elinson, S. K. Feducovich, Z. A. Starikova, A. N. Vereshchagin, P. A. Belyakov and G. I. Nikishin, Tetrahedron, 2006, 62, 3989–3996 CrossRef CAS PubMed; (b) D. Kawai, K. Kawasumi, T. Miyahara, T. Hirashita and S. Araki, Tetrahedron, 2009, 65, 10390–10394 CrossRef CAS PubMed; (c) X. Xin, Q. Zhang, Y. Liang, R. Zhang and D. Dong, Org. Biomol. Chem., 2014, 12, 2427–2435 RSC.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS.
- (a) R. Rios, H. Sundén, J. Vesely, G. Zhao, P. Dziedzic and A. Córdova, Adv. Synth. Catal., 2007, 349, 1028–1032 CrossRef CAS; (b) H. Xie, L. Zu, H. Li, J. Wang and W. Wang, J. Am. Chem. Soc., 2007, 129, 10886–10894 CrossRef CAS PubMed.
- For a detailed description of the experimental procedure, please see the ESI†.
- (a) V. Arca, C. Paradisi and G. Scorrano, J. Org. Chem., 1990, 55, 3617–3621 CrossRef CAS; (b) L. Grossi and S. Strazzari, J. Org. Chem., 2000, 65, 2748–2754 CrossRef CAS.
- (a) Y. S. Ng, C. S. Chan and K. S. Chan, Tetrahedron Lett., 2012, 53, 3911–3914 CrossRef CAS PubMed; (b) X. Zheng, L. Yang, W. Du, A. Ding and H. Guo, Chem. – Asian J., 2014, 9, 439–442 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: A picture of the setup of this reaction, experimental procedures, and characterization data for all compounds. See DOI: 10.1039/c4cc08203f |
| ‡ The authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |