
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Intrinsic formation of nanocrystalline neptunium dioxide under neutral aqueous conditions relevant to deep geological repositories†
Richard
Husar
a,
René
Hübner
b,
Christoph
Hennig
ac,
Philippe M.
Martin
d,
Mélanie
Chollet
d,
Stephan
Weiss
a,
Thorsten
Stumpf
a,
Harald
Zänker
a and
Atsushi
Ikeda-Ohno
*a
aInstitute of Resource Ecology, Helmholtz-Zentrum Dresden-Rossendorf (HZDR), Bautzner Landstrasee 400, D-01328 Dresden, Germany. E-mail: a.ikeda@hzdr.de
bInstitute of Ion Beam Physics and Materials Research, Helmholtz-Zentrum Dresden-Rossendorf (HZDR), Bautzner Landstrasee 400, D-01328 Dresden, Germany
cThe Rossendorf Beamline at the European Synchrotron Radiation Facility (ESRF), BP 220, F-38043 Grenoble, France
dCommissariat à l'énergie atomique et aux énergies alternatives (CEA), DEN, DEC, Cadarache, F-13108 Saint-Paul-Lez-Durance, France
First published on 5th December 2014
Abstract
The dilution of aqueous neptunium carbonate complexes induces the intrinsic formation of nanocrystalline neptunium dioxide (NpO2) particles, which are characterised by UV/Vis and X-ray absorption spectroscopies and transmission electron microscopy. This new route of nanocrystalline NpO2 formation could be a potential scenario for the environmental transport of radionuclides from the waste repository (i.e. under near-field alkaline conditions) to the geological environment (i.e. under far-field neutral conditions).
Owing to their high charge density, the aqueous chemistry of tetravalent actinides (An(IV)) is dominated by strong hydrolysis which often initiates the intrinsic formation of nano-sized oxide/oxyhydroxide particles.1,2 Under the geochemical conditions relevant to the repository and disposal of radioactive waste, the An(IV) nanoparticles could occur as mobile colloidal species,3,4 the potential formation of which poses serious concerns for the safety management of radioactive waste. For this reason, the An(IV) nanoparticles have attracted extensive attention over the past few years, particularly on the preparation and characterisation of pure phases of An(IV) oxide nanocrystals (NCs).1,5–7 In aqueous systems, the tetravalent state is a major concern for the early actinides, i.e. from 90Th to 94Pu,8 except for 91Pa which shows a strong oxidation tendency to form Pa(V).9 Considerable efforts by precedent researchers have yielded success in identifying the fluorite-based oxide NCs originating from aqueous solutions of Th(IV),10 U(IV),11 and Pu(IV),1,6,12 whilst no report has yet been released for Np(IV) NCs despite the longstanding prognostication of their formation.13,14 We herein report the first characterisation of nano-sized neptunium dioxide (NpO2) obtained by a novel approach based on the dilution of an aqueous bicarbonate solution of Np(IV). A major isotope of Np, 237Np (T1/2 = 2.14 × 106 years), is considered to be the predominant contributor to a potential radiation dose in radioactive waste repositories over an extremely long time frame of 1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000 years15 and, hence, it is a significant cause of concern for the long-term safety assessment of radioactive waste repository and disposal. As such, this study not only completes a series of An(IV) NCs, but would also provide fundamental information required for the long-term safety management of radioactive waste.
000000 years15 and, hence, it is a significant cause of concern for the long-term safety assessment of radioactive waste repository and disposal. As such, this study not only completes a series of An(IV) NCs, but would also provide fundamental information required for the long-term safety management of radioactive waste.
Precedent attempts to obtain aqueous-originated An(IV) NCs were based on the neutralisation of acidic An(IV) solutions.6,10,16 Our Np(IV) NCs demonstrated here were, in contrast, prepared from a weakly basic solution of Np(IV) in NaHCO3 (pH = 8.6). The background of our synthetic strategy is to dissociate the soluble Np(IV) carbonate species under weakly basic conditions to trigger olation/oxolation reactions which finally yield Np(IV) oxide clusters, whilst in the former neutralisation process the olation/oxolation reactions are induced by the hydrolysis of the hydrated An(IV) species.17 The dissociation process of the Np(IV) carbonate species was monitored by UV/Vis absorption spectroscopy. The UV/Vis spectrum of the initial colloid-free solution of Np(IV) in NaHCO3 (solution 1 in Fig. 1) was consistent with that reported for the Np(IV) carbonate species.18 A tenfold dilution of this Np(IV) carbonate solution with ultrapure water (pH = 7.0) resulted in an immediate change in UV/Vis spectra with the emergence of a new absorption maximum at 742 nm (solution 2 in Fig. 1), which indicates the formation of colloidal Np species.19 The diluted Np solution appeared to be clear for the first few hours, and later a yellow/brownish precipitate was formed (Fig. S1(a), ESI†). The redispersion of this precipitate in ultrapure water yielded an apparently transparent solution (Fig. S1(b) in ESI†). The UV/Vis spectrum of this transparent solution still showed the characteristic absorption maximum at 742 nm but with a significant increase in background (solution 3 in Fig. 1). The observed increase in background can be attributed to the light scattering from small particles,20 suggesting the formation of Np colloidal particles in solution 3. The zeta potential (ζ) of this Np colloid was determined to be 0 mV, which explains the tendency of the formed Np colloid to undergo precipitation within a few hours of redispersion. This precipitation–dispersion process was repeatable.
 | ||
| Fig. 1 UV/Vis absorption spectra of (1) the initial colloid-free Np(IV) solution in 1 M NaHCO3 at pH = 8.6, (2) tenfold dilution of solution 1 with ultrapure water, and (3) redispersion of the precipitate obtained from solution 2 in ultrapure water. The spectrum of solution 2 was recorded immediately after dilution. | ||
X-ray absorption spectroscopy, including both the X-ray absorption near-edge structure (XANES) and the extended X-ray absorption fine structure (EXAFS), was employed to characterise the Np species in the solution and precipitate samples. The dilution of the initial Np(IV) solution and subsequent formation of Np colloids resulted in no significant shift in the Np LIII-edge XANES edge position (Fig. S2 and Table S1 in ESI†), suggesting that the tetravalent state was preserved even after the colloid formation. The EXAFS structural parameters obtained for the initial Np(IV) solution (=solution 1 in Fig. 1) are well consistent with a pentacarbonate Np(IV) structure, [Np(IV)(CO3)5]6− (Table S2 in ESI†), the coordination geometry of which is comparable to those reported for [U(IV)(CO3)5]6− (ref. 21) and [Pu(IV)(CO3)5]6−.22 The EXAFS oscillation became intricate particularly in the higher k range when [Np(IV)(CO3)5]6− was transformed into a precipitate by dilution (Fig. 2(a)). The main oscillation feature of this precipitate bore a close resemblance to that of the reference NpO2 powder, although the variation of their oscillation amplitude was different (Fig. 2(a)). EXAFS structural analysis reveals that the nearest Np–O and Np–Np interatomic distances (R) obtained for the Np(IV) precipitate are well comparable to those for NpO2, whilst the coordination numbers (CNs) for the precipitate are calculated to be much lower with larger Debye–Waller factors (σ2) as compared with those for NpO2 (Table S2 in ESI†). Similar trends were also observed for UO2 NCs5 and a colloidal form of the An(IV) oxide/hydroxide species (including the polynuclear species),2,23,24 indicating that the formed Np(IV) precipitate could be identified as either small particles of crystalline NpO2 or structurally disordered Np(IV) oxide/hydroxide.
 | ||
| Fig. 2 (a) k3-weighted Np LIII-edge EXAFS spectra of Np(IV) in 1 M NaHCO3/1 M HNO3 at pH = 8.6 (red), a wet precipitate obtained by the dilution of the Np(IV) NaHCO3–HNO3 solution with ultrapure water (blue) and the reference NpO2 powder (black), and (b) their corresponding Fourier transform (FT) spectra. Solid lines, experimental data; dotted lines, theoretical fit; phase shifts (Δ) are not corrected on FTs; MS*, multiple scattering paths. The magnitude of the data for NpO2 is reduced by one fourth relative to the y axis for clear comparison of the spectra. | ||
The Np(IV) precipitate was further characterised by high-resolution transmission electron microscopy (HR-TEM). Shown in Fig. 3(a) is a TEM micrograph of the precipitate obtained from solution 3 in Fig. 1, clearly demonstrating the agglomeration of uniformly sized particles 2–5 nm in diameter. The relevant energy-dispersive X-ray (EDX) spectrum revealed the dominant presence of Np and O in the nanoparticles (Fig. S4 in ESI†). Upon zooming in on some specific particles, clear lattice images could be acquired (Fig. 3(b) and (c)), the selected area electron diffraction (SAED) patterns of which are consistent with the fluorite-type NpO2 structure (Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m)25 without the presence of secondary phases (Fig. 3(d)). Note that the gentle drying process at room temperature to prepare TEM samples would not cause the formation of NpO2 crystals, as the synthesis of crystalline NpO2 generally involves calcination at high temperature.26 Hence, it is highly likely that the observed NpO2 NCs were already present in the solution phase. Another HR-TEM image of the precipitate obtained directly from solution 2 in Fig. 1 (i.e. “non redispersed” precipitate) still showed clear lattice fringes compatible with the NpO2 structure (Fig. S5 in ESI†), confirming the formation of NpO2 NCs even in the original precipitate before the washing process. These results suggest that the dissociation of the initial soluble Np(IV) complex (i.e. [Np(IV)(CO3)5]6−) induces the intrinsic formation of nanocrystalline NpO2 in the solution phase.
m)25 without the presence of secondary phases (Fig. 3(d)). Note that the gentle drying process at room temperature to prepare TEM samples would not cause the formation of NpO2 crystals, as the synthesis of crystalline NpO2 generally involves calcination at high temperature.26 Hence, it is highly likely that the observed NpO2 NCs were already present in the solution phase. Another HR-TEM image of the precipitate obtained directly from solution 2 in Fig. 1 (i.e. “non redispersed” precipitate) still showed clear lattice fringes compatible with the NpO2 structure (Fig. S5 in ESI†), confirming the formation of NpO2 NCs even in the original precipitate before the washing process. These results suggest that the dissociation of the initial soluble Np(IV) complex (i.e. [Np(IV)(CO3)5]6−) induces the intrinsic formation of nanocrystalline NpO2 in the solution phase.
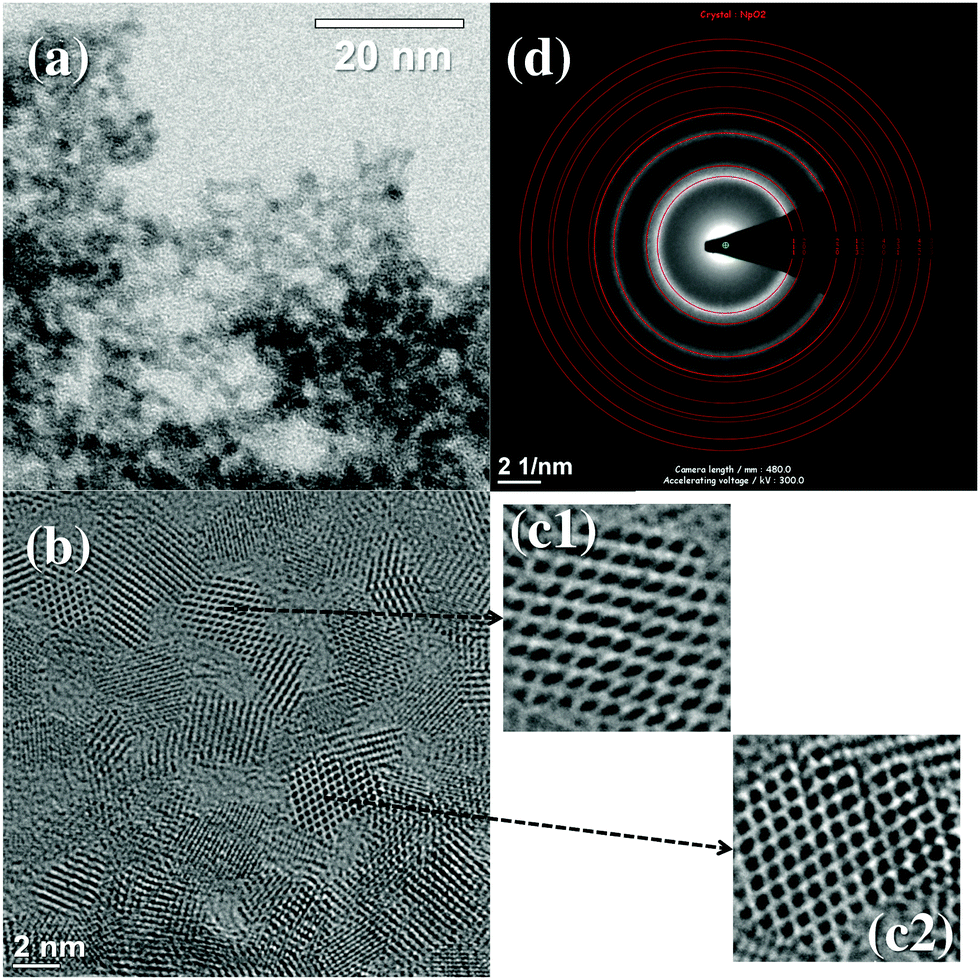 | ||
| Fig. 3 (a) Bright field TEM micrograph of the dried precipitate obtained from solution 3 in Fig. 1, (b and c) their HR-TEM images and (d) selected area electron diffraction (SAED) patterns. Red rings on (d) are the simulated diffraction patterns based on the fluorite-type NpO2 structure (ICSD card 647176), demonstrating that the precipitate is composed of a pure phase of crystalline NpO2. | ||
The precipitation of the aqueous An(IV) species under circumneutral conditions could also involve the formation of amorphous oxides/hydroxides.13,27 These amorphous phases often show a similar EXAFS pattern as observed for the pure dioxides, but their CNs for the neighbouring coordination shells (i.e. nearest An–O and An–An) are calculated to be much smaller than those for the dioxide together with larger σ2 values stemming from their amorphism.2,23,24 The CNs and σ2 values obtained for the Np(IV) wet precipitate follow this trend (Table S2 in ESI†). Therefore, the Np(IV) precipitate formed in solution can be identified as a mixture of amorphous Np(IV) oxides/hydroxides and nanocrystalline NpO2, only the latter of which could be characterised by TEM. The redispersion of the original precipitate into water removes soluble species/phases from the precipitate, finally helping in purifying NpO2 NCs from the amorphous phases. Indeed, image acquisition for the redispersed precipitate (Fig. 3) barely suffered from the presence of amorphous phases.
It has been reported that some An(IV) and Ce(IV), a chemical analogue of An(IV), have an intrinsic nature to form uniformly sized (2–3 nm) oxide NCs in solution phase,1,7,28 which is in accordance with the NpO2 NCs observed in Fig. 3. Therefore, the original precipitate (“wet precipitate” in Fig. 2) could be identified as the aggregation of 2–3 nm sized NpO2 NCs agglomerated via amorphous oxide/hydroxide phases. This amorphous phase tie is removed by the redispersion process, which finally yields uniformly sized NpO2 NCs observed in Fig. 3(a).
Soluble An(IV) species formed under alkaline conditions, such as [Np(IV)(CO3)5]6− in the present case, have been considered to be stable. This study demonstrates, however, that the hydrolysis of An(IV) is strong enough to dissociate these stable species by simple dilution and the subsequent marginal change in pH, which finally yields crystalline AnO2 nanoparticles. This study also simulates the plausible transport scenario of An(IV) from waste repositories (under near-field alkaline conditions) to the surrounding environment (under far-field neutral conditions) involving diffusion processes (dilution). The observed drastic change in the chemical form from a soluble species to stable nanocrystalline oxides would have a significant impact on understanding the behaviour of An in natural aquatic environments. This is of particular importance in terms of long-term prediction of radionuclide transport in geological environments, as AnO2 NCs are expected to be stable for a long period of time and the co-existing amorphous phases could even transform into AnO2 NCs over time.29
This study also highlights the importance of further investigation into the formation of An(IV) NCs under alkaline conditions as well as their properties as colloids, both of which are still unexplored. With a general knowledge of chemistry, one would expect the same hydrolysis product regardless of whether the initial solution is neutralized from an acidic or a basic solution. As a matter of fact, the precedent studies on An(IV) hydrolysis have focused primarily on the former neutralisation route from the acidic conditions. This would reflect the fact that the leakage of An contaminants (e.g. Pu) was often found under acidic–circumneutral conditions at shallow ground waste disposal sites (e.g. pH > 1.9 at the Maxey Flats disposal site, Kentucky, USA;30 pH = 3.9–6.8 at the Savannah River Site, South Carolina, USA;31 and pH = 5 at the Little Forest Legacy Site, New South Wales, Australia32). However, as demonstrated in Fig. S3 in the ESI,† the EXAFS spectrum for the present alkaline-originated Np(IV) precipitate is obviously different from that for the precipitate obtained under acidic conditions,33 implying that the alkaline-originated precipitate would be more amorphous than the acid-originated one. Although further studies are required to fully characterise these Np(IV) precipitates, this difference in the chemical form would finally cause a significant impact on their chemical behaviour in actual environmental and engineered systems.
In addition to the environmental implication discussed above, this study also demonstrates a new synthetic concept for producing metal dioxide (MO2) NCs via simple dilution of alkaline M(IV) solutions. The existing routes to MO2 NCs, such as TiO2 (ref. 34) or ZrO2,35 are generally based on the hydrolysis of the aquo M(IV) species. However, the alkaline-originated synthesis demonstrated in this study is potentially applicable to the production of MO2 NCs for these transition metals.36
Notes and references
- L. Soderholm, P. M. Almond, S. Skanthakumar, R. E. Wilson and P. C. Burns, Angew. Chem., Int. Ed., 2008, 47, 298–302 CrossRef CAS PubMed.
- J. Rothe, M. A. Denecke, V. Neck, R. Müller and J. I. Kim, Inorg. Chem., 2002, 41, 249–258 CrossRef CAS PubMed.
- C. Walther and M. A. Denecke, Chem. Rev., 2013, 113, 995–1015 CrossRef CAS PubMed.
- A. B. Kersting, Inorg. Chem., 2013, 52, 3533–3546 CrossRef CAS PubMed.
- E. J. Schofield, H. Veeramani, J. O. Sharp, E. Suvorova, R. Bernier-Latmani, A. Mehta, J. Stahlman, M. S. Webb, D. L. Clark, S. D. Conradson, E. S. Ilton and J. R. Bargar, Environ. Sci. Technol., 2008, 42, 7898–7904 CrossRef CAS.
- R. E. Wilson, S. Skanthakumar and L. Soderholm, Angew. Chem., Int. Ed., 2011, 50, 11234–11237 CrossRef CAS PubMed.
- C. Falaise, C. Volkringer, J. F. Vigier, A. Beaurain, P. Roussel, P. Rabu and T. Loiseau, J. Am. Chem. Soc., 2013, 135, 15678–15681 CrossRef CAS PubMed.
- N. M. Edelstein, J. Fuger, J. J. Katz and L. R. Morss, in The chemistry of the actinide and transactinide elements, ed. L. R. Morss, N. M. Edelstein, J. Fuger and J. J. Katz, Springer, Dordrecht, The Netherland, 3rd edn, 2006, vol. 3, p. 1774 Search PubMed.
- B. F. Myasoedov, H. W. Kirby and I. G. Tananaev, in The chemistry of the actinide and transactinide elements, ed. L. R. Morss, N. M. Edelstein, J. Fuger and J. J. Katz, Springer, Dordrecht, The Netherland, 3rd edn, 2006, vol. 1, p. 209 Search PubMed.
- D. J. Dzimitrowicz, P. J. Wiseman and D. Cherns, J. Colloid Interface Sci., 1985, 103, 170–177 CrossRef CAS.
- G. Rousseau, M. Fattahi, B. Grambow, L. Desgranges, F. Boucher, G. Ouvrard, N. Millot and J. C. Nièpce, J. Solid State Chem., 2009, 182, 2591–2597 CrossRef CAS PubMed.
- B. A. Powell, Z. Dai, M. Zavarin, P. Zhao and A. B. Kersting, Environ. Sci. Technol., 2011, 45, 2698–2703 CrossRef CAS PubMed.
- R. Guillaumont, T. Fanghänel, V. Neck, J. Fuger, D. A. Palmer, I. Grenthe and M. H. Rand, Update on the chemical thermodynamics of uranium, neptunium, plutonium, americium and technetium, Elsevier B.V., Amesterdam, The Netherland, 2003 Search PubMed.
- K. E. Roberts, T. J. Wolery, C. E. Atkins-Duffin, T. G. Prussin, P. G. Allen, J. J. Bucher, D. K. Shuh, R. J. Finch and S. G. Prussin, Radiochim. Acta, 2003, 91, 87–92 CrossRef CAS.
- D. C. Sassani, E. R. Siegmann, S. D. Sevougian and R. W. Andrews, Constrains on solubility-limited neptunium concentrations for use in performance assessment analyses, B00000000-01717-2200-00191, Rev. 00, US-DOE, North Las Vegas, Nevada, USA, 1998 Search PubMed.
- D. A. Costanzo, R. E. Biggers and J. T. Bell, J. Inorg. Nucl. Chem., 1973, 35, 609–622 CrossRef CAS.
- M. Henry, J. P. Jolivet and J. Livage, Struct. Bonding, 1992, 77, 135–206 CrossRef.
- D. W. Wester and J. C. Sullivan, J. Inorg. Nucl. Chem., 1981, 43, 2919–2923 CrossRef CAS.
- V. Neck, J. I. Kim, B. S. Seidel, C. M. Marquardt, K. Dardenne, M. P. Jensen and W. Hauser, Radiochim. Acta, 2001, 89, 439–446 CAS.
- C. F. Bohren and D. R. Huffman, Absorption and scattering of light by small particles, Wiley-VCH Verlag GmBH & Co. KGaA, Weinheim, Germany, 1998 Search PubMed.
- C. Hennig, A. Ikeda-Ohno, F. Emmerling, W. Kraus and G. Bernhard, Dalton Trans., 2010, 39, 3744–3750 RSC.
- D. L. Clark, S. D. Conradson, D. W. Keogh, P. D. Palmer, B. L. Scott and C. D. Tait, Inorg. Chem., 1998, 37, 2893–2899 CrossRef CAS.
- J. Rothe, C. Walther, M. A. Denecke and T. Fanghänel, Inorg. Chem., 2004, 43, 4708–4718 CrossRef CAS PubMed.
- A. Ikeda-Ohno, C. Hennig, S. Tsushima, A. C. Scheinost, G. Bernhard and T. Yaita, Inorg. Chem., 2009, 48, 7201–7210 CrossRef CAS PubMed.
- D. Taylor, Trans. J. Br. Ceram. Soc., 1984, 83, 32–37 CAS.
- J. A. Porter, Ind. Eng. Chem. Process Des. Dev., 1964, 3, 289–292 CAS.
- M. Rand, J. Fuger, I. Grenthe, V. Neck and D. Rai, Chemical Thermodynamics of Thorium, OECD-NEA, 2007 Search PubMed.
- A. Ikeda-Ohno, C. Hennig, S. Weiss, T. Yaita and G. Bernhard, Chem. – Eur. J., 2013, 19, 7348–7360 CrossRef CAS PubMed.
- R. G. Strickert, D. Rai and R. W. Fulton, ACS Symp. Ser., 1984, 246, 136–145 CrossRef PubMed.
- J. M. Cleveland and T. F. Rees, Science, 1981, 212, 1506–1509 CAS.
- M. Dai, J. M. Kelley and K. O. Buesseler, Environ. Sci. Technol., 2002, 36, 3690–3699 CrossRef CAS.
- A. Ikeda-Ohno, J. J. Harrison, S. Thiruvoth, K. Wilsher, H. K. Wong, M. P. Johansen, T. D. Waite and T. E. Payne, Environ. Sci. Technol., 2014, 48, 10045–10053 CrossRef CAS PubMed.
- R. Husar, S. Weiss, C. Hennig, R. Hübner, A. Ikeda-Ohno and H. Zänker, Environ. Sci. Technol., 2014 DOI:10.1021/es503877b.
- C. Charbonneau, R. Gauvin and G. P. Demopoulos, J. Cryst. Growth, 2009, 312, 86–94 CrossRef CAS PubMed.
- H. Cölfen, H. Schnablegger, A. Fischer, F. C. Jentoft, G. Weinberg and R. Schlögl, Langmuir, 2002, 18, 3500–3509 CrossRef.
- P. Afanasiev, Mater. Res. Bull., 2002, 37, 1933–1940 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, sample photos, XAS spectra and TEM images not shown in the main text. See DOI: 10.1039/c4cc08103j |
| This journal is © The Royal Society of Chemistry 2015 |