
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning intramolecular electron and energy transfer processes in novel conjugates of La2@C80 and electron accepting subphthalocyanines†
Lai
Feng
*ab,
Marc
Rudolf
c,
Olga
Trukhina
de,
Zdenek
Slanina
f,
Filip
Uhlik
g,
Xing
Lu
h,
Tomas
Torres
*de,
Dirk M.
Guldi
*c and
Takeshi
Akasaka
*bhij
aCollege of Physics, Optoelectronics and Energy & Collaborative Innovation Center of Suzhou Nano Science and Technology, Soochow University, 215006 Suzhou, China. E-mail: fenglai@suda.edu.cn
bLife Science Center of Tsukuba Advanced Research Alliance, University of Tsukuba, 305-8577 Tsukuba, Japan. E-mail: akasaka@tara.tsukuba.ac.jp
cDepartment of Chemistry and Pharmacy & Interdisciplinary Center for Molecular Materials, Friedrich-Alexander-Universität Erlangen-Nürnberg, 91058 Erlangen, Germany. E-mail: dirk.guldi@fau.de
dDepartment of Organic Chemistry, Autonoma University of Madrid, 28049 Madrid, Spain. E-mail: tomas.torres@uam.es
eIMDEA Nanoscience, 9 Faraday, 28049 Madrid, Spain
fDepartment of Chemistry and Biochemistry, National Chung-Cheng University, 62117 Chia-Yi, Taiwan, Republic of China
gDepartment of Physical and Macromolecular Chemistry, Charles University in Prague, 6 Albertov, 12843 Praha 2, Czech Republic
hCollege of Materials Science and Engineering, Huazhong University of Science and Technology, 430074 Wuhan, China
iFoundation for Advancement of International Science, 305-0821 Tsukuba, Japan
jDepartment of Chemistry, Tokyo Gakugei University, 184-8501 Koganei, Japan
First published on 6th November 2014
Abstract
A series of two conjugates with La2@C80 and subphthalocyanine (SubPc) have been prepared and characterized by means of cyclic voltammetry, absorption, fluorescence, and femtosecond resolved transient absorption spectroscopy. The strong electron-donating character of La2@C80 is essential to power an intramolecular electron-transfer in the La2@C80–SubPc conjugates upon photoexcitation.
Mimicking photosynthesis has been of a great interest owing to the increasing need for an efficient and sustainable conversion of solar energy.1,2 One of the key factors in the reproduction of natural photosynthesis is to understand electron-transfer events between different electron donors and acceptors of the photosynthetic apparatus. Importantly, the nature of electron donors and acceptors determines the magnitude of electron transfer and, in turn, affects the overall yield of photosynthesis. Extensive studies have been carried out to design, to synthesize, and to probe electron donor and electron acceptor materials with improved performances in solar energy conversion.3,4 For photovoltaic applications, fullerenes stand out among the myriad of electron accepting materials.5 Owing to their unique structural and redox features, fullerenes have been widely integrated into a wide facet of electron donor–acceptor systems.6 It is also well known that the use of empty fullerenes C60 and C70 as electron donors in molecular photovoltaics is limited by their poor electron-donating ability. The generation of radical cations of, for example, C60 or C70 requires rather harsh conditions.7 Up to now, none of them have been employed as electron donors either in artificial photosynthesis or in photovoltaics.
Filling empty fullerenes with metals or metallic clusters affords endohedral metallofullerenes (EMFs), whose physical, chemical and electrochemical properties are different from those of empty fullerenes.8 For example, La@C82, M2@C80 (M = La, Ce), and M3N@C80 (M = Sc, Lu) undergo easier oxidations and feature stronger absorption throughout the visible part of the solar spectrum when compared to C60 and C70. These characteristics render EMFs p-type materials for photovoltaics. Very recently, the idea of using EMFs as electron donors has been verified in photophysical assays with Lu3N@C80 or La2@C80 as the electron donor and PDI or TCAQ as the electron acceptor, respectively.9,10
Subphthalocyanines (SubPcs) are aromatic chromophores, which absorb light throughout most of the visible part of the spectrum featuring (i) high extinction coefficients, (ii) excitation energies above 2.0 eV, and (iii) low reorganization energies in electron transfer reactions.11 Importantly, they do not aggregate owing to their conical shape and are known as strong fluorophores. SubPcs bearing on their periphery electron-withdrawing substituents are well-known electron acceptors.11b Thus, electron-deficient SubPcs have been considered as promising complements to fullerenes.
In the present work, we report on the synthesis and the properties of conjugates 1a and 1b (Scheme 1) that comprise La2@C80 as electron donor and (dodecafluoro)/hexa(pentylsulfonyl)SubPc as electron acceptors. We will demonstrate that, despite negligible interactions in the ground-state, electron-transfer events occur between SubPc and La2@C80 in the excited state. However, in conjugates 2a and 2b, only energy transfer events take place.
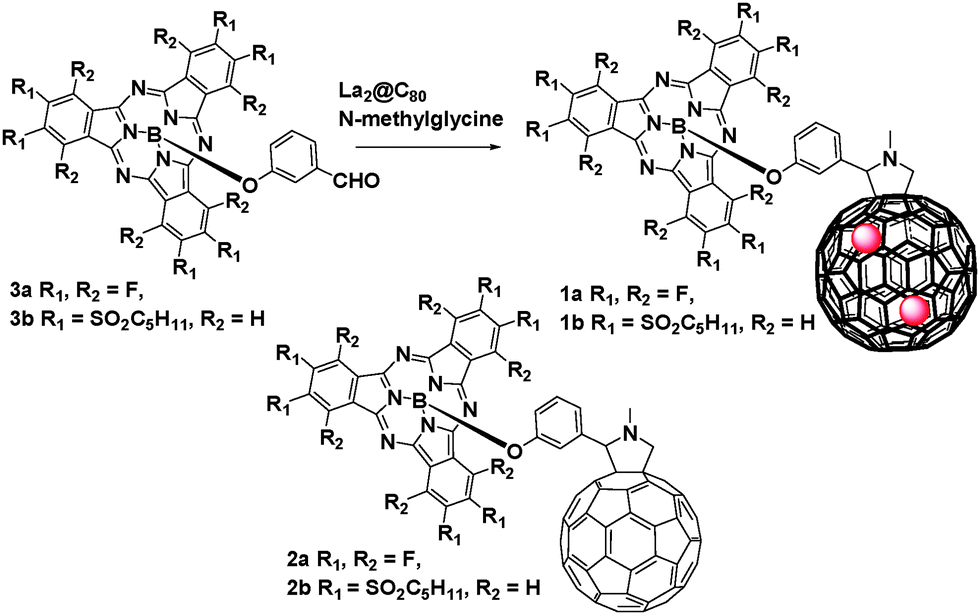 | ||
| Scheme 1 Synthesis of La2@C80–SubPc (1a and 1b) from subphthalocyanines (3a, 3b). | ||
Conjugates 1a, 1b were synthesized according to the procedure previously reported for 2a.12 Briefly, 1a, 1b were obtained in 25% or less via Prato reaction of La2@C80 and SubPc 3a, b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 13 (Scheme 1). The formation of 1a or 1b as major products was revealed by HPLC and they were isolated via a multi-step HPLC procedure (Fig. S1–S3, ESI†).14 The compositions of 1a and 1b were confirmed using MALDI-TOF mass spectrometry (Fig. S5–S7, ESI†).
13 (Scheme 1). The formation of 1a or 1b as major products was revealed by HPLC and they were isolated via a multi-step HPLC procedure (Fig. S1–S3, ESI†).14 The compositions of 1a and 1b were confirmed using MALDI-TOF mass spectrometry (Fig. S5–S7, ESI†).
To shed light onto the structural feature of 1a, VT1H NMR studies were conducted. As shown in Fig. S8 (ESI†), the 1H NMR spectrum measured at 283 K exhibits two sets of signals: those originating from pyrrolidine protons at 5–2 ppm and those from phenyl protons at 8–5.5 ppm, indicating the presence of at least two conformers of 1a in a 3:1 ratio. In each set, a pair of doublets, which are assigned to the geminal protons on the pyrrolidine, is discernible. Confirmation for this assignment was obtained by COSY experiments, revealing a reasonable cross-coupling relationship between the doublets (Fig. S9, ESI†). In addition, the chemical shifts as well as the chemical shift differences (Δδ = 1.2 to 1.3 ppm) of these geminal protons are in a good agreement with those reported previously.15 An increase of the temperature from 283 to 313 K resulted only in a broadening of the NMR signals in 1a (Fig. S8, ESI†) ruling out the possibility of conformational conversion in this temperature range. Characterization of 1b using 1H NMR was, however, hampered by the small amounts of the isolated material and its poor stability.
To further investigate the structural and electronic features of 1a, DFT-calculations were performed using a Gaussian 09 package.16 Owing to the [5,6]-addition pattern and the unsymmetrical pyrrolidine substitution, 1a adopts at most four different conformations – Fig. S13 (ESI†). Among them, conformer I, in which both, the pyrrolidine and the substituted phenoxy unit, are approaching a 5-member ring, has the lowest formation energy (Table S1, ESI†) at the M06-2X/3-21G∼6-31G*∼sdd level.17–19 In comparison, conformer II, in which both the pyrrolidine and the substituted phenoxy group are aligned close to a 6-member ring, is 2.3–3.3 kcal mol−1 less stable than conformer I. Conformers III and IV possess the least stability, namely 5.3 kcal mol−1 less than conformer I. Therefore, we hypothesize the presence of conformers I and II in purified 1a with a ratio of 3:1.
Electrochemical studies with 1a and 2a were carried out by means of CV and DPV. In the range from −2.0 to 1.2 V, 2a reveals four reversible one-electron reductions at −1.06, −1.18, −1.57, and −1.79 V and two irreversible one-electron oxidations at +0.99 and +1.08 V (Table 1 and Fig. S12, ESI†). The first oxidation, and the first and third reductions are C60 centered, while the second oxidation, and the second and forth reductions are centered on SubPc. As for 1a, the electrochemical patterns of the [5,6]-pyrrolidine La2@C80 and perfluorinated SubPc are clearly distinguishable.12,15 In particular, the first and second reductions, which are seen as a one-electron process at −0.47 and −1.12 V, coincide well with the reductions of La2@C80 and SubPc, respectively. The third reduction at −1.80 V appears as a two-electron process, involving the second reduction of La2@C80 and the second reduction of SubPc. In addition, three oxidations are visible. The first and second are fully reversible one-electron processes, which agree well with those of La2@C80. The third oxidation is a two-electron process, corresponding to the first oxidation of SubPc and the third oxidation of La2@C80. Our electrochemical assays point to the fact that ground state interactions between the electroactive constituents of 1a are negligible. The remarkable oxidative features of 1a underline the strong electron donor character of La2@C80, which is lacking in C60 in 2a.
Complementary DFT-calculations further underline the electrochemical data. As shown in Fig. S14 (ESI†), the calculated HOMO of 1a is mainly delocalized on La2@C80, while the LUMO and LUMO + 1 are localized on the endohedral La2 cluster and on the perfluorinated SubPc, respectively.
To gain further insight into the ground state features of 1a–b, we turned to absorption spectroscopy. At a first glance, the absorption spectrum of 1a–b is best described as a simple superimposition of the spectra of the individual components, namely SubPc and [5,6]-pyrrolidine La2@C80 (Fig. S10 and S11, ESI†). Detailed comparison between 1a and 1b suggests that the absorption maxima of 1b are 4 nm red-shifted relative to that of 1a, thus, confirming the stronger electron–acceptor properties of sulfonated SubPcs than of the fluorinated one. Despite the presence of [5,6]-pyrrolidine La2@C80, the absorption maximum of SubPc undergoes no shift as compared with that of SubPc 3a and 3b, indicating the lack of ground-state interaction between the individual components.
In fluorescence experiments, a solvent independent fluorescence quantum yield of 0.17 was noted for 3a. In stark contrast, fluorescence assays with 1a point to a rather marked quenching with fluorescence quantum yields of 0.005 (toluene), 0.006 (THF), and 0.006 (benzonitrile).
To attribute our spectral observation, spectroelectrochemical experiments on (F12SubPc)˙− and ((SO2C5H11)6SubPc)˙− as well as (La2@C80)˙+ were deemed important – Fig. S15 (ESI†). On the one hand, the differential absorption spectra of the electrochemically reduced 3a reveal two broad features with maxima at 455 and 655 nm, which are accompanied by shoulders at around 475 and 610 nm, as well as a minimum at 570 nm. Upon spectroelectrochemical reduction of 3b, spectral characteristics including maxima at 480, 545, 620, and 735 nm complemented by minima at 534 and 581 nm evolved. Notably, pulse radiolytic reductions with 3a or 3b in deaerated toluene/2-propanol/acetone mixtures (8:1:1 v/v) results in quantitatively similar spectra with characteristic fingerprints at 610 and 620 nm, respectively. On the other hand, a characteristic maximum at 900 nm and a broad near infrared tail evolve as spectroscopic characteristics upon spectroelectrochemical oxidation of 4.
Insights into the excited state deactivation in 4, 3a, 3b, 2a, 2b, 1a, and 1b, in general, and into the corresponding photoproducts, in particular, came from transient absorption measurements following femtosecond and nanosecond excitation. Excitation of 4 at 387 nm leads to the population of the La2@C80 singlet excited state (1.4 ± 0.2 eV), which features ground state bleaching at 465 nm and well-resolved fine structure with maxima at 516, 466, 614, 735, 800, and 900 nm. The latter is subject to a fast intersystem crossing – 60 ± 30 ps – to the triplet manifold due to the presence of the (La2)6+ cluster, which promotes efficient spin–orbit coupling. Following the singlet excited state decay, a weak and broad absorption in the 800–1200 nm region, along with broad features that taper at 550 nm, are discernible. These features relate to the La2@C80 triplet excited state (1.0 ± 0.1 eV).
F12SubPc 3a reveals upon excitation at 530 nm differential absorption changes, which include transient maxima at 440 and 600 nm as well as transient minima at 514, 575, and 635 nm – Fig. S16 (ESI†). In addition, a broad near-infrared feature spans from 650 to 1200 nm, which peaks around 710 nm. These features relate to the singlet excited state (2.16 eV) of 3a, which transforms with 1.9 ± 0.1 ns into the corresponding triplet excited state (1.4 eV). Transient absorption spectra of the latter maximize at 470 and 610 nm and minimize at 532 and 570 nm.
Commencing with the conclusion of the 530 nm excitation, SubPc 3b reveals differential absorption changes in the form of transient maxima at 424, 474, 623, 660 nm, a broad tail extending far into the near infrared, as well as transient minima at 533 and 583 nm – Fig. S17 (ESI†). These SubPc singlet excited state (2.12 eV) related transient absorption features undergo intersystem crossing to the corresponding triplet excited state (1.4 eV), which exhibits a broad transient in the visible part of the spectrum. The latter maximizes at 470 and 620 nm and minimizes at 533 and 583 nm. Owing to the presence of sulfur, which facilitates spin–orbit coupling, the intersystem crossing is accelerated relative to what is seen for 3a with lifetimes of 1220 ± 20 ps, 420 ± 10 ps, 415 ± 10 ps in toluene, THF, and benzonitrile, respectively.
Conjugate 1a gives rise upon 530 nm excitation to differential absorption changes in the form of transient maxima at 450, 600, and 720 nm as well as transient minima at 575 and 635 nm – Fig. 1. In line with the reference experiments, namely with 3a, we assign these changes to the F12SubPc singlet exited state. Instead of seeing the slow intersystem crossing to the SubPc triplet state, the SubPc singlet excited state decays ultrafast with lifetimes of 3.0 ± 0.4 ps (toluene), 2.2 ± 0.2 ps (THF), and 2.0 ± 0.2 ps (benzonitrile). Simultaneously, new transitions develop in the visible and near-infrared regions. Importantly, the new transients do not match the signature of the SubPc triplet excited state. Instead, maxima at 480 and 590 nm as well as minima at 515 and 570 nm are discernible. Please note that these features bear great resemblance to the pulse radiolytic findings in the context of reducing SubPc and, as such, relate to its π-radical anion – (F12SubPc)˙−. In the near-infrared region, a broad tail is attributable to the La2@C80 π-radical cation, that is, (La2@C80)˙+. Taking the aforementioned into consideration, we conclude that an energetically low lying radical ion pair state (1.32 eV), namely (La2@C80)˙+–(F12SubPc)˙−, is formed. Both fingerprints served as reliable probes to determine the lifetime of the metastable (La2@C80)˙+–(F12SubPc)˙− radical ion pair state. All decays were well fitted by a single exponential fitting function throughout the femtosecond time scale. In particular, lifetimes of 34 ± 2 ps (toluene), 32 ± 2 ps (THF), and 35 ± 2 ps (benzonitrile) were derived.
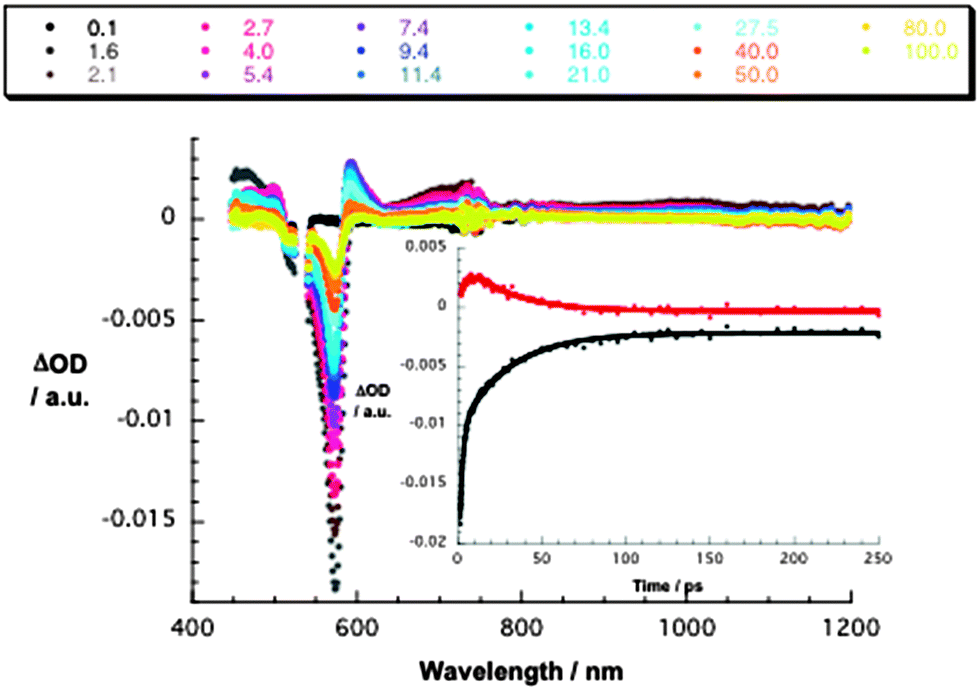 | ||
| Fig. 1 Differential absorption spectra (visible and near-infrared) obtained upon femtosecond flash photolysis (530 nm) of 1a (10−5 M) in argon-saturated THF with several time delays between 0.1 and 100 ps at room temperature. Inset: time-absorption profiles of the spectra shown at the top at 573 and 592 nm monitoring the charge separation and the charge recombination. | ||
Singlet oxygen quantum yields for 1a are as low as 0.010 in THF and support the assignment that any other state than the F12SubPc triplet excited state evolves as the product of charge recombination. Please note the singlet oxygen yields in 3a of 0.31 (toluene), 0.10 (THF), and 0.35 (benzonitrile).
Laser excitation of 1b in benzonitrile at 568 nm results immediately after excitation in differential absorption maxima at 485 and 623 nm and a minimum at 583 nm – Fig. S18 (ESI†). Although these transient features relate to the singlet excited state of SubPc they decay ultrafast with a lifetime of 1.4 ± 0.1 ps. New transients evolve, which maximize at 480 and 610 nm and minimize 583 nm. A spectral comparison with the results from the spectroelectrochemical/pulse radiolytic investigations supports the notion that the new transients are attributed to the π-radical anion – ((SO2C5H11)6SubPc)˙−. Taking the aforementioned into consideration, we postulate an electron transfer from La2@C80 to the SubPc singlet excited state to afford (La2@C80)˙+–((SO2C5H11)6SubPc)˙− (1.36 eV). It is worth mentioning that the detection of (La2@C80)˙+ in the near infrared is hampered by the thermal decomposition of SubPc. This is seen to form a product with absorptions in the 700–850 nm range. Nevertheless, the metastable (La2@C80)˙+–((SO2C5H11)6SubPc)˙− radical ion pair state decays with 28 ± 2 ps to the singlet ground state.
Likewise, 530 nm excitation of F12SubPc-C602a results in the exclusive formation of the SubPc singlet excited state – Fig. S19 (ESI†). In particular, transient maxima at 450, 600, and 720 nm as well as transient minima at 515, 575, and 635 nm are formed and decay rapidly with 1.5 ± 0.3 ps (toluene), 1.5 ± 0.3 ps (THF), and 1.4 ± 0.3 ps (benzonitrile). As the SubPc singlet excited state decay comes to an end a broad near-infrared transient, which maximizes at 910 nm, is noted, suggesting a C60 singlet excited state. Interestingly, we did not find the characteristic C60 triplet feature at 700 nm at the end of the C60 singlet excited state deactivation. In contrast, maxima at 470 and 615 nm as well as a minimum at 575 nm were found, pointing to the SubPc triplet excited state.12 From this we infer that the C60 triplet excited state (1.5 eV) undergoes a thermodynamically allowed transfer of triplet excited state energy to SubPc (1.4 eV). The kinetics at the 470 and 615 nm maxima further furnishes the kinetic assignment, namely the rate-determining step in the SubPc triplet excited state formation is the C60 centered intersystem crossing. A global analysis reveals kinetics that are very similar (1.6 ± 0.1 ns) to that of the inherent intersystem crossing dynamics seen for C60. In this context, it is reassuring that the transients seen at the end of the femtosecond experiments matches that at the beginning of the nanosecond experiment. Moreover, maxima at 470 and 610 nm, minima at 532 and 570 nm, and an excited state lifetime of 36 μs without oxygen perfectly agree with the SubPc triplet excited state of 3a. Likewise, singlet oxygen quantum yields of 2a were found to be as high as 0.28 (toluene), 0.13 (THF), and 0.41 (benzonitrile) and support the assignment that the triplet excited state evolves as the product of charge recombination. Please note that the singlet oxygen yields in 3a are as high as 0.35.
When turning to 2b, excitation at 530 nm is accompanied by the formation of its singlet excited state – Fig. S20 (ESI†). Evidence stems from monitoring maxima at 427, 474, 623, and 660 nm and minima at 535 and 585 nm. These decay in the presence of C60 rapidly with 1.5 ± 0.3 ps (toluene), 1.5 ± 0.3 ps (THF), and 1.4 ± 0.3 ps (benzonitrile) to form accordingly the C60 singlet excited state with its 910 nm maximum. Like for 2a, we did not find the characteristic C60 triplet feature. Instead, maxima at 470 and 625 nm as well as minima at 535 and 585 nm of the SubPc triplet excited state were concluded. In other words, the triplet excited state of SubPc (1.4 eV) evolves from a thermodynamically allowed transfer of triplet excited state. The kinetics at the 470 and 625 nm maxima and the 585 nm minimum document that the rate-determining step is the C60 centered intersystem crossing (1.6 ± 0.1 ns). In the absence of oxygen, the SubPc triplet excited state lifetime is 20 ± 5 μs in agreement with what was found for 3b.
Electron accepting SubPcs have been used in combination with La2@C80 to prepare a series of novel La2@C80–SubPc electron donor–acceptor conjugates to mimic the photosynthetic apparatus. Our results in terms of electrochemical and steady-state absorption reveal no appreciable electronic interactions between SubPc and La2@C80 in the ground state. This changed in the excited state, where an intramolecular electron-transfer evolves from La2@C80 to photoexcited SubPc. In comparison, reference conjugates of C60 and SubPcs feature only a singlet–singlet energy transfer from SubPc to C60. Thus, replacing C60 by La2@C80 provides a promising way to tune energy-transfer versus electron-transfer. Furthermore, considering the short separations between the electron donors and acceptors, optimizing the charge-separated state lifetimes seems achievable via the tailored design of linkers between La2@C80 and SubPc.
This work is financially supported by the NSFC (51372158), the NSF of Jiangsu Province (BK2012611), the Jiangsu Specially Appointed Professor Program (SR10800113), Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), the Project for Jiangsu Scientific and Technological Innovation Team (2013), Spanish MEC and MICINN (CTQ2011-24187/BQU and PRI-PIBUS-2011-1128), the Deutsche Forschungsgemeinschaft (GU 517/14-1) and the Comunidad de Madrid (S2013/MIT-2841, FOTOCARBON).
Notes and references
- A. A. Boghossian, M. H. Ham, J. H. Choi and M. S. Strano, Energy Environ. Sci., 2011, 4, 3834–3843 CAS.
- (a) S. Hammes-Schiffer, Acc. Chem. Res., 2009, 42, 1881–1889 CrossRef CAS PubMed; (b) M. R. Wasielewski, Chem. Rev., 1992, 92, 435–461 CrossRef CAS.
- G. de la Torre, G. Bottari, M. Sekita, A. Hausmann, D. M. Guldi and T. Torres, Chem. Soc. Rev., 2013, 42, 8049–8105 RSC.
- G. Bottari, G. de la Torre, D. M. Guldi and T. Torres, Chem. Rev., 2010, 110, 6768–6816 CrossRef CAS PubMed.
- S. Kirner, M. Sekita and D. M. Guldi, Adv. Mater., 2014, 26, 1482–1493 CrossRef CAS PubMed.
- (a) J. L. Delgado, P. A. Bouit, S. Filippone, M. A. Herranz and N. Martín, Chem. Commun., 2010, 46, 4853–4865 RSC; (b) D. M. Guldi, Chem. Soc. Rev., 2002, 31, 22–36 RSC.
- (a) K. Ohkubo, J. Ortiz, L. Martin-Gomis, F. Fernandez-Lazaro, A. Sastre-Santos and S. Fukuzumi, Chem. Commun., 2007, 589–591 RSC; (b) C. A. Reed, K. C. Kim, R. D. Bolskar and L. J. Mueller, Science, 2000, 289, 101–104 CrossRef CAS; (c) S. Fukuzumi, H. Mori, H. Imahori, T. Suenobu, Y. Araki, O. Ito and K. M. J. Kadish, J. Am. Chem. Soc., 2001, 123, 12458–12465 CrossRef CAS PubMed.
- (a) A. A. Popov, S. Yang and L. Dunsch, Chem. Rev., 2013, 113, 5989–6113 CrossRef CAS PubMed; (b) X. Lu, L. Feng, T. Akasaka and S. Nagase, Chem. Soc. Rev., 2012, 41, 7723–7760 RSC.
- (a) M. Rudolf, L. Feng, Z. Slanina, T. Akasaka, S. Nagase and D. M. Guldi, J. Am. Chem. Soc., 2013, 135, 11165–11174 CrossRef CAS PubMed; (b) L. Feng, M. Rudolf, S. Wolfrum, A. Troeger, Z. Slanina, T. Akasaka, S. Nagase, N. Martín, T. Ameri, C. J. Brabec and D. M. Guldi, J. Am. Chem. Soc., 2012, 134, 12190–12197 CrossRef CAS PubMed.
- Y. Takano, S. Obuchi, N. Mizorogi, R. García, M. A. Herranz, M. Rudolf, D. M. Guldi, N. Martín, S. Nagase and T. Akasaka, J. Am. Chem. Soc., 2012, 134, 19401–19408 CrossRef CAS PubMed.
- (a) C. G. Claessens, D. Gonzalez-Rodriguez, M. S. Rodriguez-Morgade, A. Medina and T. Torres, Chem. Rev., 2014, 114, 2192–2277 CrossRef CAS PubMed; (b) C. Romero-Nieto, A. Medina, A. Molina-Ontoria, C. G. Claessens, L. Echegoyen, N. Martin, T. Torres and D. M. Guldi, Chem. Commun., 2012, 48, 4953–4955 RSC.
- D. Gonzalez-Rodriguez, T. Torres, D. M. Guldi, J. Rivera, M. A. Herranz and L. Echegoyen, J. Am. Chem. Soc., 2004, 126, 6301–6313 CrossRef CAS PubMed.
- M. Ince, A. Medina, J. H. Yum, A. Yella, C. G. Claessens, M. V. Martinez-Diaz, M. Graetzel, M. K. Nazeeruddin and T. Torres, Chem. – Eur. J., 2014, 20, 2016–2021 CrossRef CAS PubMed.
- In the reaction mixture, besides 1a, a minor product 1a′ was also isolated and identified as a [6,6]-pyrrolidine adduct using MALDI-TOF mass and UV-vis-NIR absorption spectra (Fig. S6 and S9, ESI†).
- (a) M. Yamada, M. Okamura, S. Sato, I. C. Someya, N. Mizorogi, T. Tsuchiya, T. Akasaka, T. Kato and S. Nagase, Chem. – Eur. J., 2009, 15, 10533–10542 CrossRef CAS PubMed; (b) T. Cai, Z. Ge, E. B. Lezzi, T. E. Glass, K. Harich, H. W. Gibson and H. C. Dorn, Chem. Commun., 2005, 3594–3596 RSC.
- M. J. Frisch, et al., GAUSSIAN 09, Revision A. 02, Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- X. Y. Cao and M. Dolg, THEOCHEM, 2002, 581, 139–147 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, complete characterizations of 1a, 1b, and selected transient absorption spectra. See DOI: 10.1039/c4cc08072f |
| This journal is © The Royal Society of Chemistry 2015 |