
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A robust and modular synthesis of ynamides†
Steven J.
Mansfield
,
Craig D.
Campbell
,
Michael W.
Jones
and
Edward A.
Anderson
*
Chemistry Research Laboratory, University of Oxford, 12 Mansfield Road, Oxford, UK. E-mail: edward.anderson@chem.ox.ac.uk; Web: http://anderson.chem.ox.ac.uk Fax: +44 (0)1865 285002
First published on 6th November 2014
Abstract
A flexible, modular ynamide synthesis is reported that uses trichloroethene as an inexpensive two carbon synthon. A wide range of amides and electrophiles can be converted to the corresponding ynamides, importantly including acyclic carbamates, hindered amides, and aryl amides. This method thus overcomes many of the limitations of other approaches to this useful functionality.
Ynamides (1, Scheme 1) are versatile functionalities that are finding use in an increasing array of transformations.1,2 Their popularity can be attributed to the high regioselectivity of reactions of the polarized and nucleophilic alkyne, and also to the advent of several methods for their synthesis over the last decade. At the forefront of these are copper-catalyzed couplings of amides with alkynes3 or their derivatives,4 or dibromoalkenes (Scheme 1, eqn (1)).5 Despite these important advances, limitations remain for the preparation of certain ynamide derivatives. For instance, whilst oxazolidinones are excellent substrates for copper-catalyzed ynamide synthesis, acyclic carbamates and amides typically are not.6 Furthermore, these reactions are usually ineffective for poorly nucleophilic amides, including those with branched substituents adjacent to the nitrogen centre, and aniline derivatives. Here we report a solution to these problems, in the form of a modular and flexible ynamide synthesis that proceeds in short reaction times and displays extensive substituent diversity, and thus opens up new possibilities for the wider use of ynamides in organic synthesis.
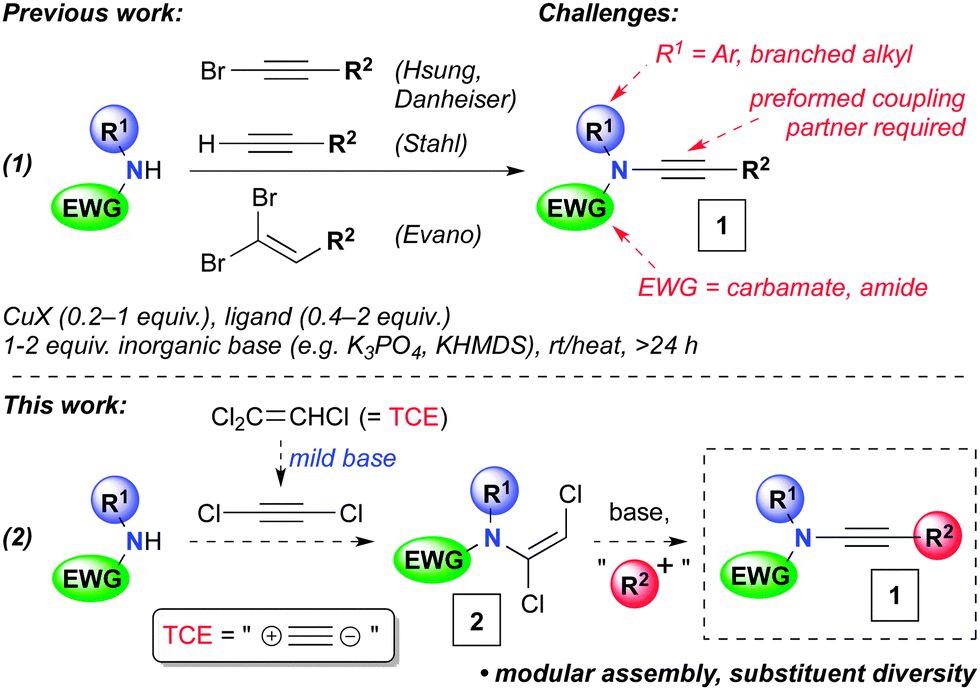 | ||
| Scheme 1 Copper-catalyzed ynamide formations, and the planned modular approach to ynamides 1via dichloroenamides 2. | ||
We hypothesized that dichloroacetylene, generated from inexpensive trichloroethene (TCE) under mildly basic reaction conditions, could serve as an excellent two carbon synthon for ynamide synthesis (eqn (2), Scheme 1).7,8 Addition of an amide to dichloroacetylene would lead to a dichloroenamide 2, a direct precursor to an ynamide via elimination. This route would benefit from a late-stage diversifying introduction of the alkyne substituent (R2) in the course of ynamide synthesis, using readily available electrophiles (i.e.2 → 1). Notably, this obviates the need for a preformed (halo)alkyne or dihaloalkene, as required in most ynamide synthesis methodology, which can restrict product scope.
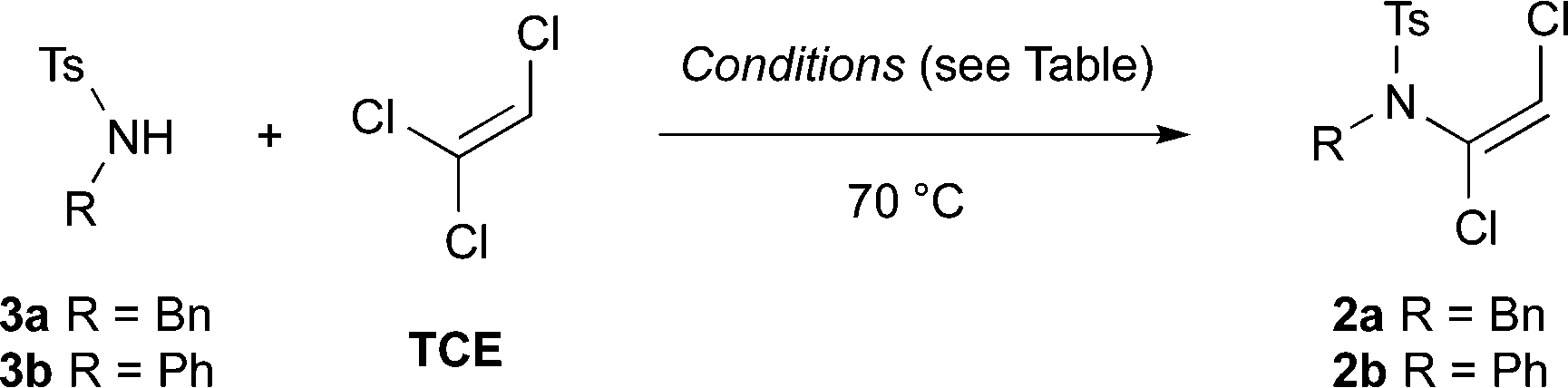
Investigations into dichloroenamide synthesis began with sulfonamides 3a and 3b, the latter of which is a challenging substrate for ynamide formation using other methods (Table 1). An initial screen of inorganic bases afforded good results with a slight excess of LiOH and TCE in DMF at 70 °C (entries 1 and 2), albeit after extended reaction times; other base–solvent combinations proved less effective (entries 3 and 4) or inconsistent (entries 5 and 6). The reaction time was significantly shortened by increasing the concentration, and the amount of base and TCE, which gave excellent yields of dichloroenamide (entries 7 and 8).9 Pleasingly, these conditions were further improved by using just 1.1 equiv. of TCE with Cs2CO3 (1.5 equiv.) in DMF at 50 °C, which effected rapid and high yielding formation of both enamides 2a and 2b (entries 9 and 10). The reaction remained efficient on multigram scale (4.1 g of 3a, 95% yield, entry 11).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | TCE equiv. | Base (equiv.), solvent ([M]) | t (h) | Yielda [%] (conversion [%]) |
| a Isolated yield; conversion determined by 1H NMR spectroscopic analysis of the crude reaction mixture. b Not determined. c Anhydrous DMF used. d Reaction conducted at 50 °C. e Conducted with 4.1 g (15.7 mmol) of 3a. | |||||
| 1 | 3a | 1.5 | LiOH (1.2), DMF (0.8) | 48 | 79 (100) |
| 2 | 3b | 1.5 | LiOH (1.2), DMF (0.8) | 48 | 63 (100) |
| 3 | 3a | 1.5 | KOH (1.2), DMSO (0.8) | 72 | n.d.b (<10) |
| 4 | 3a | 1.5 | K2CO3 (1.2), DMF (0.8) | 48 | n.d.b (<60) |
| 5 | 3a | 1.5 | KOH (1.2), MeCN (0.8) | 48 | 90 (100) |
| 6 | 3b | 1.5 | KOH (1.2), MeCN (0.8) | 48 | n.d.b (<10) |
| 7c | 3a | 3.0 | K2CO3 (3.0), DMF (1.33) | 5 | 93 (100) |
| 8c | 3b | 3.0 | K2CO3 (3.0), DMF (1.33) | 7 | 92 (100) |
| 9d | 3a | 1.1 | Cs2CO3 (1.5), DMF (1.33) | 0.75 | 90 (100) |
| 10d | 3b | 1.1 | Cs2CO3 (1.5), DMF (1.33) | 2 | 90 (100) |
| 11d,e | 3a | 1.1 | Cs2CO3 (1.5), DMF (1.33) | 1.5 | 95 (100) |
With optimized conditions in hand, we investigated the scope of dichloroenamide formation (Table 2). We were delighted to find that both unhindered and hindered N-alkyl sulfonamides were highly competent substrates, affording dichloroenamides 2c–h in excellent yields. Similarly, sulfonamides of electron-rich, electron-poor, and sterically hindered anilines also proved to be outstanding substrates (2i–m), including the particularly challenging but highly efficient reaction of 2,6-diisopropylaniline sulfonamide (2m). The N-vinylation of a range of aromatic azacycles was also successful, with indoles 2n–2q, benzotriazole 2r and imidazole 2s all obtained in good yields; for 2q, the decreased acidity of this indole starting material necessitated the use of NaH as base.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :
: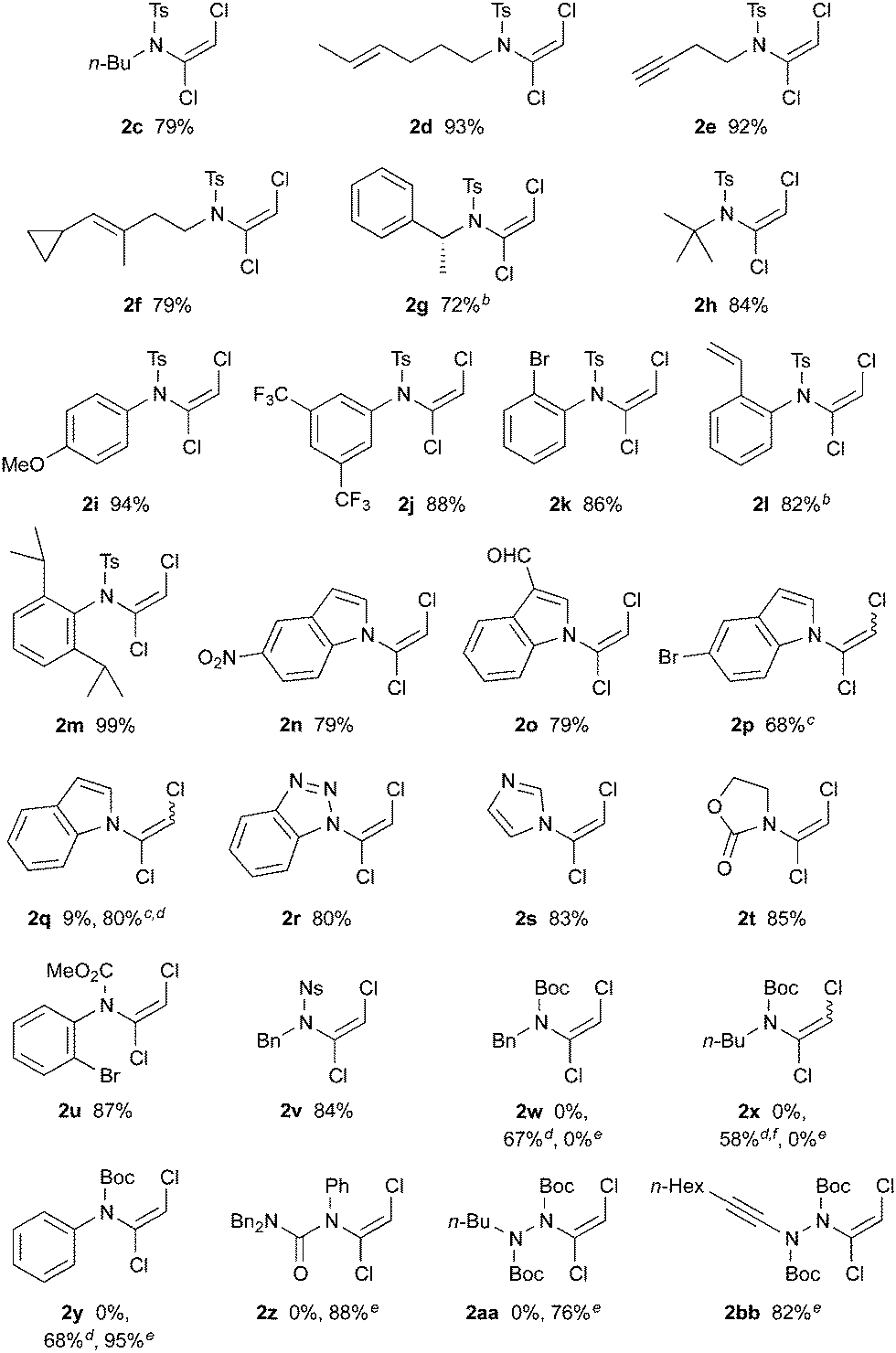
This latter example suggested that a subtle balance of pKa and nucleophilicity determines the reactivity of the substrate towards dichlorovinylation. This indeed turned out to be the case for the reactions of other amide derivatives: where unhindered carbamate derivatives such as oxazolidinone 2t and methoxycarbamate 2u were readily obtained using Cs2CO3-promoted vinylation conditions, substrates that are both less nucleophilic and weakly acidic (such as ureas, hydrazides and tert-butyl carbamates) required the use of either phase-transfer conditions,10 or NaH as base for formation of the desired dichloroenamides (2w–2bb), with all obtained in good to excellent yields. In virtually all cases, only the (E)-stereoisomer of these bench-stable dichloroenamide products was obtained (see Table 2), as determined through X-ray crystallographic analysis of products 2a (Fig. 1) and 2t, with others assigned by analogy.11
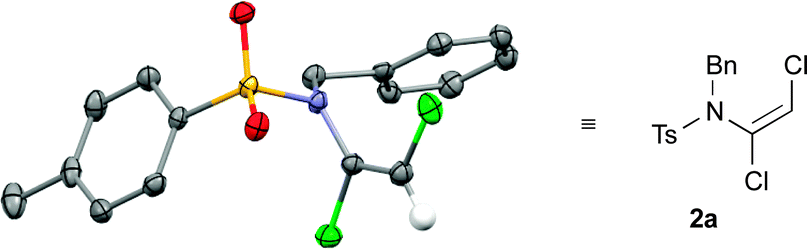 | ||
| Fig. 1 ORTEP diagram for (E)-dichloroenamide 2a (the alkene hydrogen is retained for clarity). | ||
Having established mild, general and high yielding routes to 1,2-dichloroenamides, we next surveyed conditions to effect their conversion to functionalised ynamides (Table 3).12 After some optimisation,13 we found either n-butyllithium or phenyllithium to be efficient bases for this elimination process, with the former of these requiring a more controlled addition of the base at −78 °C to avoid side-reactions. Following trapping of the alkynyllithium intermediate 4 with iodomethane, ynamide 1a was isolated in 85% yield.
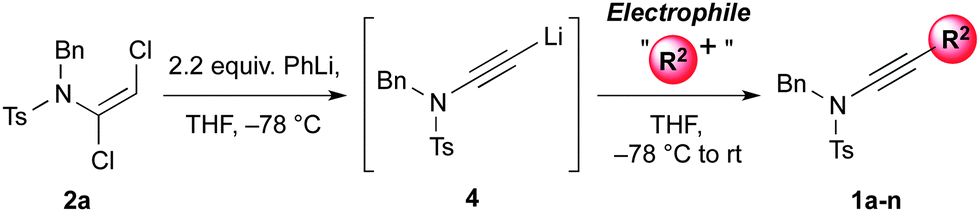
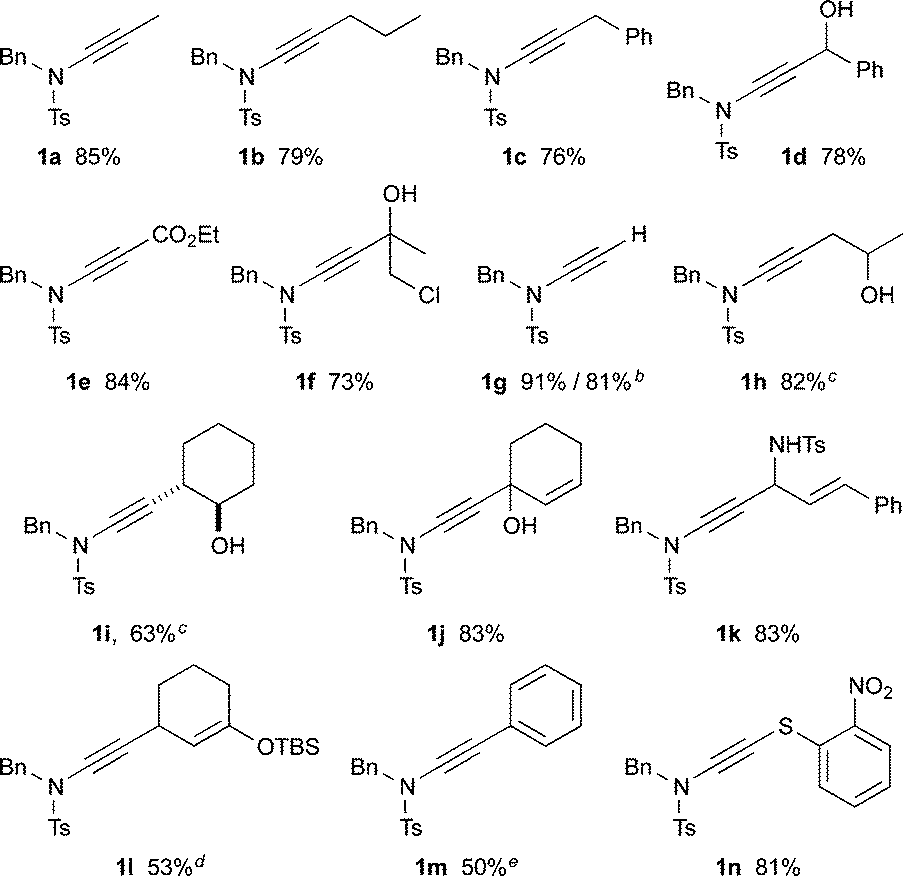
This lithiated ynamide proved to be an extremely competent nucleophile, giving high yields of a wide range of ynamides. Products arising from reaction with primary alkyl iodides/bromides (1a–c), and various carbonyl derivatives including aldehydes, ketones and chloroformates (1d–f), were all formed successfully.14 The terminal ynamide 1g was obtained in excellent yield, including on gram scale, via direct protonation of 4. The use of epoxides as electrophiles offers a convenient entry to β-hydroxy ynamides, with products 1h and 1i isolated in good yield. We also investigated the selectivity of 1,2- versus 1,4-addition of the amidoacetylide anion to α,β-unsaturated carbonyl derivatives; exclusive 1,2-addition was observed with α,β-unsaturated ketones and aldimines (1j,k), whereas clean 1,4-addition was accomplished following transmetallation to zinc (1l).15 This transmetallation also enabled acetylenic arylation to be achieved in a single step from the dichloroenamide by in situ Negishi coupling (1m).16 Finally, we were gratified to observe smooth formation of thioynamide 1n, a markedly stable double heteroatom-substituted alkyne, upon trapping with a disulfide.
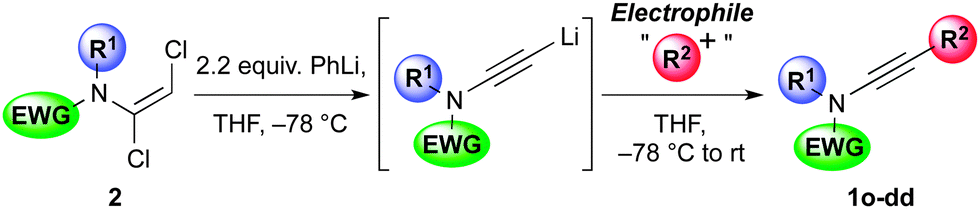
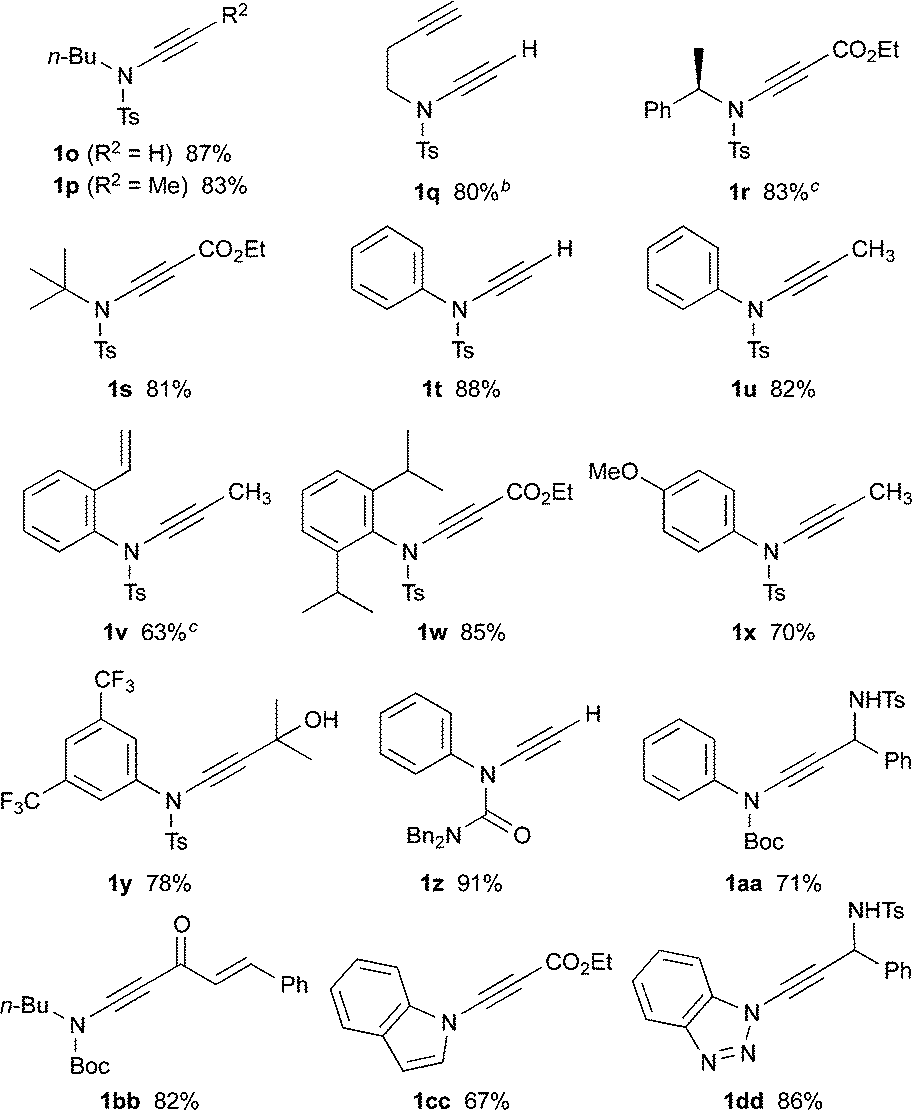
Following the success of this strategy, the generality of the process was explored, with a particular focus on the formation of ynamides inaccessible by other means. Most pleasingly, the protocol met with wide success, providing access to an array of ynamides (Table 4). Whilst these reactions could all be performed using PhLi, in a few instances (1r, 1v) n-BuLi gave superior results. Thus, N-alkyl ensulfonamides were smoothly converted to various ynamides 1o–s, including the efficient syntheses of the N-tert-butyl ynamide 1s, for which no other method has been reported. N-Aryl ynamides 1t–y were formed in excellent yields, with a wide tolerance of electronic and steric effects, the latter emphasised by the high yield of the 2,6-diisopropylphenyl ynamide 1w (85%). In addition to sulfonyl ynamides, the chemistry is equally effective with other electron-withdrawing functionalities: N-aryl urea ynamide 1z, and the Boc-protected N-aryl and N-alkyl ynamides 1aa and 1bb, were all produced in high yields. The successful application of this chemistry extended to the formation of azacyclic ynamines, as illustrated by indole 1cc and benzotriazole 1dd, which further underlines the broad scope of the methodology.
In conclusion, a rapid, economic and modular route to ynamides has been developed that overcomes limitations encountered with other ynamide-forming procedures, and offers a reliable means to prepare a diverse range of ynamides with extensive variation of all substituents. The reaction is tolerant of a wide range of functionality on the amide component, including successful reactions of carbamates, hindered amides, and aryl amides. Combined with a veritable diversity of electrophile partners, this strategy can access previously challenging or unattainable ynamides, without requiring preformed alkyne or alkene coupling partners.
We thank the EPSRC (EP/H025839/1) for support.
Notes and references
- For recent reviews of ynamide chemistry, see: (a) X.-N. Wang, H.-S. Yeom, L.-C. Fang, S. He, Z.-X. Ma, B. L. Kedrowski and R. P. Hsung, Acc. Chem. Res., 2013, 47, 560–578 CrossRef PubMed; (b) G. Evano, K. Jouvin and A. Coste, Synthesis, 2013, 17–26 CAS; (c) Y. Zhang, K. A. DeKorver, H. Y. Li, A. G. Lohse, R. Hayashi, Z. J. Lu and R. P. Hsung, Chem. Rev., 2010, 110, 5064–5106 CrossRef PubMed; (d) G. Evano, A. Coste and K. Jouvin, Angew. Chem., Int. Ed., 2010, 49, 2840–2859 CrossRef CAS PubMed; (e) For recent examples of ynamide methodology, see: C. Theunissen, B. Métayer, N. Henry, G. Compain, J. Marrot, A. Martin-Mingot, S. Thibaudeau and G. Evano, J. Am. Chem. Soc., 2014, 136, 12528–12531 CrossRef CAS PubMed; (f) S. N. Karad and R.-S. Liu, Angew. Chem., Int. Ed., 2014, 53, 9072–9076 CrossRef CAS PubMed; (g) B. Peng, X. Huang, L.-G. Xie and N. Maulide, Angew. Chem., Int. Ed., 2014, 53, 8718–8721 CrossRef CAS PubMed; (h) H. A. Laub, G. Evano and H. Mayr, Angew. Chem., Int. Ed., 2014, 53, 4968–4971 CrossRef CAS PubMed; (i) Z. Xin, S. Kramer, J. Overgaard and T. Skrydstrup, Chem. – Eur. J., 2014, 20, 7926–7930 CrossRef CAS PubMed; (j) H. V. Adcock, T. Langer and P. W. Davies, Chem. – Eur. J., 2014, 20, 7262–7266 CrossRef CAS PubMed; (k) P. Zhang, A. M. Cook, Y. Liu and C. Wolf, J. Org. Chem., 2014, 79, 4167–4173 CrossRef CAS PubMed; (l) M. D. Santos and P. W. Davies, Chem. Commun., 2014, 50, 6001–6004 RSC; (m) A. M. Cook and C. Wolf, Chem. Commun., 2014, 50, 3151–3154 RSC; (n) S. K. Pawar, D. Vasu and R.-S. Liu, Adv. Synth. Catal., 2014, 356, 2411–2416 CrossRef CAS.
- (a) For work from our group, see: C. D. Campbell, R. L. Greenaway, O. T. Holton, H. A. Chapman and E. A. Anderson, Chem. Commun., 2014, 50, 5187–5189 RSC; (b) P. R. Walker, C. D. Campbell, A. Suleman, G. Carr and E. A. Anderson, Angew. Chem., Int. Ed., 2013, 52, 9139–9143 CrossRef CAS PubMed; (c) R. L. Greenaway, C. D. Campbell, H. A. Chapman and E. A. Anderson, Adv. Synth. Catal., 2012, 354, 3187–3194 CrossRef CAS; (d) R. L. Greenaway, C. D. Campbell, O. T. Holton, C. A. Russell and E. A. Anderson, Chem. – Eur. J., 2011, 17, 14366–14370 CrossRef CAS PubMed.
- (a) T. Hamada, X. Ye and S. S. Stahl, J. Am. Chem. Soc., 2008, 130, 833–835 CrossRef CAS PubMed; (b) X. J. Jin, K. Yamaguchi and N. Mizuno, Chem. Commun., 2012, 48, 4974–4976 RSC.
- (a) M. O. Frederick, J. A. Mulder, M. R. Tracey, R. P. Hsung, J. Huang, K. C. M. Kurtz, L. C. Shen and C. J. Douglas, J. Am. Chem. Soc., 2003, 125, 2368–2369 CrossRef CAS PubMed; (b) J. R. Dunetz and R. L. Danheiser, Org. Lett., 2003, 5, 4011–4014 CrossRef CAS PubMed; (c) Y. Zhang, R. P. Hsung, M. R. Tracey, K. C. M. Kurtz and E. L. Vera, Org. Lett., 2004, 6, 1151–1154 CrossRef CAS PubMed; (d) X. Zhang, Y. Zhang, J. Huang, R. P. Hsung, K. C. M. Kurtz, J. Oppenheimer, M. E. Petersen, I. K. Sagamanova, L. Shen and M. R. Tracey, J. Org. Chem., 2006, 71, 4170–4177 CrossRef CAS PubMed; (e) K. Dooleweerdt, H. Birkedal, T. Ruhland and T. Skrydstrup, J. Org. Chem., 2008, 73, 9447–9450 CrossRef CAS PubMed; (f) F. M. Istrate, A. K. Buzas, I. D. Jurberg, Y. Odabachian and F. Gagosz, Org. Lett., 2008, 10, 925–928 CrossRef CAS PubMed; (g) K. Jouvin, F. Couty and G. Evano, Org. Lett., 2010, 12, 3272–3275 CrossRef CAS PubMed; (h) W. Jia and N. Jiao, Org. Lett., 2010, 12, 2000–2003 CrossRef CAS PubMed; (i) K. Jouvin, J. Heimburger and G. Evano, Chem. Sci., 2012, 3, 756–760 RSC; (j) For an iron-catalyzed approach, see: B. Yao, Z. Liang, T. Niu and Y. Zhang, J. Org. Chem., 2009, 74, 4630–4633 CrossRef CAS PubMed; (k) Y. Tokimizu, S. Oishi, N. Fujii and H. Ohno, Org. Lett., 2014, 16, 3138–3141 CrossRef CAS PubMed; (l) For large scale applications, see: A. L. Kohnen, J. R. Dunetz and R. L. Danheiser, Org. Synth., 2007, 84, 88–101 CrossRef CAS; (m) I. K. Sagamanova, K. C. M. Kurtz and R. P. Hsung, Org. Synth., 2007, 84, 359–367 CrossRef CAS; (n) A. Coste, F. Couty and G. Evano, Org. Synth., 2010, 87, 231–244 CrossRef CAS; (o) For pioneering work using alkynyliodonium salts, see: B. Witulski and T. Stengel, Angew. Chem., Int. Ed., 1998, 37, 489–492 CrossRef CAS.
- (a) A. Coste, G. Karthikeyan, F. Couty and G. Evano, Angew. Chem., Int. Ed., 2009, 48, 4381–4385 CrossRef CAS PubMed; (b) Y. Yang, X. Zhang and Y. Liang, Tetrahedron Lett., 2012, 53, 6557–6560 CrossRef CAS PubMed; (c) K. Jouvin, A. Coste, A. Bayle, F. Legrand, G. Karthikeyan, K. Tadiparthi and G. Evano, Organometallics, 2012, 31, 7933–7947 CrossRef CAS; (d) M. G. Wang, J. Wu and Z. C. Shang, Synlett, 2012, 589–594 CAS.
- For limitations of bromoalkyne and 1,1-dibromoalkene methods, see ref. 4d and 5c. Danheiser’s combination of carbamate anions and bromoalkynes successfully affords ynamides at room temperature (20 h), albeit requiring a full equivalent of CuI and KHMDS; see ref. 4b.
- For the synthesis of relatively unstable ynamines from dialkylamide anions and dihaloacetylenes, see: (a) H. G. Viehe and M. Reinstein, Angew. Chem., Int. Ed., 1964, 3, 506 CrossRef; (b) H. G. Viehe, Angew. Chem., Int. Ed., 1967, 6, 767–778 CrossRef CAS; (c) R. van der Heiden and L. Brandsma, Synthesis, 1987, 76–77 CrossRef CAS; (d) R. V. Joshi, Z. Q. Xu, M. B. Ksebati, D. Kessel, T. H. Corbett, J. C. Drach and J. Zemlicka, J. Chem. Soc., Perkin Trans. 1, 1994, 1089–1098 RSC; (e) J. Balsells, J. Vázquez, A. Moyano, M. A. Pericàs and A. Riera, J. Org. Chem., 2000, 65, 7291–7302 CrossRef CAS PubMed; (f) L. M. Geary and P. G. Hultin, Org. Lett., 2009, 11, 5478–5481 CrossRef CAS PubMed.
- For the equivalent chemistry of potassium alkoxides: A. Moyano, F. Charbonnier and A. E. Greene, J. Org. Chem., 1987, 52, 2919–2922 CrossRef CAS.
- For reaction of phenols under these conditions, see: Z. S. Sales and N. S. Mani, J. Org. Chem., 2009, 74, 891–894 CrossRef CAS PubMed.
- (a) For the formation of dichloroacetylene under these conditions, see: A. Jonczyk and K. Michalski, Synlett, 2002, 1703–1705 CrossRef CAS PubMed; (b) J. Pielichowski and D. Bogdal, J. Prakt. Chem., 1989, 331, 145–148 CrossRef CAS.
- The structures of 2a and 2t were determined by single crystal X-ray diffraction. See the ESI† (CIF) for more details; CCDC 1022574 and 1022575.
- (a) Although 2,2-dichloroenamides have been shown to be precursors to ynamides, their synthesis (via dichloroolefination of formamides using excess PPh3/CCl4) is significantly more challenging; see: D. Brückner, Synlett, 2000, 1402–1404 Search PubMed; (b) D. Brückner, Tetrahedron, 2006, 62, 3809–3814 CrossRef PubMed; (c) S. Couty, M. Barbazanges, C. Meyer and J. Cossy, Synlett, 2005, 905–910 CAS.
- See the ESI† for details of reaction optimisation for ynamide synthesis, and for the isolation of a chloroynamide intermediate from treatment of 2a with 1.2 equiv. PhLi or LiHMDS.
- (a) D. Rodríguez, M. F. Martínez-Esperón, L. Castedo and C. Saá, Synlett, 2007, 1963 Search PubMed; (b) R. Qi, X. N. Wang, K. A. DeKorver, Y. Tang, C. C. Wang, Q. Li, H. Li, M. C. Lv, Q. Yu and R. P. Hsung, Synthesis, 2013, 1749–1758 CAS; (c) X. N. Wang, R. P. Hsung, R. Qi, S. K. Fox and M. C. Lv, Org. Lett., 2013, 15, 2514–2517 CrossRef CAS PubMed ; see also ref. 1k and m.
- (a) S. Kim and M. L. Joo, Tetrahedron Lett., 1990, 31, 7627–7630 CrossRef CAS; (b) For a review, see: S. Fujimori, T. F. Knopfel, P. Zarotti, T. Ichikawa, D. Boyall and E. M. Carreira, Bull. Chem. Soc. Jpn., 2007, 80, 1635–1657 CrossRef CAS.
- (a) M. F. Martínez-Esperón, D. Rodríguez, L. Castedo and C. Saá, Tetrahedron, 2006, 62, 3843–3855 CrossRef PubMed; (b) D. Rodriguez, L. Castedo and C. Saa, Synlett, 2004, 783–786 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterisation data, copies of 1H and 13C NMR spectra for dichloroenamides and ynamides, and X-ray crystallographic data. CCDC 1022574 and 1022575. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc07876d |
| This journal is © The Royal Society of Chemistry 2015 |