
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly reversible capacity at the surface of a lithium-rich manganese oxide: a model study using an epitaxial film system†
S.
Taminato
a,
M.
Hirayama
*a,
K.
Suzuki
a,
N. L.
Yamada
b,
M.
Yonemura
b,
J. Y.
Son
c and
R.
Kanno
a
aDepartment of Electronic Chemistry, Interdisciplinary Graduate School of Science and Engineering, Tokyo Institute of Technology, 4259 Nagatsuta, Midori-ku, Yokohama 226-8502, Japan. E-mail: hitrayama@echem.titech.ac.jp; Fax: +81-45-924-5403; Tel: +81-45-924-5403
bNeutron Science Division, Institute of Materials Structure Science, High Energy Accelerator Research Organization (KEK), 203-1 Shirakata, Tokai, Ibaraki 319-1106, Japan
cJapan Synchrotron Radiation Research Institute (JASRI), 1-1-1 Koto, Sayo, Hyogo 679-5198, Japan
First published on 4th December 2014
Abstract
Epitaxial films of Li2MnO3 were synthesized using pulsed laser deposition. A 12.6 nm film exhibited a high discharge capacity of over 300 mA h g−1 following its fiftieth cycle and better stability than 29.8 and 47.8 nm films. The surfaces of such films are intrinsically active at the electrochemical interface.
Lithium-rich manganese oxide (Li2MnO3) is one of the most promising candidates for a lithium battery cathode material, due to its high charge–discharge capacity of over 250 mA h g−1.1–4 Although Li2MnO3 has been reported to be intrinsically inactive with regard to lithium deintercalation as a result of the high valence state of Mn4+,5 nanosized Li2MnO3 is activated upon initial charging cycle by the release of oxygen from the lattice to the electrolyte.6 The resulting oxygen-deficient phase is transformed to a phase that exhibits reversible lithium (de)intercalation. However, the use of Li2MnO3 presents a significant challenge due to the significant reduction in discharge capacity and discharge voltage upon cycling. The key to addressing this issue, and thus obtaining Li2MnO3 exhibiting both high capacity and high stability, is to elucidate the mechanism of the electrochemical activation that is initiated at the electrode/electrolyte interface.
Cathodes intended for practical applications in lithium batteries are complicated systems that consist of a polycrystalline active material, conductive carbon and a binder. A simple system with no additives is more desirable with regard to investigation of the interfacial reactions. Epitaxial film electrodes fabricated by pulsed laser deposition (PLD) have a two-dimensional interface that provides a homogeneous reaction field suitable for interfacial reaction analysis.7–11 Furthermore, these films can be readily made into model electrodes having different thicknesses, with no significant variations in composition, crystallinity or surface area. Despite these advantages, there have been no reports to date concerning the synthesis of epitaxial Li2MnO3 film electrodes. In the present study, we therefore attempted to fabricate an epitaxial Li2MnO3 film electrode using PLD. The crystal structure, composition and valence state of the Mn in the resulting films were evaluated using X-ray diffraction (XRD), neutron reflectometry, inductively coupled plasma-mass spectrometry (ICP-MS), X-ray absorption near edge structure (XANES) and hard X-ray photoemission spectroscopy (HAXPES). The electrochemical activity of epitaxial Li2MnO3 film electrodes with thicknesses of 47.8, 29.8 and 12.6 nm was investigated using charge–discharge measurements, and the Li2MnO3 activation process is discussed herein based on the dependence of the electrochemical activity on the film thickness.
Li2MnO3 films were synthesized by employing a PLD system composed of a KrF excimer laser (Lambda Physik, COMPex102 and 201) and a vacuum chamber (AOV Inc., PSAD-3000). Prior to Li2MnO3 deposition, a 30 nm thick SrRuO3(111) film was deposited on 0.5% Nb-doped SrTiO3(111) substrates as a current collector.12 The synthetic conditions consisted of a Li3.2MnO3 target, a substrate temperature of 923 K, a substrate–target distance of 60 mm, a laser frequency of 1–5 Hz, a deposition time of either 10 or 30 min, a laser energy of either 0.8 or 1.1 J cm−2 and an oxygen pressure of 75 Pa.
Film orientations were characterized by XRD using an X-ray diffractometer (Rigaku, ATX-G) with CuKα1 radiation. Elemental Li and Mn ratios were determined from ICP-MS (Agilent Technologies, 7500cs) analyses of samples dissolved in aqua regia diluted with ultrapure water at 373 K for 30 min. XANES measurements were performed in the fluorescence mode using a germanium single-element solid-state detector installed at SPring-8 BL14B2 and data were collected at an oblique incidence angle of 4°. Pre-edge background and post-edge normalizations of spectra to unity were performed using the ATHENA software package.13 The oxidation states of Mn ions in the films were determined from Mn 3s HAXPES spectra acquired at SPring-8 BL46XU using a hemispherical electron energy analyzer (SCIENTA, R-4000) with an incident photon energy level of approximately 7940 eV and a photoelectron take-off angle of 80°. The binding energy was calibrated according to the Au 4f7/2 core level spectrum. Neutron reflectometry was performed using a time-of-flight reflectometer (SOFIA) installed at J-PARC BL16.14,15 The Parratt32 program, which applies Parratt's method,16 was used for reflectivity data analysis.17
Charge–discharge characteristics were examined using 2032-type coin cells assembled in an argon-filled glovebox.
The counter/reference electrodes were lithium metal and the electrolyte was 1 mol dm−3 LiPF6 in an ethylene carbonate–diethyl carbonate mixture (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7, v/v). All cell tests were conducted at 298 K. The charge–discharge capacities were calculated from the deposited area (10 × 7 mm), and the thickness and density of the Li2MnO3 film estimated from the neutron reflectometry results.
:7, v/v). All cell tests were conducted at 298 K. The charge–discharge capacities were calculated from the deposited area (10 × 7 mm), and the thickness and density of the Li2MnO3 film estimated from the neutron reflectometry results.
Fig. 1 shows the out-of-plane XRD patterns of a Li2MnO3 film. The film generated diffraction peaks at 18.8°, 38.1° and 81.3° along the out-of-plane [111] direction, which are attributed to the 00l diffraction peaks of Li2MnO3 with the C2/m space group. The 020 and 060 diffraction peaks were observed along the in-plane [1–10] direction at 20.7° and 65.3°, respectively (Fig. S1 in ESI†). The 020 peak is attributed to a superlattice structure with honeycomb Li/Mn ordering in the transition metal layer.18 No diffraction peaks of other phases were observed in the XRD patterns, confirming the single phase formation of a lithium-rich layered Li2MnO3. The rocking curve of the 020 reflection indicates six-fold symmetry at intervals of 60° (Fig. S2 in ESI†). Although the 020 reflection should have two-fold symmetry, the six-fold symmetry demonstrates that the Li2MnO3(001) film is composed of 60°-rotated domains. The cell parameters are calculated to be a = 4.953(14) Å, b = 8.579(4) Å, c = 5.0095(8) Å, and β = 109.235(5)°.19 No significant changes in the crystal structure or the composition were observed when varying the film thicknesses (Fig. S3 and S4 in ESI†).
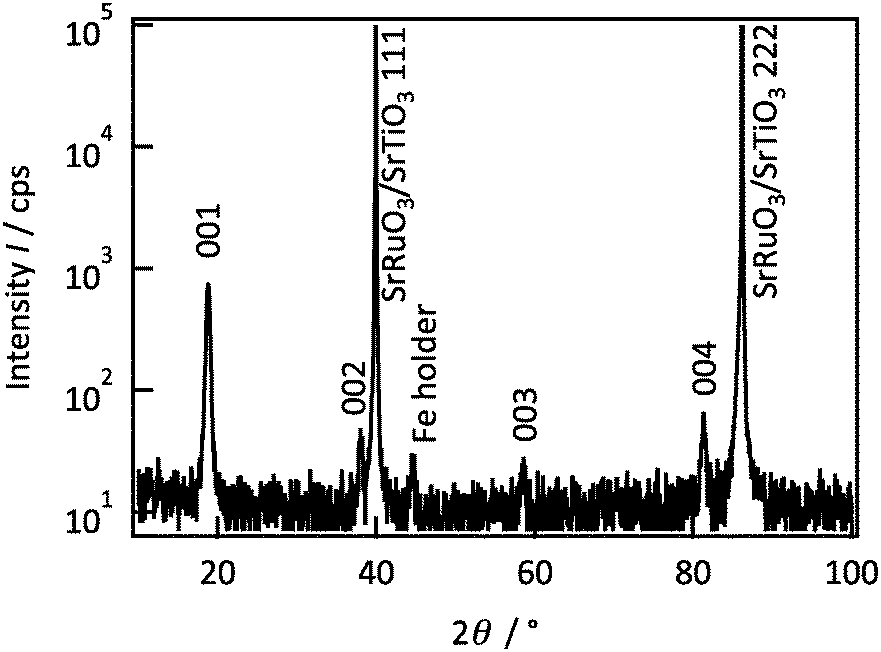 | ||
| Fig. 1 Out-of-plane XRD pattern for a Li2MnO3 film on SrRuO3(111)/Nb:SrTiO3(111). | ||
The ratio of lithium to manganese atoms in the Li2MnO3 film was estimated by ICP-MS analysis to be Li:Mn = 1.85(7):1. The origin of the compositional deviation was investigated by XANES, HAXPES and neutron reflectometry analyses. Fig. 2a shows the XANES spectrum obtained from a Li2MnO3(001) film and from polycrystalline Li2MnO3 powder. The XANES spectrum and the E0 value of the film indicate that its structure and oxidation state are identical to those of polycrystalline Li2MnO3. The oxidation state of the Mn can be determined from the difference in the binding energies of the two separated Mn 3s peaks in the HAXPES spectrum.20,21 The HAXPES spectrum in Fig. 2b shows a difference in the peak positions of 4.3 eV, corresponding to MnIV (4.4 eV). Both the XANES and HAXPES results show that tetravalent Mn is present in the film; therefore, there were no significant changes in the valence state of Mn. This result demonstrates that the film is deficient in both lithium and oxygen and thus has the composition Li2−2xMn(IV)O3−x as a result of charge compensation.
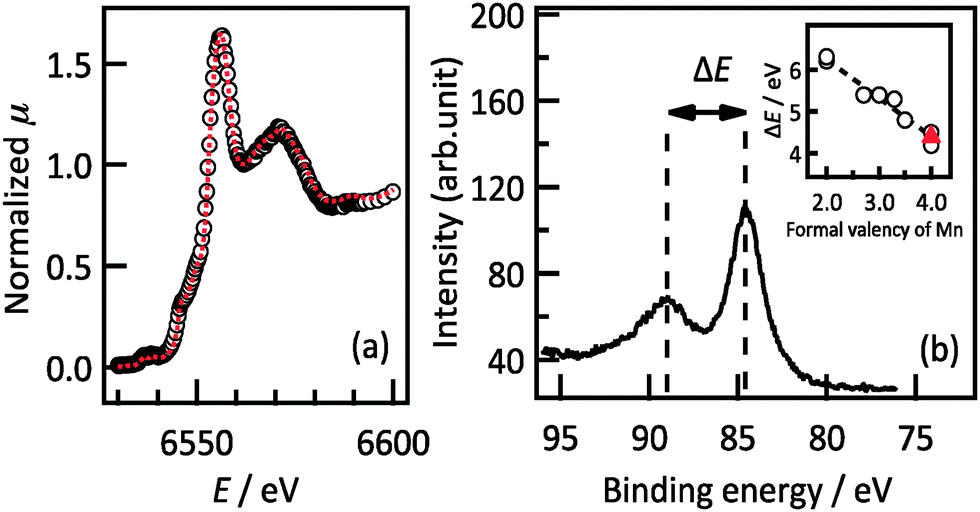 | ||
Fig. 2 (a) Mn-K edge XANES and (b) Mn 3s HAXPES spectrum of a Li2MnO3(001) film. The red dashed line in (a) depicts the XANES spectrum of polycrystalline Li2MnO3. The inset of (b) shows the splitting width of the Li2MnO3(001) film (denoted as  ), polycrystalline Mn2+O, Mn2.67+3O4, LiMn3+O2, La0.9Mn3.3+O3, La0.5Ca0.5Mn3.5+O3, Li2Mn4+O3, and SrMn4+O3 (denoted as ○) as a function of the formal Mn valency. ), polycrystalline Mn2+O, Mn2.67+3O4, LiMn3+O2, La0.9Mn3.3+O3, La0.5Ca0.5Mn3.5+O3, Li2Mn4+O3, and SrMn4+O3 (denoted as ○) as a function of the formal Mn valency. | ||
Neutron reflectometry was employed to analyse the extent of the lithium and oxygen deficiencies in the films, due to its high sensitivity for light elements.17Fig. 3 presents the result of neutron reflectivity analysis and a scattering length density (SLD) profile of a Li2MnO3(001) film. A three-layer model composed of Li2MnO3/SrRuO3/SrTiO3 generated the best fit to the reflectivity curve. The thickness, surface roughness and SLD values of the film were 45.1 nm, 5.4 nm and 1.97 × 10−4 nm−2, respectively. The SLD is defined according to the following equation.
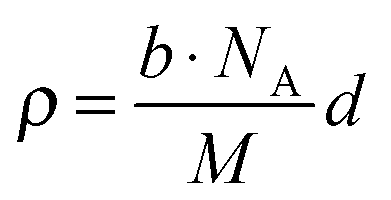 | (1) |
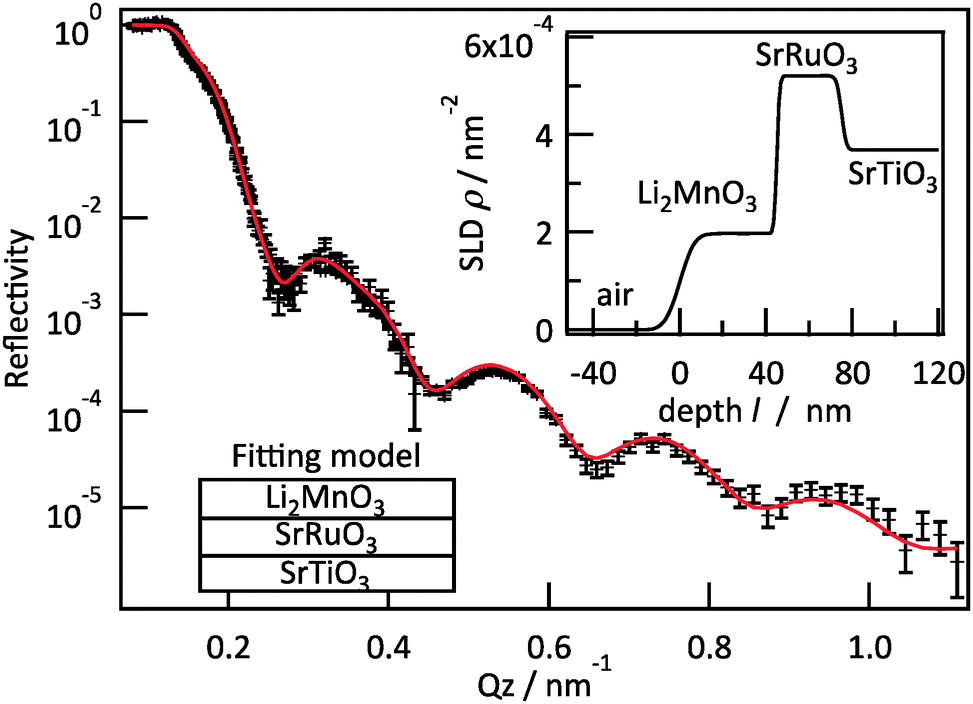 | ||
| Fig. 3 Neutron reflectometry results for an epitaxial Li2MnO3(001) film. The insets show the fitting model and the SLD profile calculated using the fitting parameters. | ||
Here ρ is the SLD, M is the molecular weight, b is the overall scattering length in the unit cell and NA is Avogadro's number. The lithium and oxygen deficiencies in the film were estimated using the SLD value, together with the formula Li2−2xMn(IV)O3−x and a lattice volume of 200.9(14) Å3 determined from neutron reflectometry, HAXPES and XRD. In this manner, the value of x was determined to be 0.05. A Li/Mn ratio of 1.90 is consistent with the results of ICP analysis (1.86(7)). Thus, we may conclude that the films had the chemical formula Li1.90Mn(IV)O2.95. The synthesis of Li2−xMnO3−y by the chemical treatment of nanosized Li2MnO3 with LiH has been reported.22 In the present study, the direct synthesis of lithium and oxygen deficient lithium-rich manganese oxide was successfully achieved using PLD. The formation of a deficient phase is expected, given the low partial pressures of lithium and oxygen and the high temperature applied during the synthesis.23
Fig. 4 presents charge–discharge curves of 47.8, 29.8 and 12.6 nm thick Li2MnO3(001) films. Upon the initial charging, the voltage increased gradually from 3.8 to 4.6 V and then plateaued. A potential slope is often observed with nanosized film electrodes because of side reactions that are quite prominent due to the small amount of active material.24 The plateau region corresponds to the activation of Li2MnO3 accompanied by the extraction of lithium and oxygen, leading to structural transformation to an electrochemically active phase.3,25–28 All the films exhibited reversible charge–discharge reactions upon subsequent cycling. The initial discharge capacities increased with decreasing film thickness from 47.8 nm (120 mA h g−1) through 29.8 nm (194 mA h g−1) to 12.6 nm (279 mA h g−1). Furthermore, the discharge capacity of the 12.6 nm thick film gradually increased with subsequent cycles to reach 318 mA h g−1 in the fiftieth cycle. The 3 V region in the discharge curves of the 12.6 nm thick film showed an increase in the capacity during the cycling, indicating a transition to another phase. The average discharge potential was 3.26 V in the first cycle and remained relatively stable at 3.24 V by the fiftieth cycle. The phase transition in the 12.6 nm thick film resulted in the increase in the discharge capacity with no significant fading of the discharging potential. The discharge capacities and cycling stability of the 12.6 nm thick film were superior to those previously reported for Li2MnO3 with particle sizes of 70 nm (140 mA h g−1 in the tenth cycle)6 and 50 nm (240 mA h g−1 in the tenth cycle).
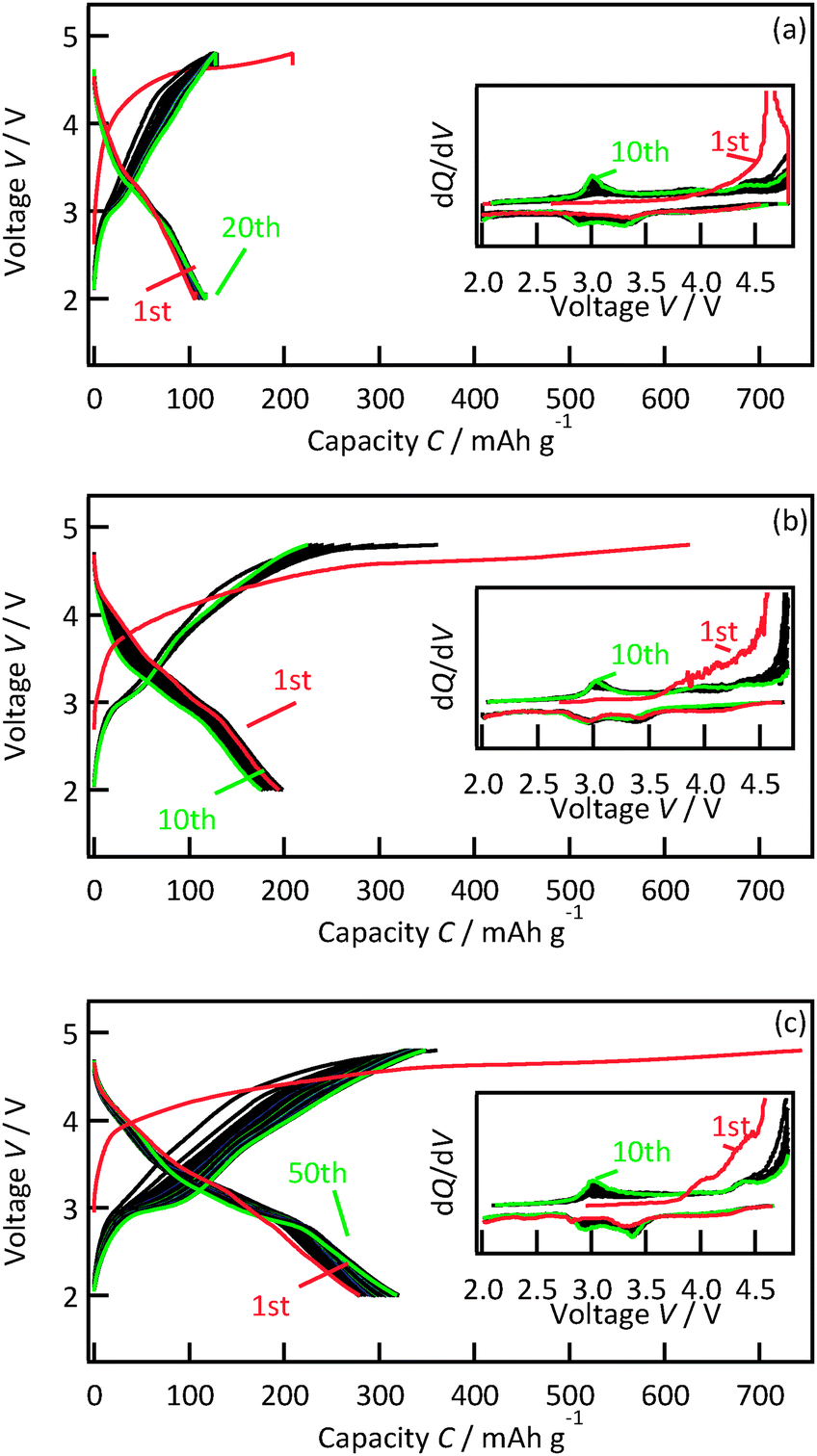 | ||
| Fig. 4 Charge–discharge curves and differential capacity plots for Li2MnO3(001) films with thicknesses of (a) 47.8 nm, (b) 29.8 nm and (c) 12.6 nm, measured over the range from 2.0 to 4.8 V with current densities of 602.5, 103.3 and 516.5 mA g−1, respectively. | ||
To clarify the thickness dependence of the capacities, the reaction potentials of the active phase were investigated. The differential capacity plots during the initial ten cycles are shown as the inset in Fig. 4. After the electrochemical activation at the first charging, all films exhibited two cathodic peaks at around 3.4 V and 2.9 V and two anodic peaks at around 3.0 and 4.4 V. No significant differences in the potentials were observed depending on the film thickness, which reveals that the similar active phase was formed in the 12.6, 29.8, and 47.8 nm thick films. Hence the high capacity observed for the 12.6 nm thick film demonstrates the high electrochemical activity at the surface region of the Li2MnO3.
The transformation from pristine Li2MnO3 to the active phase is followed by the extraction of lithium and oxygen at the electrode/electrolyte interface, so that oxygen ions must diffuse from the Li2MnO3 bulk to the interface during the initial activation process. Considering the low diffusion coefficient of oxygen in Li2MnO3 at room temperature, it is reasonable to conclude that oxygen release occurs primarily in the surface region of the electrode. An influence of the high electric field at the interface could be another driving force for the release of oxygen from the electrode surface. As a detailed activation mechanism, it has been proposed that the extraction of lithium in the transition metal layer causes migration of transition metals to the lithium layer and tetrahedral lithium formation, which causes a phase transition at the first charging.29 A first-principles study for Li2MnO3 has suggested that the lithium ions in the lithium layer participate in the lithium extraction at the beginning of charging, followed by the lithium removal from the transition metal layer.30 In contrast, lithium ions in the transitional layer should participate in the lithium extraction at the beginning of charging in our Li2MnO3 films with the restricted reaction plane of (001).31,32 The facile formation of lithium deficiency in the transition metal layers could be related to the higher electrochemical activity of the Li2MnO3(001) film electrodes than that of polycrystalline Li2MnO3 previously reported. Further investigation based on structural analyses is needed to clarify the detailed mechanism.
Previous reports have suggested various strategies to promote the initial activation. Specifically, nanosizing enlarges the contact area of the Li2MnO3 particles with the electrolyte, which enhances the activation frequency,6,33 stepwise charging could promote structural rearrangement under mild conditions,34 leading to good reversibility of the lithium (de)intercalation into the reconstructed Li2MnO322 and, lastly, a large number of stacking faults are formed in nanosized Li2MnO3 when applying a low temperature during synthesis to reduce the particle size, implying that stacking faults lead to high electrochemical activity.6 Based on these mechanisms, pre-reduction to form a disordered arrangement prior to electrochemical cycling is accepted as an effective means of achieving the facile activation of the layered Li2MnO3−d phase.22 However, the higher capacity and better stability of the 12.6 nm thick film compared to the 47.8 and 29.8 nm thick films cannot be explained based on these factors, since these films all had the same contact area, the electrochemical tests were performed under the same conditions with no stepwise charging, and the films had similar ordering structures and lithium and oxygen deficiencies. Our results obtained using these model electrodes offer new insight into the activation process of lithium-rich layered cathodes; the surface region of the electrode is intrinsically active with regard to the electrochemically induced transformation to a high capacity phase exhibiting high electrochemical stability.
In summary, epitaxial Li2MnO3(001) films were successfully synthesized on SrRuO3/SrTiO3(001) substrates using PLD. The films generated 00l and 0k0 XRD peaks showing a monoclinic lattice along the SrTiO3[111] and [1–10] directions. Lithium and oxygen vacancies were determined in these films, which were assigned the chemical composition Li1.90MnIVO2.95. The 12.6 nm thick film showed higher capacity over 300 mA h g−1 and better stability compared to the 29.8 and 47.8 nm thick films. The dependence of the electrochemical properties on the film thickness demonstrates that the surface region is actively reconstructed to generate a high-capacity phase with high electrochemical stability.
This work was supported by the Research and Development Initiative for Scientific Innovation of New Generation Battery (RISING Project) of the New Energy and Industrial Technology Development Organization (NEDO; Japan). The synchrotron X-ray experiments were performed as projects approved by the Japan Synchrotron Radiation Research Institute (2013B1530 and 2013B1711), and the neutron experiments were performed as projects approved by the Japan Proton Accelerator Research Complex (2013B0166).
Notes and references
- C. S. Johnson, J. S. Kim, C. Lefief, N. Li, J. T. Vaughey and M. M. Thackeray, Electrochem. Commun., 2004, 6, 1085–1091 CrossRef CAS PubMed.
- T. Ohzuku, M. Nagayama, K. Tsuji and K. Ariyoshi, J. Mater. Chem., 2011, 21, 10179–10188 RSC.
- N. Yabuuchi, K. Yoshii, S.-T. Myung, I. Nakai and S. Komaba, J. Am. Chem. Soc., 2011, 133, 4404–4419 CrossRef CAS PubMed.
- H. Koga, L. Croguennec, M. Ménétrier, P. Mannessiez, F. Weill and C. Delmas, J. Power Sources, 2013, 236, 250–258 CrossRef CAS PubMed.
- B. Ammundsen and J. Paulsen, Adv. Mater., 2001, 13, 943–956 CrossRef CAS.
- D. Y. W. Yu, K. Yanagida, Y. Kato and H. Nakamura, J. Electrochem. Soc., 2009, 156, A417–A424 CrossRef CAS PubMed.
- M. Hirayama, N. Sonoyama, T. Abe, M. Minoura, M. Ito, D. Mori, A. Yamada, R. Kanno, T. Terashima, M. Takano, K. Tamura and J. Mizuki, J. Power Sources, 2007, 168, 493–500 CrossRef CAS PubMed.
- K. Sakamoto, M. Hirayama, N. Sonoyama, D. Mori, A. Yamada, K. Tamura, J. Mizuki and R. Kanno, Chem. Mater., 2009, 21, 2632–2640 CrossRef CAS.
- K. Sakamoto, M. Hirayama, H. Konishi, N. Sonoyama, N. Dupré, D. Guyomard, K. Tamura, J. Mizuki and R. Kanno, Phys. Chem. Chem. Phys., 2010, 12, 3815–3823 RSC.
- M. Hirayama, H. Ido, K. Kim, W. Cho, K. Tamura, J. Mizuki and R. Kanno, J. Am. Chem. Soc., 2010, 132, 15268–15276 CrossRef CAS PubMed.
- Y. Zheng, S. Taminato, Y. Xu, K. Suzuki, K. Kim, M. Hirayama and R. Kanno, J. Power Sources, 2012, 208, 447–451 CrossRef CAS PubMed.
- K. Suzuki, K. Kim, S. Taminato, M. Hirayama and R. Kanno, J. Power Sources, 2013, 226, 340–345 CrossRef CAS PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- N. L. Yamada, N. Torikai, K. Mitamura, H. Sagehashi, S. Sato, H. Seto, T. Sugita, S. Goko, M. Furusaka, T. Oda, M. Hino, T. Fujiwara, H. Takahashi and A. Takahara, Eur. Phys. J. Plus, 2011, 126, 1–13 Search PubMed.
- K. Mitamura, N. L. Yamada, H. Sagehashi, N. Torikai, H. Arita, M. Terada, M. Kobayashi, S. Sato, H. Seto, S. Goko, M. Furusaka, T. Oda, M. Hino, H. Jinnai and A. Takahara, Polym. J., 2013, 45, 100–108 CrossRef CAS.
- L. G. Parratt, Phys. Rev., 1954, 95, 359–369 CrossRef.
- M. Hirayama, M. Yonemura, K. Suzuki, N. Torikai, H. Smith, E. Watkinsand, J. Majewski and R. Kanno, Electrochemistry, 2010, 78, 413–415 CrossRef CAS PubMed.
- J. Breger, M. Jiang, N. Dupré, Y. S. Meng, Y. Shao-Horn, G. Ceder and C. P. Grey, J. Solid State Chem., 2005, 178, 2575–2585 CrossRef CAS PubMed.
- Y. S. Meng, G. Ceder, C. P. Grey, W. S. Yoon, M. Jiang, J. Bréger and Y. Shao-Horn, Chem. Mater., 2005, 17, 2386–2394 CrossRef CAS.
- V. Galakhov, M. Demeter, S. Bartkowski, M. Neumann, N. Ovechkina, E. Kurmaev, N. Lobachevskaya, Y. Mukovskii, J. Mitchell and D. Ederer, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 113102 CrossRef.
- X. Wang, Q. Cui, Y. Pan and G. Zou, J. Alloys Compd., 2003, 354, 91–94 CrossRef CAS.
- K. Kubota, T. Kaneko, M. Hirayama, M. Yonemura, Y. Imanari, K. Nakane and R. Kanno, J. Power Sources, 2012, 216, 249–255 CrossRef CAS PubMed.
- S. Kim, M. Hirayama, W. Cho, K. Kim, T. Kobayashi, R. Kaneko, K. Suzuki and R. Kanno, CrystEngComm, 2014, 16, 1044 RSC.
- H. Konishi, K. Suzuki, S. Taminato, K. Kim, S. Kim, J. Lim, M. Hirayama and R. Kanno, J. Power Sources, 2014, 246, 365–370 CrossRef CAS PubMed.
- A. D. Robertson and P. G. Bruce, Chem. Mater., 2003, 15, 1984–1992 CrossRef CAS.
- Y.-S. Hong, Y. J. Park, K. S. Ryu and S. H. Chang, Solid State Ionics, 2005, 176, 1035–1042 CrossRef CAS PubMed.
- M. M. Thackeray, C. S. Johnson, J. T. Vaughey, N. Li and S. A. Hackney, J. Mater. Chem., 2005, 15, 2257–2267 RSC.
- A. R. Armstrong, M. Holzapfel, P. Novák, C. S. Johnson, S.-H. Kang, M. M. Thackeray and P. G. Bruce, J. Am. Chem. Soc., 2006, 128, 8694–8698 CrossRef CAS PubMed.
- B. Xu, C. R. Fell, M. Chi and Y. S. Meng, Energy Environ. Sci., 2011, 4, 2223–2233 CAS.
- Y. Koyama, I. Tanaka, M. Nagao and R. Kanno, J. Power Sources, 2009, 189, 798–801 CrossRef CAS PubMed.
- S. Taminato, M. Hirayama, K. Suzuki, K. Kim, Y. Zheng, K. Tamura, J. i. Mizuki and R. Kanno, J. Mater. Chem. A, 2014, 2, 17875–17882 CAS.
- Y. Zheng, S. Taminato, K. Suzuki, M. Hirayama and R. Kanno, Thin Solid Films, 2012, 520, 4889–4893 CrossRef CAS PubMed.
- J. Lim, J. Moon, J. Gim, S. Kim, K. Kim, J. Song, J. Kang, W. B. Im and J. Kim, J. Mater. Chem., 2012, 22, 11772–11777 RSC.
- A. Ito, D. Li, Y. Ohsawa and Y. Sato, J. Power Sources, 2008, 183, 344–346 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: XRD and XANES results of Li2MnO3 films. See DOI: 10.1039/c4cc07758j |
| This journal is © The Royal Society of Chemistry 2015 |