
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective rearrangement of terminal epoxides into methylketones catalysed by a nucleophilic rhodium–NHC–pincer complex†
Eva
Jürgens
a,
Barbara
Wucher
a,
Frank
Rominger
b,
Karl W.
Törnroos
c and
Doris
Kunz
*a
aInstitut für Anorganische Chemie, Eberhard Karls Universität Tübingen, Auf der Morgenstelle 18, 72076 Tübingen, Germany. E-mail: Doris.Kunz@uni-tuebingen.de
bInstitut für Organische Chemie, Heidelberg University, Im Neuenheimer Feld 250, 69120 Heidelberg, Germany
cDepartment of Chemistry, University of Bergen, Allégaten 41, 5007 Bergen, Norway
First published on 22nd December 2014
Abstract
An efficient RhI–NHC–pincer catalyst for the highly regioselective Meinwald rearrangement of monoalkylated epoxides into methylketones under mild conditions is presented. The nucleophilic epoxide opening is assisted by Lewis acids.
Epoxides are widely used as substrates in organic synthesis,1 as they can be transformed under ring opening into various functional groups. One well-documented reaction is the so-called Meinwald rearrangement, i.e. the rearrangement into aldehydes or ketones usually catalysed by Lewis acids.2 Selectivity is determined by formation of the most stable carbenium intermediate followed by an alkyl or hydride shift.3 Therefore, aldehydes are the major product when using terminal epoxides. A number of Lewis acid catalysts1 are known for internal epoxides3,4 while catalysts for the rearrangement of monoalkyl-substituted terminal epoxides are less common. Only a few catalysts are known to selectively convert monoalkylated epoxides into methylketones, e.g. Pd(OAc)2,5a,b MnI2 or Co2(CO)8 (ref. 5c) In those cases a nucleophilic ring opening can explain the inverse product selectivity. In the following, we describe the first rhodium catalysed Meinwald rearrangement of terminal epoxides to methylketones (Scheme 1).
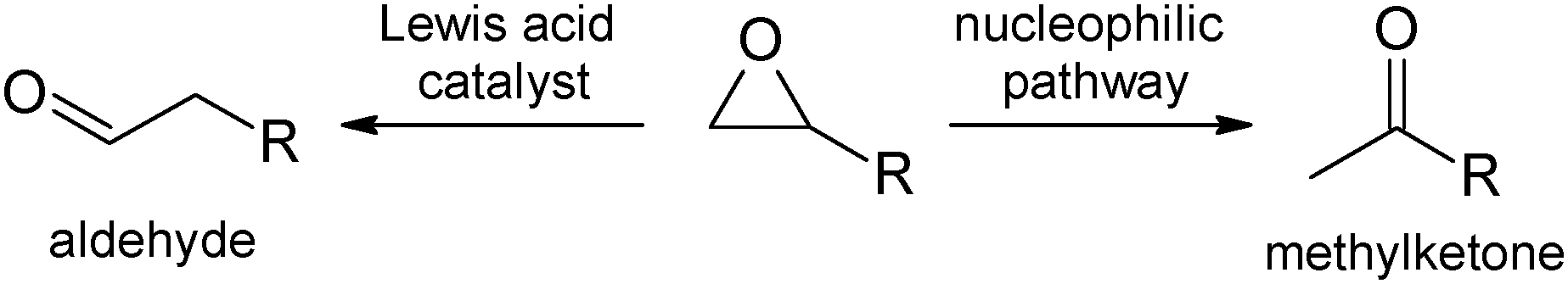 | ||
| Scheme 1 Rearrangement of terminal epoxides (Meinwald reaction). | ||
In 2006 we reported the highly nucleophilic character of rhodium–pincer-complex 16 that is caused by the two electron-donating N-heterocyclic carbene moieties (Fig. 1).7 Therefore, complex 1 seems to be a promising candidate for catalysing the nucleophilic epoxide rearrangement. Initially, we carried out the reaction with various epoxides in the presence of 10 mol% of pure complex 1, but no reaction could be achieved. However, upon addition of a stoichiometric amount of lithium chloride in tetrahydrofuran a rearrangement product was detected in low yields.
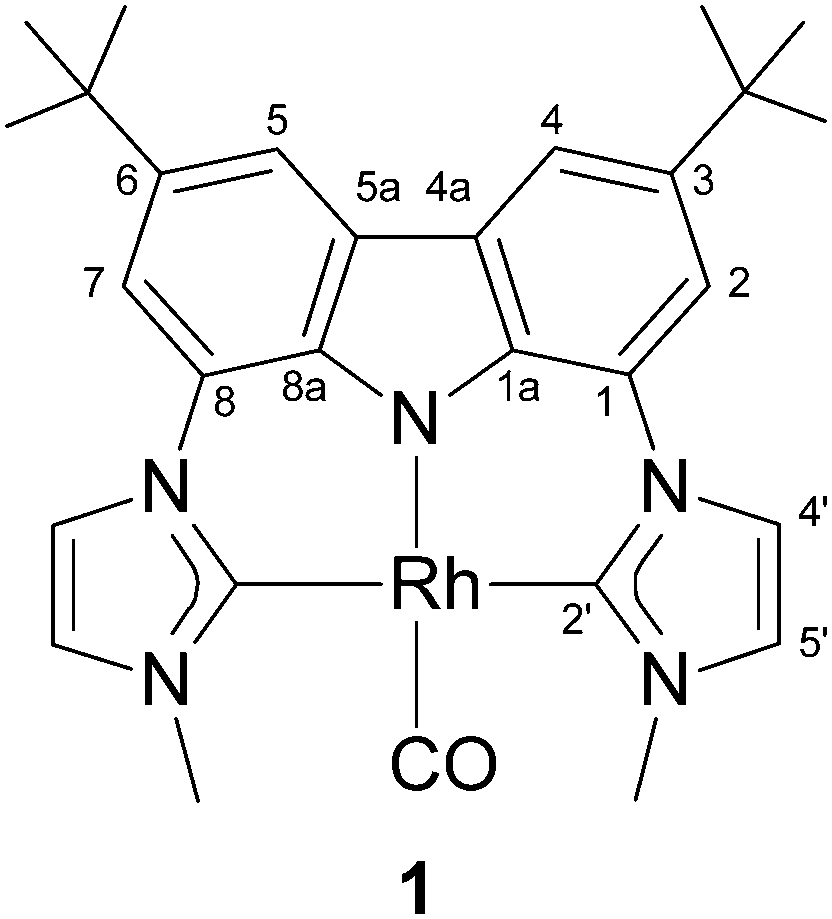 | ||
| Fig. 1 The highly nucleophilic NHC–pincer rhodium complex 1. | ||
Using dichloromethane or acetonitrile suppresses the rearrangement to the methylketone and results in formation of new organometallic species (vide infra),8 while benzene improved the reaction rate remarkably. In addition, lithium salts of weakly coordinating anions such as lithium tetrakis(pentafluorophenyl)borate or lithium bis(trifluoromethanesulfonimide) (LiNTf2) led to very high reaction rates at 60 °C (see ESI† for optimisation details).
As strong Lewis acids can act as catalysts for the epoxide rearrangements themselves, we checked their individual reactivity towards 1,2-epoxyhexane, but none of the Lewis acid additives used catalyses the rearrangement on its own, not even at elevated temperatures of up to 120 °C in thf and 80 °C in benzene. We then optimised the amount of catalyst, lithium salt additive as well as the reaction temperature by analysing the reaction mixture after 100 min (Table 1). The rearrangement proceeded almost quantitatively after this time when using 30–50 mol% of LiNTf2 and 10 mol% of 1 (Rh![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Li = 1:3–1:5) at 60 °C (entries 3–5) or only 10 mol% of the Li additive at 80 °C (entry 6), but already at room temperature or 40 °C slow rearrangement is observed (entries 1 and 2). Reducing the catalyst loading to 1 mol% decreases the reaction rate affording only 18% of the methylketone after 2 h (entry 7). Good results are still achieved using 5 mol% of 1 and 20 mol% LiNTf2 at 60 °C (95% yield after 2 h; entry 8). All experiments with 1,2-epoxyhexane gave the methylketone as the sole rearrangement product; the respective aldehyde was never detected.
:Li = 1:3–1:5) at 60 °C (entries 3–5) or only 10 mol% of the Li additive at 80 °C (entry 6), but already at room temperature or 40 °C slow rearrangement is observed (entries 1 and 2). Reducing the catalyst loading to 1 mol% decreases the reaction rate affording only 18% of the methylketone after 2 h (entry 7). Good results are still achieved using 5 mol% of 1 and 20 mol% LiNTf2 at 60 °C (95% yield after 2 h; entry 8). All experiments with 1,2-epoxyhexane gave the methylketone as the sole rearrangement product; the respective aldehyde was never detected.
|
|
|||||
|---|---|---|---|---|---|
| Entry | T [°C] | 1 [mol%] | LiNTf2 [mol%] | Time [min] | Yieldb,c [%] |
| a Reaction conditions: 1 (10 mol%), 1,2-epoxyhexane (35 μL). 0.5 mL benzene, 100 min, all reactions were carried out using a J. Young NMR tube. b The ketone was the only observed reaction product. c Yield was determined by 1H NMR using 1,3,5-trimethoxybenzene as internal standard. | |||||
| 1 | 25 | 10 | 10 | 100 | 3 |
| 2 | 40 | 10 | 30 | 100 | 7 |
| 3 | 60 | 10 | 10 | 100 | 51 |
| 4 | 60 | 10 | 30 | 100 | 94 |
| 5 | 60 | 10 | 50 | 100 | 98 |
| 6 | 80 | 10 | 10 | 100 | 98 |
| 7 | 60 | 1 | 30 | 120 | 18 |
| 8 | 60 | 5 | 20 | 120 | 95 |
The best results for this reaction have been reported by Kagan as well as Kulawiec with a combination of Pd(OAc)2 and PBu3 that resulted in the selective formation of the ketone at 120 °C in toluene without the use of any additive.5a,b Using SmI2, MnI2 or Co2(CO)8 also formed the corresponding ketones with a selectivity of above 95% and reaction times between 2 h (MnI2, 70 °C) and 24 h (Co2(CO)8, 40 °C, MeOH), but only yields between 70–80% were obtained.5c To probe the nucleophilic effect of our rhodium catalyst, we then tested commercially available Wilkinson catalyst [Rh(PPh3)3Cl] and [Rh(μ-Cl)(COD)]2 under our optimized conditions (100 min, 60 °C, benzene, 30 mol% LiNTf2) with 1,2-epoxyhexane, but observed only inferior results (Table 2). About 35% of the methylketone could be obtained when heating the reaction mixture with [Rh(PPh3)3Cl] at 85 °C for 60 h. In the case of [Rh(μ-Cl)(COD)]2 no catalytic activity was observed applying these conditions.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Amount [mol%] | Temp. [°C] | Time [h] | Yield [%] |
| a Reaction conditions: LiNTf2 (30 mol%), 60 °C, C6D6 (0.5 mL), all reactions were carried out in a J. Young NMR tube with 1,2-epoxyhexane (35 μL) as substrate. b The ketone was observed as sole reaction product. c Yield was determined by 1H NMR using 1,3,5-trimethoxybenzene as internal standard. | |||||
| 1 | 1 | 10 | 60 | 2 | 98a,b,c |
| 2 | Rh(PPh3)3Cl | 10 | 60 | 2 | 0a,b,c |
| 3 | Rh(PPh3)3Cl | 10 | 85 | 60 | 35a,b,c |
| 4 | [Rh(μ-Cl)(COD)]2 | 10 | 60 | 2 | 0a,b,c |
| 5 | [Rh(μ-Cl)(COD)]2 | 10 | 85 | 60 | 0a,b,c |
| 65c | Co2(CO)8 (ref. 5c) | 4 | 40 | 24 | 71 |
| 75a,b | Pd(OAc)2, PBu3 (ref. 5a and b) | 5–10 | 120 | 3 | 88 |
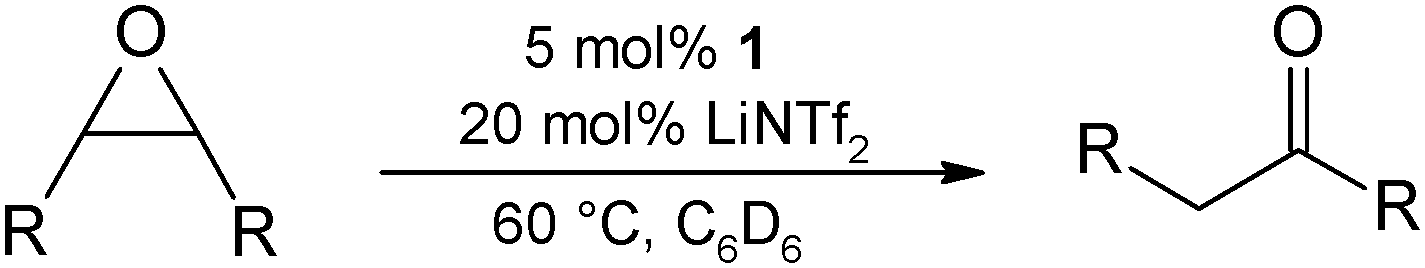
As a first substrate scope, we found that propylenoxide is rearranged to acetone in excellent yields (Table 3, entry 1), whereas styrene oxide gave a mixture of acetophenone and 2-phenylacetaldehyde at 60 °C in a 3:2 ratio in an only overall 10% yield after 2 h (entry 3). A blank test revealed that LiNTf2 itself reacts with styrene oxide leading exclusively to the aldehyde at 60 °C. This side reaction can be suppressed completely when lowering the temperature to 30 °C, however, only 5% of acetophenone were obtained after 16 h (entry 4). Using LiCl as a Lewis acid additive (60 °C) did not lead to any rearrangement product. Cyclohexene oxide, a 1,2-disubstituted epoxide, can be rearranged to cyclohexenone in 80% yield (80 °C, entry 5). As expected 2,2-dimethyloxirane did not rearrange into a ketone as the reaction is blocked by the additional methyl substituent (entry 6). Traces of the aldehyde were formed due to Lewis-acidic epoxide opening by LiNTf2 (blank test).
| Entry | Substrate | Product | Time [h] | Yielda,b [%] |
|---|---|---|---|---|
| a Reaction conditions: 1 (5 mol%), LiNTf2 (20 mol%), 60 °C, all reactions were carried out in a J. Young NMR tube in C6D6 (0.4 mL). b Yield was determined by 1H NMR using 1,3,5-trimethoxybenzene as internal standard. c At 30 °C. d At 80 °C. | ||||
| 1 |
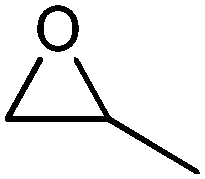
|
Acetone | 3 | 93 |
| 2 |
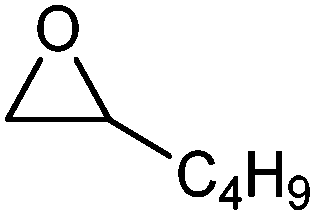
|
2-Hexanone | 2 | 95 |
| 3 |
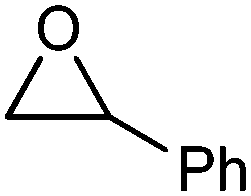
|
Acetophenone:2-phenylacetaldehyde | 2 | 6:4 |
| 4 |
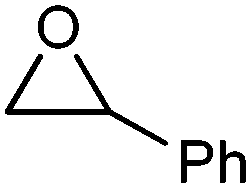
|
Acetophenone | 16 | 5c |
| 5 |
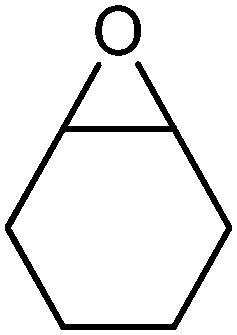
|
Cyclohexanone | 24 | 80d |
| 6 |
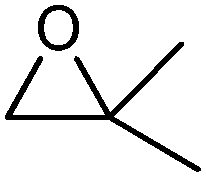
|
No ketone traces of aldehyde | 24 | 0 |
A nucleophilic mechanism for the Meinwald reaction was suggested for Co2(CO)8 in methanol that involves in situ formation of the nucleophile [Co(CO)4]− as well as cationic [Co(MeOH)6]+ to activate the epoxide upon coordination.5c–e Therefore, a plausible mechanism for our 16 e− rhodium complex 1 starts with preactivation of the epoxide by the Lewis acid additive (Scheme 2, (A)) followed by a nucleophilic attack of the RhI centre at the most electrophilic side of the epoxide which is also the least hindered one. In C6D6 RhIII intermediate 2 (B) is obtained which was confirmed by in situ formation of 2 in a stoichiometric reaction at RT. The metal bound CO ligand was identified by its 13C NMR chemical shift of δ = 207 ppm and an IR stretching frequency (benzene) at 2046 cm−1.9 Subsequent β-hydride migration (C) could lead to the RhIII hydrido complex I that releases the ketone by reductive elimination (D) under regeneration of RhI complex 1. So far, no metal hydrido complex was observed during reaction, which could be due to a fast reductive elimination process. Alternatively, intermediate 2 could release the product directly by a concerted 1,2-hydride shift-SNi reaction via transition state II (E) to release the product and close the catalytic cycle.
 | ||
| Scheme 2 Proposed reaction pathway for the conversion of monoalkyl-substituted epoxides to methylketones catalyzed by 1. | ||
In thf-d8 however, only formation of 3 upon CO insertion (F) was observed during the NMR spectroscopic monitoring of the catalytic reaction.9 The identical product 3 could be synthesized independently by a stoichiometric reaction of 1 with one equivalent of the respective epoxide in the presence of the Lewis acid additive in tetrahydrofuran (room temperature) or acetonitrile (60 °C). The 1H NMR spectrum of 3 in thf-d8 solution displays a double set of resonances for the ligand backbone due to the reduced symmetry of the complex (R = CH3). A doublet at 1.16 ppm is assigned to the methyl group resulting from the reaction of the epoxide with rhodium complex 1. The other characteristic peaks of the ring-opened epoxide moiety are superimposed by the residual solvent peak (thf-d8) and epoxide signals, but could be detected by 2D NMR experiments as well as in acetonitrile-d3. In the 13C NMR spectrum (thf-d8) the doublet at 229.4 ppm (1JRhC = 43.3 Hz) strongly hints towards a CO insertion and formation of the Rh acyl complex 3. In addition a 13C DEPT-135 experiment confirms the signal for the CH2 group at 26.1 ppm (2JRhC = 30.0 Hz). Proof that compound 3 is a resting state and can react (partly) back into the catalytic cycle was obtained by removing all volatiles in vacuo after generation of 3 and redissolving the residue in thf-d8. After 2 d at room temperature the peaks of 3 cannot be detected, whilst the peak of acetone as well as the signals of isopropanol and the poorly soluble yellow species 4a appear.
The 13C DEPT-135 experiment of this species reveals the signal of a CH group at 93.6 ppm; the respective proton signal is found at δH = 3.96 ppm. All other peaks also coincide well with the formation of complex 4a by dehydrogenation (G). Single crystals suitable for X-ray diffraction were obtained from saturated solutions of the reaction mixtures at room temperature. The analyses confirm formation of the unsaturated five-membered rhodacycles in complexes 4a and b (Fig. 2, for 4a see ESI†). This also explains the formation of isopropanol from acetone during the course of the reaction. Formation of 4 can only be observed after formation of complex 3. In pure C6D6 neither complex 3 nor complex 4 is obtained. However, after generation of 3 (R = CH3) in thf-d8, removal of all volatiles in vacuo and dissolving of the residue in C6D6, the formation of both, complex 1 and acetone as well as formation of complex 4a is observed. We assume that residual thf-d8, coordinated to the Li+ cation, prevents direct observation of 2 under these conditions.
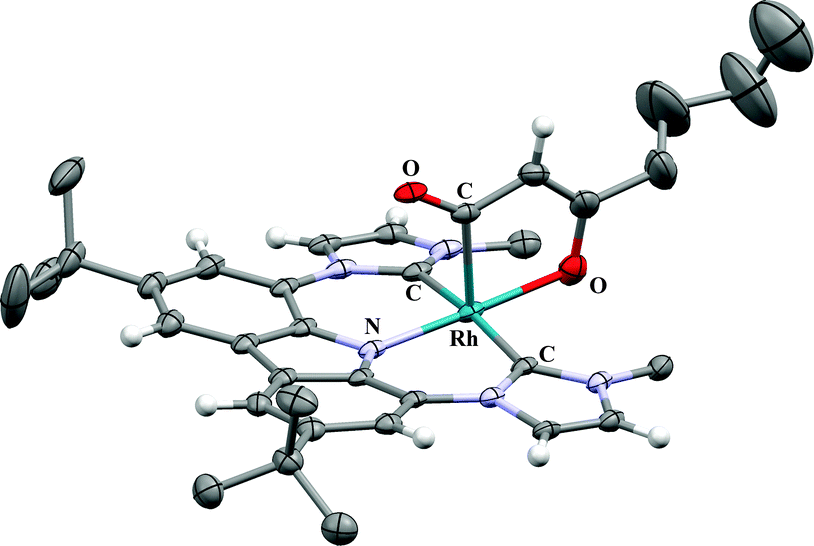 | ||
| Fig. 2 X-ray crystal structure of the side product 4b bearing an unsaturated rhodacycle. For reason of better clarity, the anisotropic displacement parameters are given at the 20% probability level and only the hydrogen atoms of sp2-hybridized C-atoms are shown and solvent molecules omitted.9 | ||
We showed that terminal epoxides can be transformed into ketones under mild conditions using the strong nucleophilic rhodium catalyst 1. To the best of our knowledge this is the most reactive and selective catalyst for this transformation and the first example of a rhodium catalyst yielding the methylketone as product.
Financial support from the Deutsche Forschungsgemeinschaft (SFB 623, GK 850) as well as the BMBF and MWK-BW (Professorinnenprogramm, LGF for E.J.) is gratefully acknowledged. We thank Lars Wesemann for a helpful discussion.
Notes and references
- J. G. Smith, Synthesis, 1984, 629 CrossRef CAS PubMed.
- (a) J. Meinwald, S. S. Labana and M. S. Chadha, J. Am. Chem. Soc., 1963, 85, 582 CrossRef CAS; (b) B. Rickborn, in Comprehensive Organic Synthesis, ed. B. M. Trost, Pergamon, Oxford, 1991, vol. 3, pp. 733–775 Search PubMed.
- (a) B. Rickborn and R. M. Gerkin, J. Am. Chem. Soc., 1971, 93, 1693 CrossRef CAS; (b) D. Milstein, O. Buchman and J. Blum, J. Org. Chem., 1977, 42, 2299 CrossRef CAS; (c) S. Kulasegaram and R. J. Kulawiec, J. Org. Chem., 1997, 62, 6547 CrossRef CAS; (d) B. C. Ranu and U. Jana, J. Org. Chem., 1998, 63, 8212 CrossRef CAS; (e) A. M. Anderson, J. M. Blazek, P. Garg, B. J. Payne and R. S. Mohan, Tetrahedron Lett., 2000, 41, 1527 CrossRef CAS; (f) M. Robinson, K. Pillinger and A. Graham, Tetrahedron Lett., 2006, 33, 5919 CrossRef PubMed; (g) D. J. Vyas, E. Larionov, C. Besnard, L. Guénée and C. Mazet, J. Am. Chem. Soc., 2013, 135, 6177 CrossRef CAS PubMed; (h) N. Humbert, D. J. Vyas, C. Besnard and C. Mazet, Chem. Commun., 2014, 50, 10592 RSC.
- (a) K. Suda, K. Baba, S. Nakajima and T. Takanami, Tetrahedron Lett., 1999, 40, 7243 CrossRef CAS; (b) A. Procopio, R. Dalpozzo, A. De Nino, M. Nardi, G. Sindona and A. Tagarelli, Synlett, 2004, 2633 CrossRef CAS PubMed; (c) H. O. J. House, J. Am. Chem. Soc., 1955, 77, 3070 CrossRef CAS; (d) Y. Kita, S. Kitagaki, Y. Yoshida, S. Mihara, D. F. Fang, M. Kondo, S. Okamoto, R. Imai, S. Akai and H. J. Fujioka, J. Org. Chem., 1997, 62, 4991 CrossRef CAS.
- (a) S. Kulasegaram and R. J. Kulawiec, Tetrahedron, 1998, 54, 1361 CrossRef CAS; (b) S. Kulasegaram and R. J. Kulawiec, J. Org. Chem., 1994, 59, 7195 CrossRef CAS; (c) J. Prandi, J. L. Namy, G. Menoret and H. B. Kagan, J. Organomet. Chem., 1985, 285, 449 CrossRef CAS; (d) J. L. Eisenmann, R. Yamartino and J. Howard Jr., J. Org. Chem., 1961, 26, 2102 CrossRef CAS; (e) J. L. Eisenmann, J. Org. Chem., 1962, 27, 2706 CAS.
- (a) M. Moser, B. Wucher, F. Rominger and D. Kunz, Organometallics, 2007, 26, 1024 CrossRef CAS; (b) B. Wucher, M. Moser, S. A. Schumacher, F. Rominger and D. Kunz, Angew. Chem., 2009, 121, 4481 ( Angew. Chem., Int. Ed. , 2009 , 48 , 4417 ) CrossRef.
- (a) D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 2000, 100, 39 CrossRef CAS PubMed; (b) W. A. Herrmann, Angew. Chem., 2002, 114, 1342 ( Angew. Chem., Int. Ed. , 2002 , 41 , 1290 ) CrossRef; (c) F. E. Hahn and M. C. Jahnke, Angew. Chem., 2008, 120, 3166 ( Angew. Chem., Int. Ed. , 2008 , 47 , 3122 ) CrossRef; (d) S. Diez-Gonzalez, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612 CrossRef CAS PubMed; (e) D. Kunz and E. Jürgens, in Molecular Catalysts: Structure and Functional Design, ed. L. H. Gade and P. Hofmann, Wiley-VCH, Weinheim, 2014, pp. 183–206 Search PubMed.
- In dichloromethane oxidative addition (trans) of the solvent to complex 1 is observed which leads to [Rh(bimca)Cl(CH2Cl)(CO)]. M. Moser, PhD thesis, Heidelberg University, Heidelberg, 2007.
- See the ESI† for details.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR spectra and crystallographic data. CCDC 1018408 (4a) and 1018409 (4b). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc07154a |
| This journal is © The Royal Society of Chemistry 2015 |