
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Covalent coupling via dehalogenation on Ni(111) supported boron nitride and graphene†
Claudius
Morchutt
ab,
Jonas
Björk
 c,
Sören
Krotzky
a,
Rico
Gutzler
*a and
Klaus
Kern
ab
c,
Sören
Krotzky
a,
Rico
Gutzler
*a and
Klaus
Kern
ab
aMax Planck Institute for Solid State Research, Heisenbergstrasse 1, 70569 Stuttgart, Germany. E-mail: r.gutzler@fkf.mpg.de
bInstitut de Physique de la Matière Condensée, Ecole Polytechnique Fédérale de Lausanne, 1015 Lausanne, Switzerland
cDepartment of Physics, Chemistry and Biology, IFM, Linköping University, 58183 Linköping, Sweden
First published on 24th December 2014
Abstract
Polymerization of 1,3,5-tris(4-bromophenyl)benzene via dehalogenation on graphene and hexagonal boron nitride is investigated by scanning tunneling microscopy experiments and density functional theory calculations. This work reveals how the interactions between molecules and graphene or h-BN grown on Ni(111) govern the surface-confined synthesis of polymers through C–C coupling.
The synthesis of surface-supported covalent organic networks has found its way into many surface science laboratories, making use of a wide range of reaction mechanisms.1–5 Pioneered by the Ullmann-type coupling of phenyl-porphyrins by Grill et al.,1 organic oligomers and polymers have been created on several different metal substrates.3,6,7 The role of the metallic surface is two-fold: it acts as a geometric constraint for polymerization in two dimensions, and it functions as a heterogeneous catalyst for the coupling reactions.8 An alternative is the use of thin decoupling layers on top of the metal substrate to electronically and spatially separate the polymer from the metal. Graphene has found widespread use as a support for self-assembled molecular monolayers9 and also atomically thin hexagonal boron-nitride (h-BN) sheets are currently investigated as templates for molecular nano-structures.10 The latter has recently been used in the coupling of halogenated hexaphenylene into covalently linked oligomers,11 albeit of small lateral extension compared to polymeric networks grown from the same parent molecule on metal surfaces.12 Multilayer films of oriented 2D covalent networks were synthesized using single-layer graphene as a growth template under solvothermal conditions.13 The synthesis of single sheets of 1D or 2D polymers on non-metallic surfaces would open the door towards physical and chemical characterization of the organic structures that cannot be achieved on metal surfaces due to strong polymer-surface interactions.
Here, we report the synthesis of covalent organic nanostructures from the brominated precursor molecule 1,3,5-tris(4-bromophenyl)-benzene (TBB) on the two interfaces h-BN/Ni(111) and graphene/Ni(111). Both graphene and h-BN function as model decoupling layers from the highly reactive Ni surface, but due to the non-negligible interaction with the Ni surface14,15 the reactivity is shown to remain sufficiently large for debromination of the precursor molecule as evidenced by STM. The scission of the C–Br bond is studied in detail by density functional theory (DFT) calculations, exhibiting strong similarities despite the different electronic structure of conductive graphene and insulating h-BN.
All experiments were carried out in an UHV chamber (base pressure < 5 × 10−10 mbar) equipped with a home-built STM operated at room temperature. (A detailed Methods section can be found in the ESI.†) STM images of defect-free surfaces of h-BN/Ni(111) and graphene/Ni(111) are provided in the ESI,† Fig. S1 and S2. TBB was purchased from Sigma-Aldrich. The molecules were evaporated from a Knudsen cell held between 200 °C and 210 °C. The deposition times varied between 15 min and 30 min. Covalent coupling was induced at 250 °C or 300 °C; post-annealing and deposition onto hot substrates resulted in similar structures. The DFT calculations were performed with the VASP code,16 using the projector-augmented wave method to describe ion–core interactions,17 and a kinetic energy cut-off of 500 eV. Exchange–correlation effects were described by the van der Waals density functional,18 with the version of Hamada,19 which gives an accurate description of related systems.20 Transition states were calculated using the nudged elastic band21 and Dimer methods.22
The precursor molecule TBB was initially sublimed onto bare Ni(111) at room temperature to investigate possible self-assembled structures and covalent coupling reactions (Scheme Fig. 1a). Fig. 1b shows a typical STM image at high coverage, in which single molecules are indicated in green. Molecules arrange in two different binding geometries with respect to the surface without long-range order. Circular bright protrusions (green circle) are observed and are tentatively attributed to bromine atoms that have split off from the molecules. Apart from dehalogenation, TBB seems to stay intact on Ni(111) at RT and no C–C bond breaking is observed. The absence of long-range order can be explained by the strong interaction between metal and surface-stabilized radicals.
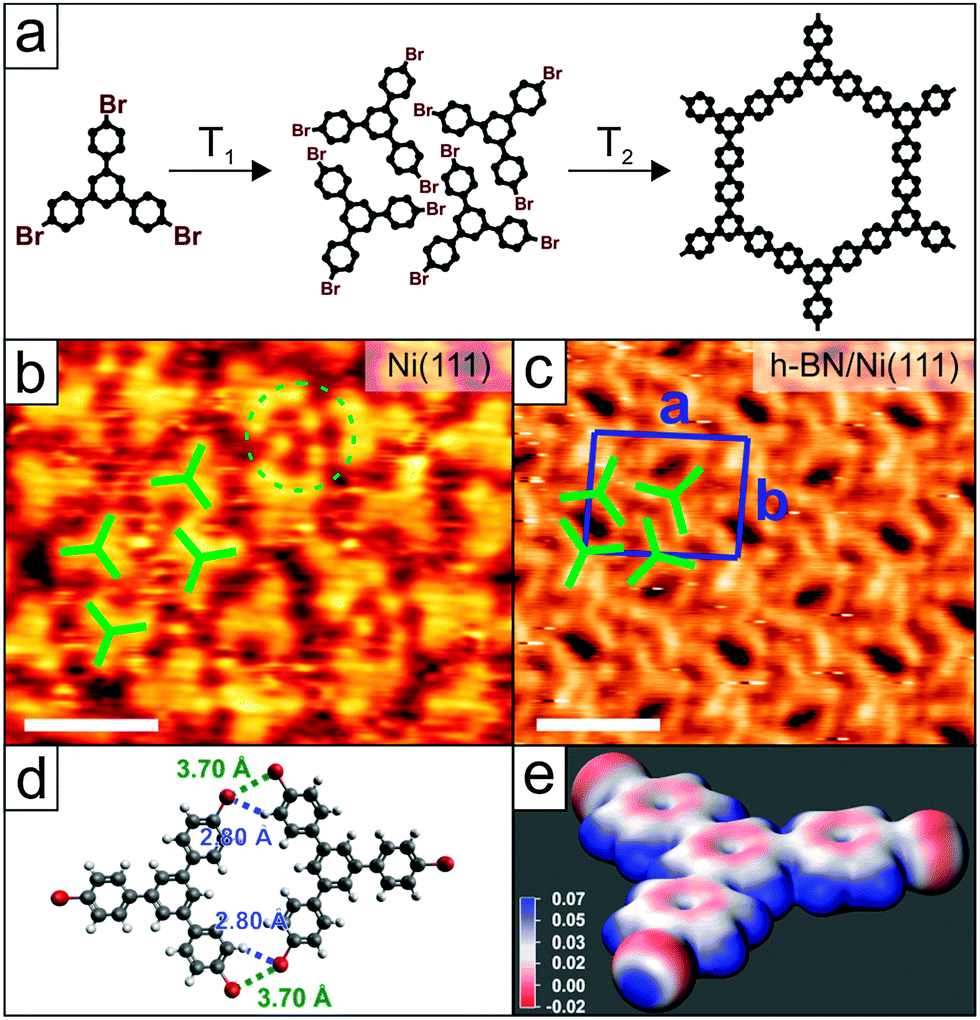 | ||
| Fig. 1 Structure of TBB and its adsorption on Ni(111) and h-BN/Ni(111). (a) Scheme of TBB (left) in the reaction from self-assembled structure (middle) to the covalently connected oligomer (right). (b) High-resolution STM image of TBB on Ni(111). The molecules order locally with the absence of long-range order (Ubias = −1.4 V, I = 52 pA). (c) High-resolution STM image of TBB self-assembled on h-BN/Ni(111) (Ubias = –0.8 V, I = 50 pA). Scale bar in (b) and (c): 2.0 nm. (d) Calculated dimer as observed in the self-assembled network with stabilizing close contacts between Br and H atoms. (e) Electrostatic potential map of the TBB monomer. Units in Rydberg e−1. | ||
In order to promote further debromination and to increase the diffusion of monomers we systematically annealed the sample. On coinage metal surfaces this leads to the formation of covalently bonded polymers.23–27 Such reaction products that arise from covalent coupling between dehalogenated TBB were not observed. However, starting at approximately 150 °C we observe the decomposition of TBB on the surface (ESI† Fig. S3).
The situation changes when TBB is deposited onto a single layer of h-BN which passivates the bare Ni surface. Fig. 1c shows a self-assembled structure of intact TBB that forms after deposition onto a hot surface (140 °C) at sub-monolayer coverage (ESI,† Fig. S4 for an overview image) with an oblique unit cell (a = 2.5 nm, b = 2.0 nm, 92° angle) containing four molecules. The packing arrangement is similar to self-assembled structures of TBB reported on Au(111).24,28 For additional insight into the intermolecular interactions, a dimer of two planar TBB molecules is calculated (Fig. 1d Gaussian 09,29 DFT, M06-2X functional, 6-31G(d,p) basis set), resembling two of the four molecules found in the unit cell. Four close contacts can be found, two Br⋯Br (3.70 Å) and two Br⋯H bonds (2.80 Å). The electrostatic potential map of the molecules (Fig. 1e) reveals the typical σ-hole at the Br atom, resulting in attractive interactions between this electropositive hole (blue) and the electronegative circumference (red) between two Br atoms of adjacent molecules, and the electronegative belt and H atoms.25,30 Upon annealing (523–573 K, 15 min) the C–Br bonds are cleaved and oligomers are observed on the surface. Fig. 2a shows an STM image of two oligomers: a heptamer and a trimer; a single TBB molecule is visible at the left end of the heptamer that is not connected to other molecules. Intermolecular distances of 1.3 nm within the oligomers and scaled molecular models confirm the formation of covalent bonds. Fig. 2b shows an STM image of a quasi-hexagon with a bright protrusion at the upper part, an overview is given in Fig. 2c, where oligomers of different sizes can be observed. Dimers, trimers, and larger oligomers are clearly visible. Long-range order and extended oligomers/polymers are not observed, and many terminal sites of the oligomers are apparent. These terminal sites are presumably dehalogenated, based on geometric considerations of the size of the monomeric units in the oligomers, and bind to the underlying graphene and h-BN layers. Bright features in the STM topograph are found close to the oligomers. As these features appear to be higher than the oligomers (ESI,† Fig. S5 and S6), we tentatively assign them to upright standing molecular fragments. The origin of the upright adsorption geometry will be discussed below. Large polymers are not observed for different annealing temperatures and times.
 | ||
| Fig. 2 Reaction products of TBB on h-BN/Ni(111). (a) High-resolution STM image of a pentagon-shaped oligomer (left) and a trimer (right). Scaled ball-and-stick models are superimposed (Ubias = −1.0 V, I = 46 pA). (b) High-resolution STM image of a quasi-hexagon (Ubias = −1.5 V, I = 330 pA). (c) Overview STM image of reaction products. Oligomers of different size and shape are visible (Ubias = −1.4 V, I = 52 pA). Scale bar in (a) and (b) 2.6 nm and in (c) 6.0 nm. | ||
Graphene was used as an alternative decoupling layer to examine whether extended polymers can be synthesized. A self-assembled structure was observed under similar conditions as on h-BN (ESI† Fig. S7). Upon annealing (523–573 K, 15 min) covalently bonded oligomers are formed. Fig. 3a shows an STM image of a kinked hexamer as well as a scaled ball-and-stick model, confirming the formation of covalent bonds between the precursor molecules. A pentamer as well as a dimer (upper part) and an isolated monomer are shown in Fig. 3b (overview in Fig. 3c). Again, no long-range order is observed and only small oligomers are formed. As on h-BN, we frequently observed irregular features appearing higher than the flat oligomers, which are tentatively assigned to upright standing molecules or fragments. The absence of long-range order and a large polymer size on h-BN and graphene stands in contrast to the coinage metals, on which the same precursor molecule forms extended polymeric networks, and on which dehalogenation and C–C coupling occur at lower temperatures. Limited mobility of the dehalogenated molecules due to a strong interaction with the surface is likely responsible for this observation, which also explains the occurrence of single molecules pinned to the surface.
 | ||
| Fig. 3 Oligomers of TBB on graphene/Ni(111). (a) High-resolution STM image of a hexamer with a superimposed scaled ball-and-stick model (Ubias = −1.3 V, I = 32 pA). Scale bar: 1.5 nm. (b) High-resolution STM image of oligomers. (Ubias = −0.8 V, I = 55 pA). Scale bar: 2.6 nm. (c) Overview STM image of reaction products. Oligomers of different sizes are visible with a lack of long-range order (Ubias = −1.3 V, I = 32 pA). Scale bar: 3.3 nm. | ||
To substantiate this hypothesis, DFT calculations were performed on the debromination of the model compound bromobenzene on the two interfaces h-BN/Ni(111) and graphene/Ni(111). Fig. 4a shows the initial state (IS), transition state (TS), intermediate state (IntS) and final state (FS) for the dehalogenation of bromobenzene on h-BN/Ni(111). Note that the actual dehalogenation takes place between the initial and intermediate state, while the steps between IntS and FS are merely the diffusion of the phenyl ring and/or the bromine atom, which were not explicitly calculated. Already in the TS a boron atom (pink) is slightly lifted out of the plane of the h-BN layer and binds to the brominated carbon atom. The plane of the phenyl ring tilts away from the surface. In the IntS the split-off bromine atom binds to a neighbouring boron atom, which is also slightly lifted. In the FS, the phenyl ring and the bromine are well separated, enabling both species to adapt a more favourable adsorption configuration. The dehalogenation reaction is exothermic (an energy of 0.41 eV is released) with an energy barrier of 1.51 eV. This value is higher than the energy values calculated for Cu(111), Ag(111) and Au(111), which range between 0.66 eV and 1.02 eV.8 The dehalogenation barrier correlates with the annealing temperatures used to induce C–C coupling of TBB on h-BN/Ni(111). Assuming a pre-exponential factor of 1013 s−1, 585 K is required to achieve a reaction rate of 1 s−1, close to the maximum annealing temperature of 573 K (using the Arrhenius equation, ESI† Fig. S8). The calculated barrier is three times larger than the experimentally obtained activation energy of TBB on Cu(111),31 presumably a consequence of the damping of the Ni d levels due to the decoupling layer. Notably, following a similar path as on the Ni-supported surface, the reaction is not possible on the free standing h-BN sheet, demonstrating the active role of the underlying metal surface in the catalytic dissociation reaction (ESI,† Fig. S19).
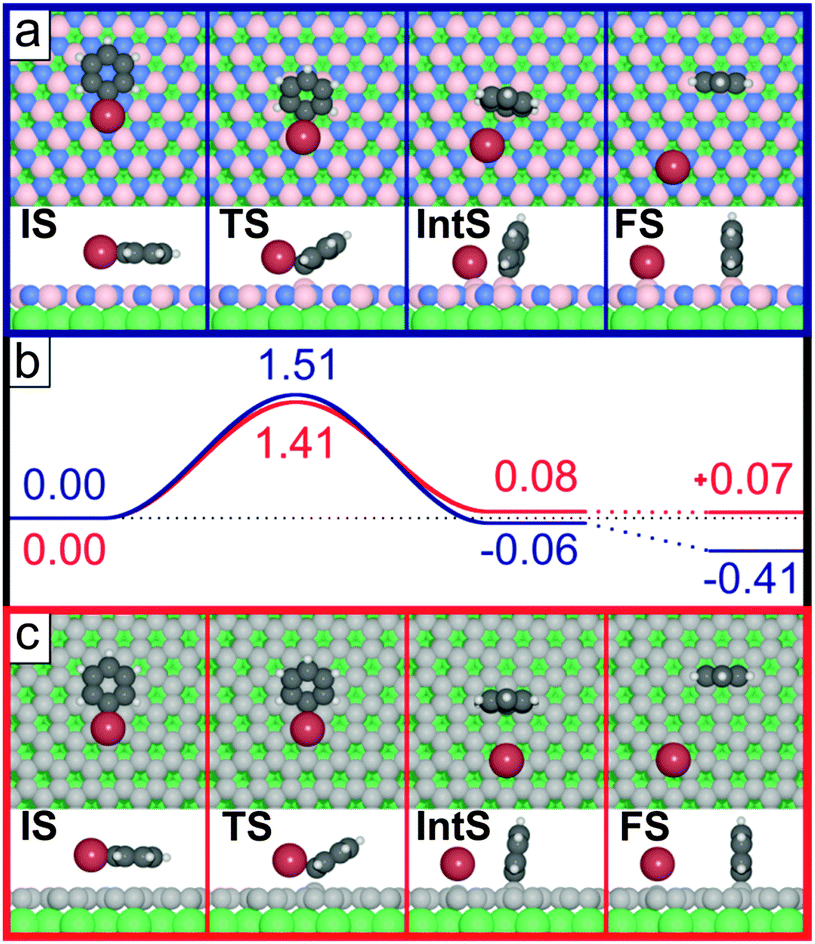 | ||
| Fig. 4 Initial state (IS), transition state (TS), intermediate state (IntS) and final state (FS) of the dehalogenation of bromobenzene on (a) h-BN/Ni(111) and (c) graphene/Ni(111). (b) Energy diagram of the reaction on h-BN/Ni(111) in blue and on graphene/Ni(111) in red. Both substrates exhibit a barrier of approximately 1.5 eV for the debromination. (Grey: carbon, white: hydrogen, brown: bromine, blue: nitrogen, light pink: boron, green: nickel.) Values in eV. | ||
The perpendicular adsorption of the dehalogenated molecule (Fig. 4a, FS) is an indicator of a strong chemical coupling between carbon and boron atoms (∼2.5 eV, see below). This strong interaction results in a significantly reduced mobility of the molecules on the surface and explains the absence of large polymers. The diffusion barrier is calculated to be 1.9 eV for the fully debrominated TBB on the h-BN/Ni(111) surface (ESI,† Fig. S16). This barrier is smaller by 0.3 eV than the barrier of the cyclohexa-m-phenylene radical on Cu(111), for which larger polymers than those observed here are formed.12 Debrominated phenyl has a diffusion barrier of 1.7 eV on h-BN/Ni(111) (ESI,† Fig. S15) and is substantially larger than on the coinage metal surfaces, which range between 0.05 eV and 0.2 eV.8 The large diffusion barrier stems from the highly unfavourable binding of the phenyl rings to B atoms in the TSs (ESI,† Fig. S15). It was previously pointed out that a balance of diffusion and coupling rates governs the formation of branched oligomers or regular polymers.12,32 In a diffusion-limited process, where the coupling rate is much larger compared to the diffusion rate,33 disordered, branched oligomers dominate. Taking into account the large diffusion barrier for the dehalogenated molecules calculated by DFT, we propose such a diffusion-limited growth process on the decoupling layer leading to small oligomers as observed in our experiments. Based on the upright adsorption geometry of the phenyl unit, the observed unusual bright protrusions can tentatively be attributed to vertically standing molecular fragments.
The DFT calculations for bromobenzene on graphene/Ni(111) show very similar results. Fig. 4c (red) sketches the reaction pathway for debromination. The carbon atom of graphene that binds to bromobenzene is slightly lifted and the reaction is endothermic by 0.07 eV. The energy barrier of 1.41 eV is comparable to the value of bromobenzene on h-BN/Ni(111), and is in line with the theoretical annealing temperature of 546 K suggested by an Arrhenius equation to achieve a reaction rate of 1 s−1. The calculations also show a strong interaction between the phenyl unit and graphene/Ni(111), which again explains the absence of larger oligomers/polymers due to the significantly reduced diffusion of debrominated TBB molecules. For the bromobenzene dissociation on both h-BN/Ni(111) and graphene/Ni(111), the phenyl ring in the TS interacts strongly with the respective surface in the absence of any substantial bromine–surface interaction (cf.Fig. 4a and c, TS side view). Reminiscing the comparable energy barriers for the two surfaces, also the adsorption energies of phenyl are very similar (−2.52 eV and −2.54 eV on h-BN/Ni(111) and graphene/Ni(111), respectively). This indicates that the strength of the phenyl–surface bond formation drives the debromination, and thus controls the energy barrier of the reaction. Furthermore, the diverging reaction energies between the surfaces can be traced to the different adsorption energy of bromine; −2.43 eV on h-BN/Ni(111) and −1.85 eV on graphene/Ni(111), illustrating the importance of the interaction of both the phenyl and the bromine with the surface in the FS (see ESI† for details). Additional calculations reveal that the cleavage of the C–Br bond on the freestanding graphene has a barrier of 2.85 eV, 1.4 eV larger than on graphene/Ni(111). This underlines the catalytic relevance of the supporting Nickel crystal for the dehalogenation reaction.
In summary, graphene and h-BN single layers on Ni(111) are presented here as substrates for the growth of covalent nanostructures in the form of oligophenylene. A strong interaction between dehalogenated molecules and decoupling layer results in limited mobility and inhibits the growth of polymers with large spatial extension. The large diffusion barrier of surface-stabilized radicals might be a fundamental limitation for the growth of (ordered) polymers on graphene and h-BN in vacuum. However, we expect that order and spatial extension of the polymers can be improved by a suitable choice of supporting metal surface for graphene and h-BN, or by switching to solution-based approaches.34
We are grateful to Dr Matthias Schreck and Michael Weinl for providing the nickel thin film samples Ni/YSZ/Si(111). Computer resources were allocated by the National Super-computer Centre, Sweden, through SNAC.
Notes and references
- L. Grill, M. Dyer, L. Lafferentz, M. Persson, M. V Peters and S. Hecht, Nat. Nanotechnol., 2007, 2, 687 CrossRef CAS PubMed.
- D. Perepichka and F. Rosei, Science, 2009, 323, 216 CrossRef CAS PubMed.
- G. Franc and A. Gourdon, Phys. Chem. Chem. Phys., 2011, 13, 14283 RSC.
- H.-Y. Gao, H. Wagner, D. Zhong, J.-H. Franke, A. Studer and H. Fuchs, Angew. Chem., Int. Ed., 2013, 52, 4024 CrossRef CAS PubMed.
- Y.-Q. Zhang, N. Kepčija, M. Kleinschrodt, K. Diller, S. Fischer, A. C. Papageorgiou, F. Allegretti, J. Björk, S. Klyatskaya, F. Klappenberger, M. Ruben and J. V Barth, Nat. Commun., 2012, 3, 1286 CrossRef PubMed.
- J. W. Colson and W. R. Dichtel, Nat. Chem., 2013, 5, 453 CrossRef CAS PubMed.
- X. Zhang, Q. Zeng and C. Wang, Nanoscale, 2013, 5, 8269 RSC.
- J. Björk, F. Hanke and S. Stafström, J. Am. Chem. Soc., 2013, 135, 5768 CrossRef PubMed.
- J. M. MacLeod and F. Rosei, Small, 2014, 10, 1038 CrossRef CAS PubMed.
- S. Joshi, F. Bischoff, R. Koitz, D. Ecija, K. Seufert, A. P. Seitsonen, J. Hutter, K. Diller, J. I. Urgel, H. Sachdev, J. V. Barth and W. Auwärter, ACS Nano, 2014, 8, 430 CrossRef CAS PubMed.
- T. Dienel, J. Gómez-Díaz, A. P. Seitsonen, R. Widmer, M. Iannuzzi, K. Radican, H. Sachdev, K. Müllen, J. Hutter and O. Gröning, ACS Nano, 2014, 8, 6571 CrossRef CAS PubMed.
- M. Bieri, M. T. Nguyen, O. Gröning, J. Cai, M. Treier, K. Aït-Mansour, P. Ruffieux, C. A. Pignedoli, D. Passerone, M. Kastler, K. Müllen and R. Fasel, J. Am. Chem. Soc., 2010, 132, 16669 CrossRef CAS PubMed.
- J. W. Colson, A. R. Woll, A. Mukherjee, M. P. Levendorf, E. L. Spitler, V. B. Shields, M. G. Spencer, J. Park and W. R. Dichtel, Science, 2011, 332, 228 CrossRef CAS PubMed.
- A. Preobrajenski, A. Vinogradov and N. Mårtensson, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 165404 CrossRef.
- K. Yamamoto, M. Fukushima, T. Osaka and C. Oshima, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 11358 CrossRef CAS.
- G. Kresse, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef.
- M. Dion, H. Rydberg, E. Schröder, D. C. Langreth and B. I. Lundqvist, Phys. Rev. Lett., 2004, 92, 246401 CrossRef CAS PubMed.
- I. Hamada, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 121103 CrossRef.
- J. Björk and S. Stafström, ChemPhysChem, 2014, 15, 2851 CrossRef PubMed.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9978 CrossRef CAS PubMed.
- J. Kästner and P. Sherwood, J. Chem. Phys., 2008, 128, 014106 CrossRef PubMed.
- R. Gutzler, H. Walch, G. Eder, S. Kloft, W. M. Heckl and M. Lackinger, Chem. Commun., 2009, 4456 RSC.
- M. O. Blunt, J. C. Russell, N. R. Champness and P. H. Beton, Chem. Commun., 2010, 46, 7157 RSC.
- H. Walch, R. Gutzler, T. Sirtl, G. Eder and M. Lackinger, J. Phys. Chem. C, 2010, 114, 12604 CAS.
- Q. Fan, C. Wang, L. Liu, Y. Han, J. Zhao, J. Zhu, J. Kuttner, G. Hilt and J. M. Gottfried, J. Phys. Chem. C, 2014, 118, 13018 CAS.
- L. Cardenas, R. Gutzler, J. Lipton-Duffin, C. Fu, J. L. Brusso, L. E. Dinca, M. Vondráček, Y. Fagot-Revurat, D. Malterre, F. Rosei and D. F. Perepichka, Chem. Sci., 2013, 4, 3263 RSC.
- J. C. Russell, M. O. Blunt, J. M. Garfitt, D. J. Scurr, M. Alexander, N. R. Champness and P. H. Beton, J. Am. Chem. Soc., 2011, 133, 4220 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 09, Revision A.02, 2009 Search PubMed.
- R. Gutzler, O. Ivasenko, C. Fu, J. Brusso, F. Rosei and D. Perepichka, Chem. Commun., 2011, 47, 9453 RSC.
- M. Chen, J. Xiao, H.-P. Steinrück, S. Wang, W. Wang, N. Lin, W. Hieringer and J. M. Gottfried, J. Phys. Chem. C, 2014, 118, 6820 CAS.
- J. Eichhorn, D. Nieckarz, O. Ochs, D. Samanta, M. Schmittel, P. J. Szabelski and M. Lackinger, ACS Nano, 2014, 8, 7880 CrossRef CAS PubMed.
- J. Björk and F. Hanke, Chem. – Eur. J., 2014, 20, 928 CrossRef PubMed.
- L. Xu, X. Zhou, W. Q. Tian, T. Gao, Y. F. Zhang, S. Lei and Z. F. Liu, Angew. Chem., 2014, 126, 9718 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, additional STM data and DFT calculations. See DOI: 10.1039/c4cc07107g |
| This journal is © The Royal Society of Chemistry 2015 |