
Herringbone to cofacial solid state packing via H-bonding in diketopyrrolopyrrole (DPP) based molecular crystals: influence on charge transport†
Joydeep
Dhar
,
Durga Prasad
Karothu
and
Satish
Patil
*
Solid State and Structural Chemistry Unit, Institute of Science, Bangalore 560012, India. E-mail: satish@sscu.iisc.ernet.in; Tel: +91 080 2293 2651
First published on 10th November 2014
Abstract
The mono-alkylation of DPP derivatives leads to cofacial π–π stacking via H-bonding unlike their di-alkylated counterparts, which exhibit a classical herringbone packing pattern. Single crystal organic field-effect transistor (OFET) measurements reveal a significant enhancement of charge carrier mobility for mono-hexyl DPP derivatives.
The performance of optoelectronic devices based on organic semiconductors is limited by poor charge transport. The charge carrier mobility (μ) in molecular semiconductors is strongly influenced by a variety of factors such as purity, traps, defects and molecular stability.1 To surmount these limitations, molecular engineering has taken a centre stage.2–4 Large numbers of new organic functional materials have been synthesized to achieve the best performance for devices such as organic photovoltaics (OPVs) and organic field effect transistors (OFETs).5–7 In this regard, diketopyrrolopyrrole (DPP) based π-conjugated systems have shown promise as versatile materials with excellent charge transport properties.8,9 Yun-Hi Kim and co-workers have reported one of the highest p-type charge carrier mobilities for a DPP-based polymer, surpassing the mobility of amorphous silicon.10 Our group has also observed one of the best electron mobilities for a DPP based π-conjugated low-band gap polymer.11 Inspired by these results, we envisioned to investigate the structure–property correlation of DPP monomers to explore the full potential as a molecular material for organic electronics.
The DPP-based monomers were synthesized by the pseudo-Stobbe condensation reaction followed by di-alkylation of the amide groups present in the DPP chromophore.12 While following this procedure a minor product was also separated along with the expected di-hexyl DPP derivative. The highly crystalline minor product was identified as mono-alkylated DPP by 1H, 13C and ESI mass spectroscopy (ESI†). To our surprise, single crystal structure determination reveals a classical herringbone12 and cofacial layered structure for di- and mono-hexyl DPP, respectively. This encouraged us to enhance the yield of mono-hexyl DPP by controlling the reaction conditions, shown in Scheme 1, and pursue further with an aim of studying the comparative charge transport properties of this important class of materials which was ignored previously. Herein, we report the synthesis and characterization of mono-alkylated DPP for the first-time. Detailed studies have unraveled several unusual features including the role of H-bonding in influencing favourable packing for enhanced charge transport in single crystals of mono-hexyl DPP than its di-alkylated counterpart.
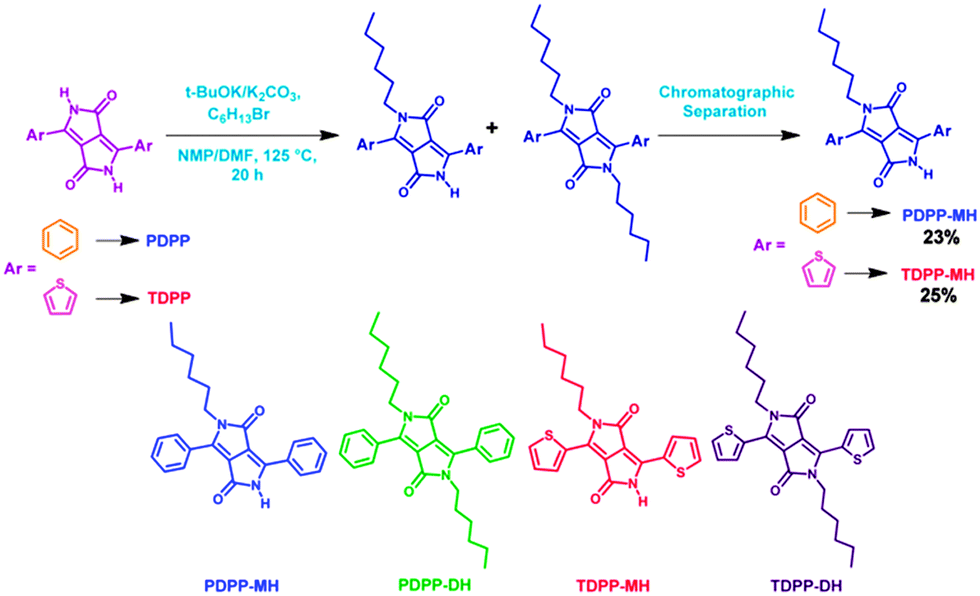 | ||
| Scheme 1 Synthesis of PDPP-MH and TDPP-MH. The chemical structures of four different DPP molecules are also shown. | ||
The optical properties of mono-hexyl DPP molecules were almost similar to their di-alkylated analogues with the appearance of the dual band structure in absorption and emission spectra (Fig. 1(a) and (b)). The low energy band in the absorption spectrum showed vibronic splitting for both mono-hexyl derivatives. But there was a minor blue shift (10 nm) of the lowest energy band for TDPP-MH (λmax at 500, 540 nm) as compared to TDPP-DH (λmax at 510, 550 nm) indicating an increase in the band gap while the optical band gap remained almost the same for both the phenyl coupled DPP (PDPP) derivatives. Both PDPP-MH and TDPP-MH showed a blue-shifted vibronic emissive band in comparison with the di-hexyl DPP molecules. The hypsochromic shift was measured to be ∼10 nm for TDPP-MH while it was 25 nm for PDPP-MH. As a result, we observe a significant decrease in the Stokes shift for PDPP-MH (0.19 eV) as compared to PDPP-DH (0.32 eV), but for TDPP-MH and TDPP-DH the Stokes shift was almost similar (0.4–0.5 eV). The reduction in the Stokes shift suggests a decrease in torsion in PDPP-MH. Furthermore, density functional theory (DFT) calculations have corroborated our assumption. The detailed optical properties of mono-hexyl derivatives are given in Table S1 in the ESI.† Theoretical studies have shown that in the absence of an alkyl chain the repulsive interaction between ortho-hydrogen of the phenyl group and a nearby hydrogen atom attached to the alkyl group vanishes and the phenyl group appears almost in the same plane with the DPP core with a torsion angle of 8° enhancing electronic coupling between donor and acceptor moieties (Fig. 1d). On the other hand, the other phenyl ring remains twisted as in PDPP-DH by 36° due to the above said repulsive interaction. Calculations on TDPP-MH predict the trans-orientation of two thiophene units with respect to each other and both the donor groups lie in the same plane with the acceptor having a minimum torsion angle of less than 1°.
 | ||
| Fig. 1 The normalized absorption (a) and emission (b) spectra and fluorescence excited state decay profile (c) of mono- and di-hexyl DPP derivatives. (d) Energy minimized ground state geometry of PDPP-MH and TDPP-MH obtained by density functional theory (DFT) calculations. The torsion between donor and acceptor units has also been mentioned. | ||
The consequence of higher planarity in TDPP-MH is the stronger intramolecular electronic coupling between donor and acceptor moieties resulting in a higher molar extinction coefficient for TDPP-MH (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 L mol−1 cm−1) than the PDPP-MH (15000 L mol−1 cm−1) as measured experimentally. Fluorescence quantum yield decreases from 80% in PDPP-MH to 72% in TDPP-MH, which is attributed to the heavy atom effect facilitating the inter-system crossing rates.13 On the similar line, the fluorescence excited state lifetime also decreases from PDPP-MH to TDPP-MH as shown in Fig. 1c. The frontier molecular orbital energies were measured by cyclic voltammetry (CV) experiments. The HOMO and LUMO energies are summarized in Table S1 (ESI†). The HOMO energy level destabilized by 0.1 eV in TDPP-MH from PDPP-MH while LUMO is stabilized by ∼0.2 eV by replacing phenyl with thiophene at the donor moiety.
300 L mol−1 cm−1) than the PDPP-MH (15000 L mol−1 cm−1) as measured experimentally. Fluorescence quantum yield decreases from 80% in PDPP-MH to 72% in TDPP-MH, which is attributed to the heavy atom effect facilitating the inter-system crossing rates.13 On the similar line, the fluorescence excited state lifetime also decreases from PDPP-MH to TDPP-MH as shown in Fig. 1c. The frontier molecular orbital energies were measured by cyclic voltammetry (CV) experiments. The HOMO and LUMO energies are summarized in Table S1 (ESI†). The HOMO energy level destabilized by 0.1 eV in TDPP-MH from PDPP-MH while LUMO is stabilized by ∼0.2 eV by replacing phenyl with thiophene at the donor moiety.
The single crystal X-ray diffraction study reveals the molecular arrangements and their interactions in the solid state. Both the molecules crystallize in the monoclinic space group P21/n z = 4. The unique difference between di- and mono-hexyl DPP derivatives lies in their crystal packing arrangements. The free amide group of mono-alkylated DPP molecules plays a pivotal role in turning around the molecular packing in the unit cell. PDPP-MH exhibits a cofacial layered structure while TDPP-MH demonstrates cofacial herringbone packing due to the intermolecular H-bonding between two amide functionalities in the DPP chromophore locking the orientation of the molecules. Thus, intermolecular H-bonding influences the packing pattern of DPP-MH deviating from their di-hexyl counterparts which show exclusively classical edge-to-face herringbone packing. Both PDPP-MH and TDPP-MH showed slip-stacked packing arrangement with the donor thiophene/phenyl group appearing on top of the acceptor DPP chromophore, which is a common feature of D–A–D molecular crystals presumably due to the charge transfer interaction. The shortest π–π packing distances were measured to be 3.48 Å and 3.71 Å for PDPP-MH and TDPP-MH, respectively. Similar to our theoretical findings, we see the variation in the degree of torsion between the acceptor moiety and two donor groups in both the molecules due to relief from steric repulsion in the absence of an alkyl chain. Torsion varies from 2° to as high as 24° in PDPP-MH while they were 1° and 10° in TDPP-MH. The N–H⋯O H-bond distances were calculated to be 2.04 Å and 1.94 Å in PDPP-MH and TDPP-MH, respectively. Weak C–H⋯O interaction was also found in both the crystals with the distances of 2.43 Å and 2.27 Å in PDPP-MH and TDPP-MH (Fig. 2).
 | ||
| Fig. 2 Cofacial packing via H-bonding in PDP-MH and TDPP-MH. | ||
The transmission electron microscopy (TEM) study shows that the nano-to micron sized structures deposited on a copper grid are single crystalline in nature (Fig. 3). The mono-hexyl DPP derivatives form fiber like single crystals while di-hexyl analogues form small plate like crystallites (Fig. S9 and S10, ESI†). We believe that the presence of H-bonding interaction is responsible for variation in morphologies for mono-hexyl DPP derivatives. The ordered bright diffraction spots in the selected area electron diffraction (SAED) pattern confirm the single crystallinity of microstructures. The bright field TEM images and corresponding SAED patterns obtained from single crystal di-hexyl derivatives have been provided in the ESI.†
 | ||
| Fig. 3 Bright field TEM images (a, c) and corresponding SAED patterns (b, d) of the single crystalline microwires of PDPP-MH (a, b) and TDPP-MH (c, d). | ||
The H-bonding interaction was further supported by differential scanning calorimetry (DSC). For TDPP-MH, we have observed the appearance of two endo- and exothermic transitions during heating and cooling cycles, respectively (Fig. 4). The first endothermic peak was positioned at 207 °C with an enthalpy change of ∼2.3 kJ mol−1 which matches quite well with the H-bonding energy. This compound melted at around 250 °C. The change in enthalpy during melting transition was measured to be 21.5 kJ mol−1. But dual peaks were not observed for PDPP-MH. In both heating and cooling cycles only one peak was observed, though the enthalpy change during the melting process at around 250 °C was higher i.e. ∼26.8 kJ mol−1. The phase transitions observed in DSC measurements were further confirmed by optical polarising microscopy. For TDPP-MH crystals, we have observed a thermal transition at around 208 °C associated with the change in the crystal texture followed by an isotropic phase transition at 255 °C. But PDPP-MH crystals turn into an isotropic phase at 247 °C with a single thermal transition.
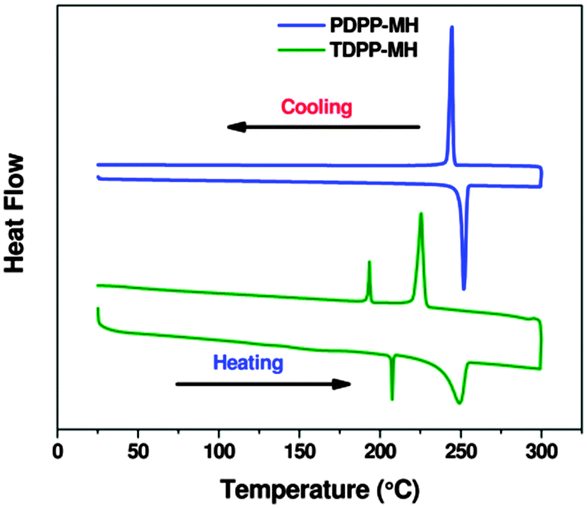 | ||
| Fig. 4 DSC thermograms of PDPP-MH and TDPP-MH. | ||
OFET devices were fabricated using Si/SiO2 as a gate and dielectric material in bottom-gate bottom-contact (BGBC) device geometry. A photo-lithographically patterned gold (Au) source and drain contacts were developed on a hexamethyldisilazane (HMDS) treated 190 nm thick dielectric layer of SiO2. The solutions of respective organic materials were drop-casted on the substrate and crystals were grown by the slow evaporation method in the channel. The mono-hexyl derivatives form thin and sharp needle like crystals whereas their di-hexyl analogues form bigger crystallites in between the source and drain electrodes. The measurements were carried out under ambient conditions at room temperature. Fig. 5 shows the output characteristics and corresponding optical images of the devices fabricated from single crystalline PDPP-MH and TDPP-MH microwires. The device data is summarized in Table 1.
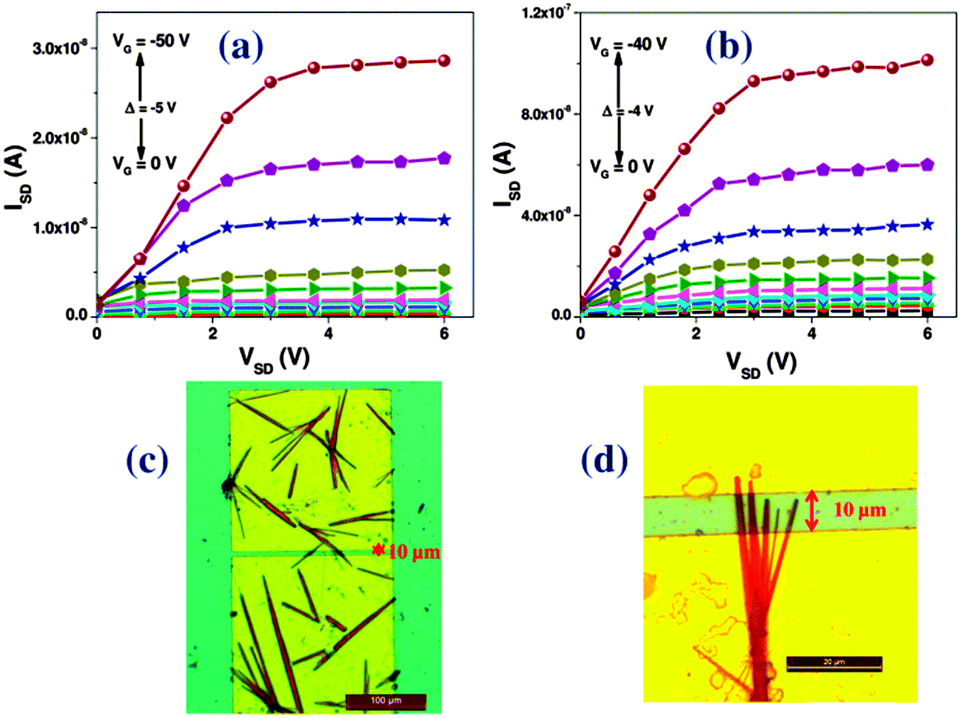 | ||
| Fig. 5 Output characteristics (a, b) and optical images of the corresponding devices (c, d) fabricated from single crystalline PDPP-MH (a, c) and TDPP-MH (b, d) microwires. | ||
| Compounds | Structure | p-type mobility (μaverage) (cm2 V−1 s−1) | On/Off |
|---|---|---|---|
| Devices were fabricated in bottom-gate bottom-contact geometry. | |||
| PDPP-MH | Cofacial layered | 1.6 × 10−2 | 102 |
| TDPP-MH | Cofacial herringbone | 0.3 | 102 |
| PDPP-DH | Classical herringbone | 2 × 10−4 | 102 |
| TDPP-DH | Classical herringbone | 4 × 10−3 | 103 |
Significant differences in charge transport characteristics were observed between mono- and di-hexyl DPP derivatives, which are related to the intrinsic nature of the molecular semiconductors. We have observed higher p-type mobilities for mono-hexyl DPPs as compared to their di-hexyl counterparts. The average p-type mobilities for PDPP-MH and TDPP-MH was recorded to be 1.6 × 10−2 cm2 V−1 s−1 and 0.3 cm2 V−1 s−1, respectively. Whereas the average mobility values were two orders less for their di-hexyl analogues. The best mobility of 2 cm2 V−1 s−1 was measured for TDPP-MH. The highest mobilities obtained from PDPP-DH and TDPP-DH were 3 × 10−3 cm2 V−1 s−1 and 5 × 10−2 cm2 V−1 s−1, respectively. On/off ratios of these devices were ∼102–103. The current shows saturation behaviour with an increase in the source–drain voltage for almost all the devices prepared from DPP-MH but this was not observed for devices fabricated from DPP-DH. This may be related to the crystal quality or contact resistance at the semiconductor-electrode interface.
The cofacial π-stacking improves the transfer integral as compared to edge-to-face herringbone packing according to Marcus theory, hence predicting better carrier transport through the π-stacked molecules if other factors remain the same.14,15 Zhenan Bao and co-workers have also reported the same for tetracene derivatives.16
To correlate the experimental observation theoretically, the transfer integral (t) has been calculated for all the four crystals in the three crystallographic directions (Fig. S15 and S16, ESI†).1 Employing the wB97xD function and 6-31g* basis set, the single point energy of the dimeric structure (obtained from the crystal structure) was calculated. Half of the energy difference between the HOMO and HOMO − 1 gives an estimate of the transfer integral for hole transport while for electron transport, it is the energy difference between LUMO and LUMO + 1. PDPP-DH has the t values in the z-direction, which corresponds to a herringbone arrangement. The t values (0.095 eV and 0.10 eV) predict equally good hole and electron mobilities. Along the y-direction smaller t values (0.02 eV and 0.015 eV) indicate reduced transport. Charge transfer integrals are zero in the x-direction. Removing one hexyl group (PDPP-MH) results in the cofacial crystal structure with hydrogen bonding in the y-direction. However, very small (0.005 eV and 0.005 eV) t values were observed in this direction. Charge transfer integrals are about equally large in the other two directions (0.05 eV and 0.07 eV (x-direction); 0.065 and 0.065 eV (z-direction)) for hole and electron transport, respectively. But, for both mono- and di-hexyl TDPP, we observe that all the directions can take part in charge transport with a relatively high t value for the DPP-MH system. The TDPP-DH system has the largest t values (0.075 eV and 0.14 eV) along the y-direction predicting good electron mobility. Upon removing one hexyl group, hydrogen bonding leads to rearrangement of the crystal structure. For TDPP-MH the best mobility is predicted in the z-direction, where the t value for electron and hole transport reaches 0.125 eV. The t values of all the four molecules in three crystallographic directions are summarized in Table S2 (ESI†). Since, all the FET measurements were carried out under ambient conditions, experimentally no electron transport was observed.
The overall results indicate that DPP-DH systems conduct mainly in one direction with high t values, while in DPP-MH systems charge transport occurs in two or three directions with considerable t values. Therefore, the total charge transport predicted for mono-substituted systems is increased as compared to di-substituted ones. The hydrogen bonding direction does, however, contribute very little to the charge transport. Transport mainly occurs along π-stacks or along herringbone arrangements and appears to be influenced by the exact distance and orientation of the molecules.
In summary, two novel mono-hexyl DPP derivatives with phenyl and thiophene as donor groups have been synthesized and their properties are comprehensively characterized by single crystal X-ray analysis and charge transport measurements. The single crystal structure determination reveals the uniqueness of mono-hexyl DPP from its di-hexyl analogues. While di-hexyl PDPP and TDPP exhibit herringbone packing arrangements, the mono-hexyl derivatives show cofacial layered structure. The deviation is due to the intermolecular H-bonding by the free amide group in mono-hexyl DPP molecules. Higher p-type charge carrier mobilities were observed for PDPP-MH and TDPP-MH as compared to the di-hexyl DPP molecules. Theoretical predictions based on transfer integral calculations corroborate the experimental observation of superior charge transport properties of mono-hexyl DPP derivatives.
The authors acknowledge the Ministry of Communication and Information Technology under a grant for the Centre of Excellence in Nanoelectronics, Phase II. The authors greatly acknowledge Prof. Ulrike Salzner, Bilkent University for her assistance with theoretical calculations. Joydeep Dhar thanks Council of Scientific and Industrial Research (CSIR) for senior research fellowship (SRF). The authors also acknowledge Centre for Nano Science and Engineering and Indian Institute of Science for providing the facilities for the device fabrication and FETEM measurements.
Notes and references
- V. Coropceanu, J. Cornil, D. A. da Silva Filho, Y. Olivier, R. Silbey and J.-L. Bredas, Chem. Rev., 2007, 107, 926–952 CrossRef CAS PubMed.
- M. Gsaenger, J. H. Oh, M. Koenemann, H. W. Hoeffken, A.-M. Krause, Z. Bao and F. Wuerthner, Angew. Chem., 2010, 49, 740–743 CrossRef CAS PubMed.
- A. Putta, J. D. Mottishaw, Z. Wang and H. Sun, Cryst. Growth Des., 2014, 14, 350–356 CAS.
- M. C. R. Delgado, E.-G. Kim, D. A. da Silva Filho and J.-L. Bredas, J. Am. Chem. Soc., 2010, 132, 3375–3387 CrossRef PubMed.
- A. W. Hains, Z. Q. Liang, M. A. Woodhouse and B. A. Gregg, Chem. Rev., 2010, 110, 6689–6735 CrossRef CAS PubMed.
- C. L. Wang, H. L. Dong, W. P. Hu, Y. Q. Liu and D. B. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed.
- Y. J. Cheng, S. H. Yang and C. S. Hsu, Chem. Rev., 2009, 109, 5868–5923 CrossRef CAS PubMed.
- Y. N. Li, P. Sonar, L. Murphy and W. Hong, Energy Environ. Sci., 2013, 6, 1684–1710 CAS.
- C. B. Nielsen, M. Turbiez and I. McCulloch, Adv. Mater., 2013, 25, 1859–1880 CrossRef CAS PubMed.
- I. Kang, H.-J. Yun, D. S. Chung, S.-K. Kwon and Y.-H. Kim, J. Am. Chem. Soc., 2013, 135, 14896–14899 CrossRef CAS PubMed.
- C. Kanimozhi, N. Yaacobi-Gross, K. W. Chou, A. Amassian, T. D. Anthopoulos and S. Patil, J. Am. Chem. Soc., 2012, 134, 16532–16535 CrossRef CAS PubMed.
- J. Dhar, N. Venkatramaiah, A. Anitha and S. Patil, J. Mater. Chem. C, 2014, 2, 3457–3466 RSC.
- R. D. Pensack, Y. Song, T. M. McCormick, A. A. Jahnke, J. Hollinger, D. S. Seferos and G. D. Scholes, J. Phys. Chem. B, 2014, 118, 2589–2597 CrossRef CAS PubMed.
- J. D. Mottishaw and H. Sun, J. Phys. Chem. A, 2013, 117, 7970–7979 CrossRef CAS PubMed.
- X.-K. Chen, J.-F. Guo, L.-Y. Zou, A.-M. Ren and J.-X. Fan, J. Phys. Chem. C, 2011, 115, 21416–21428 CAS.
- H. Moon, R. Zeis, E. J. Borkent, C. Besnard, A. J. Lovinger, T. Siegrist, C. Kloc and Z. N. Bao, J. Am. Chem. Soc., 2004, 126, 15322–15323 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, experimental details, and additional spectra. CCDC 992180 and 1015521. For ESI and crystallographic data in CIF or other electronic format see see DOI: 10.1039/c4cc06063f |
| This journal is © The Royal Society of Chemistry 2015 |