
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Live cell tracking of symmetry break in actin cytoskeleton triggered by abrupt changes in micromechanical environments†
S.
Inoue‡§
a,
V.
Frank§
a,
M.
Hörning§
b,
S.
Kaufmann
a,
H. Y.
Yoshikawa
c,
J. P.
Madsen
d,
A. L.
Lewis
e,
S. P.
Armes
d and
M.
Tanaka
*ab
aPhysical Chemistry of Biosystems, Institute of Physical Chemistry, University of Heidelberg, D69120 Heidelberg, Germany. E-mail: tanaka@uni-heidelberg.de
bInstitute for Integrated Cell-Material Sciences (WPI iCeMS), Kyoto University, Kyoto 606-8501, Japan
cDepartment of Chemistry, Saitama University, Saitama 338-8570, Japan
dDepartment of Chemistry, Dainton Building, University of Sheffield, Brook Hill, Sheffield, South Yorkshire S3 7HF, UK
eBiocompatibles UK Ltd, Lakeview, Riverside Way, Watchmoor Park, Camberley, Surrey GU15 3YL, UK
First published on 8th September 2015
Abstract
With the aid of stimulus-responsive hydrogel substrates composed of ABA triblock copolymer micelles, we monitored the morphological dynamics of myoblast (C2C12) cells in response to an abrupt change in the substrate elasticity by live cell imaging. The remodeling of actin cytoskeletons could be monitored by means of transient transfection with LifeAct-GFP. Dynamic changes in the orientational order of actin filaments were characterized by an order parameter, which enables one to generalize the mechanically induced actin cytoskeletons as a break of symmetry. The critical role that acto-myosin complexes play in the morphological transition was verified by the treatment of cells with myosin II inhibitor (blebbistatin) and the fluorescence localization of focal adhesion contacts. Such dynamically tunable hydrogels can be utilized as in vitro cellular micro-environments that can exert time-dependent stimuli to mechanically regulate target cells.
Introduction
There is compelling literature evidence that biological cells can sensitively detect not only biochemical stimuli, but also the mechanical properties of their microenvironment.1,2 To date, chemically cross-linked hydrogels have been widely utilized as artificial extracellular matrix (ECM) models.3 The substrate elasticity can be readily adjusted ex situ by varying the concentration of bifunctional cross-linker and the reaction time,4,5 hence such materials have been used to regulate morphology2,3,6 and motility7–9 of contractile cells, as well as lineage specific differentiation of mesenchymal stem cells (MSCs).10,11 However, recent in vivo studies (and also experiments using organ cultures) suggest that various cell behaviours during development and disease are correlated with dynamic changes in the stiffness of cellular microenvironments. For example, transplanted stem cells exhibited remarkably enhanced bone regeneration upon the degradation of alginate scaffolds.12 Moreover, Wolf et al. reported that tumor cells change their mode of migration according to the matrix density during metastasis.13 These findings inspired the design of ECM models whose mechanical properties can be altered in a time-dependent manner.14 Several recent studies demonstrated that the stiffness of thiolated ECMs based on hyaluronic acid15 or gelatin16,17 is enhanced by the formation of disulfide bonds and reduced on addition of disulfide cleaving reagents such as dithiothreitol. However, such materials have fundamental drawbacks: the kinetics of stiffening and softening cannot be easily fine-tuned, and it is not possible to reversibly switch the substrate stiffness.Previously, we proposed the use of physically cross-linked hydrogels composed of an inter-connected micellar network of ABA triblock copolymer chains comprising a biocompatible central B block of poly(2-(methacryloyloxy)ethyl phosphorylcholine) (PMPC, mean degree of polymerization, n = 250) and two outer A blocks of poly(2-(diisopropylamino)ethyl methacrylate) (PDPA, n = 50 for each). Since the PDPA block is a weak polycation with a variable degree of ionization at around physiological pH, (de)protonation of these chains alters the micelle structure. Therefore, the Young's gel modulus could be modulated by pH titration without any adverse effect on cellular functions. In our previous account, we demonstrated the reversible switching of morphology and adhesion strength of myoblast (C2C12) cells without compromising cell viability.18
In this study, we further extended this strategy to investigate the in situ spatio-temporal correlation between morphological dynamics and cytoskeletal remodeling. To quantitatively assess the extent of orientation of actin microfilaments, C2C12 cells were transiently tranfected by LifeAct-GFP. This enabled us to monitor the temporal evolution of the order parameter <S> of actin cytoskeletons19,20 from live cells that experience abrupt changes in substrate stiffness. The kinetics of non-equilibrium relaxation of <S> in response to an “elasticity jump” was correlated with three morphological parameters: maximum projected area, aspect ratio, and global contact angle.
Experimental section
Materials
Deionized water (Genpure, TKA Niederelbern, Germany) was used throughout this study. Unless stated otherwise, all other chemicals were purchased either from Sigma-Aldrich (Munich, Germany) or Carl Roth (Karlsruhe, Germany), and were used without further purification. The synthesis of the PDPA50-PMPC250-PDPA50 triblock copolymer (Mn = 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500; Mw/Mn = 1.43; as judged by aqueous gel permeation chromatography studies conducted at low pH using a series of near-monodisperse poly(2-vinylpyridine) calibration standards) was achieved by atom transfer radical polymerization (ATRP), as reported previously.21,22
500; Mw/Mn = 1.43; as judged by aqueous gel permeation chromatography studies conducted at low pH using a series of near-monodisperse poly(2-vinylpyridine) calibration standards) was achieved by atom transfer radical polymerization (ATRP), as reported previously.21,22
Cell culture and transfection
The mouse myoblast cell line (C2C12, <20 passages) purchased from DSMZ GmbH (Braunschweig, Germany) was maintained in polystyrene flasks in a 37 °C incubator, and cultured in RPMI-1640 media modified with HEPES (Sigma-Aldrich, Munich, Germany) supplemented with 10 wt% of fetal bovine serum (PAA laboratories, Cölbe, Germany) 100 U ml−1 penicillin and 100 μg ml−1 streptomycin (PAA laboratories).To visualize actin filaments, C2C12 (lineage <20 passages) was transfected with LifeAct-TagGFP2 (ibidi, Munich, Germany), a mammalian expression vector encoding LifeAct-TagGFP2 fusion protein, a 17 amino acid actin filament binding domain fused with eGFP. This vector was introduced by lipofection using Torpedo® lipofection reagent (ibidi, Munich, Germany) according to the manufacturer's protocol. Briefly, C2C12 cells were seeded at a density of 1 × 105 cells per μ-dish (ø = 35 mm) and cultured in RPMI-1640 media (PAA laboratories, Cölbe, Germany) supplemented with 10% of fetal bovine serum (PAA laboratories, Cölbe, Germany) one day before transfection. No antibiotics were included in the medium. Only samples with 90–100% confluency were used for transfection. For lipoplex formation, a DNA solution (1 μg in 100 μl PBS) was added to a solution of Torpedo® (3 μl Torpedo dissolved in 100 μl PBS) and incubated for 20 min at room temperature. Cells were transfected by incubation with the lipoplex solution for 24 h. C2C12 cells were seeded on PDPA50-PMPC250-PDPA50 hydrogels after mechanical detachment in order to avoid proteolytic degradation of extracellular adhesion proteins.
Sample preparation
Copolymer hydrogel films were prepared by spin-coating of a methanolic solution of PDPA50-PMPC250-PDPA50 (50 mg ml−1) for 60 s at 4000 rpm, resulting in a dry film thickness of 0.50 μm.18 Each sample was annealed at 80 °C under nitrogen flow for 1 h, dried in a vacuum oven at 80 °C for 48 h, and soaked in RPMI-1640 medium overnight to remove residual methanol. Prior to the cell seeding, the triblock copolymer hydrogel films were immersed overnight in RPMI-1640 media with a solution pH that was adjusted to 8.0. Then cells were seeded on top of the triblock copolymer hydrogel films in custom-made petri dishes with glass cover slip bottom (diameter 37 mm, thickness 0.17 mm), and cultured with a fresh medium with pH 8.0.Morphological analysis
Cell adhesion behavior on the triblock copolymer hydrogel film or other substrates was monitored using an Axioobserver (Carl Zeiss, Oberkochen, Germany) and a Perkin-Elmer Ultraview spinning disk confocal set-up mounted on an inverted microscope (Nikon, Düsseldorf, Germany). The maximum projected area A and the aspect ratio AR (the ratio between major and minor axis) of the adherent cells were calculated using Matlab (Mathworks).In addition, we measured the global contact angle Φ and cytoskeletal order parameter <S> (Fig. 3). The global contact angle was determined by a conventional sessile droplet assumption.23 The order parameter <S> was calculated using custom automated algorithms written in Matlab (Mathworks) as described previously.19,20 First, n = 15 elongated Laplace of Gaussian (eLoG) kernels were generated by convoluting a Laplacian filter

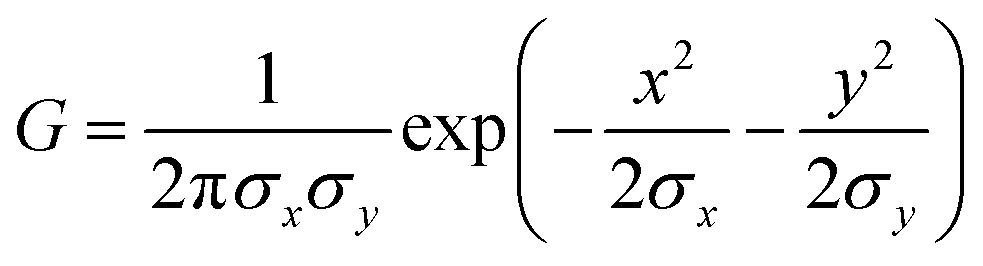
rotated from 0 to π–π/n in steps of π/n. The elongated shape of the Gaussian was fixed to σy = 3σx with σx = 2.0. For each pixel the maximum response of the n eLoG kernels convoluted with the original image was obtained and set to a single maximum response image,
| Imax(x, y) = max[eLoG(n) × I(x, y)]. |
I
max was processed by an intensity-threshold to yield the binary mask of the segmented stress fibers,24 and round bodies shorter than 50 pixels were removed. Finally, the order parameter, <S> = <cos2θ> was calculated from the histogram of pixel numbers multiplied by the corresponding fluorescence intensities and thus the local amount of actin filaments.
Inhibition of myosin II, localisation of focal adhesion
(−)-Blebbistatin (Sigma-Aldrich, Germany) was dissolved in DMSO at a concentration of 3.4 mM. The effects of myosin inhibition were studied by the incubation of (−)-blebbistatin at a final concentration of 20 μM. To avoid undesired interference with cell functions, the total volume concentration of DMSO in the cell culture medium was maintained at 0.006 wt%. To visualize focal adhesions, cells were fixed with 4.0 wt% paraformaldehyde (Riedel-de-Häen, Germany) and treated with 0.05 wt% Triton X 100 (Carl Roth, Karlsruhe, Germany) in PBS. After passivation with 1.0 wt% bovine serum albumin (BSA, Roth) in PBS, integrin β1 was labeled with rat anti-mouse CD29 antibody (1:50, BD Bioscience) followed by Alexa Fluor 488 goat anti-rat IgG (1:100, Life Technologies). Actin was stained with Atto-647 phalloidin (1:100, Sigma), and cell nuclei were stained with DAPI (1:500, Sigma).
Results and discussion
Fig. 1 represents (a) phase contrast and (b) fluorescence images of C2C12 cells on a glass substrate after transient transfection with LifeAct-GFP. An average transfection efficiency of about 45% was achieved by carefully optimizing the preparative parameters. The high transfection efficiency and strong fluorescence signals allow for the precise determination of orientational orders of actin cytoskeletons. | ||
| Fig. 1 (a) Phase contrast and (b) fluorescence images of live C2C12 cultured on a glass substrate after transient transfection with LifeAct-GFP. | ||
Fig. 2 represents confocal fluorescence images obtained for a live C2C12 cell experiencing an abrupt change in its substrate elasticity from E ∼ 40 kPa to 2 kPa. On a stiff substrate (E ∼ 40 kPa), the cell was significantly flattened, exhibiting a pronounced stress fiber formation. After the substrate elasticity was reduced to E ∼ 2 kPa, the cell began to change its shape at t ∼ 30 min. It should be noted that there was a lag time of 15 or 30 min before cells underwent morphological transitions, corresponding how fast the focal contacts (pinning centres) were detached. However, once the cells began to change their morphology, this process was complete within 10 min. In fact, we found that some cells even detached from the substrates as a result of the abrupt decrease in the contact area.
 | ||
| Fig. 2 Confocal fluorescence images obtained for a live C2C12 cell experiencing a dynamic change in its substrate elasticity from E ∼ 40 kPa to 2 kPa. After t ∼ 30 min the cell began to change its shape, reaching to the steady state in ∼10 min. | ||
To parameterize the dynamic response of C2C12 cells experiencing abrupt changes in micromechanical environments, we first assessed the maximum projection area A and aspect ratio AR at each time point. In this study, we also introduced two more quantitative measures, namely the global contact angle Φ and the cytoskeletal order parameter <S>.
Fig. 3 represents (a) side views and (b) top views of the reconstructed three-dimensional profiles of a live C2C12 cell before (t = 0 min, left panels) and after (t = 61 min, right panels) the morphological transition. As presented in Fig. 3a, the global shape of a cell could well be fitted according to a sessile drop approximation. The dynamic change in substrate elasticity from 40 kPa to 2 kPa led to a significant increase in the contact angle from Φ40 kPa = 29° to Φ2 kPa = 120°. Despite apparently good agreement on both stiff and soft substrates, it should be noted that the contact angle analysis used here is based on a crude assumption that the shape deformation of a cell is dominated by tension and is thus plastic. This is, strictly speaking, invalid for a biological cell membranes, because the mode of deformation is not only plastic but also elastic.25 In fact, the height profile of a lipid membrane in the vicinity of a contact substrate (h ≤ 100 nm) is dominated by its elasticity, which is characterized by a capillary length.26,27 However, it should be noted that the determination of the capillary length and thus the adhesion free energy is not possible from the fluorescence images, because the detected fluorescence signals originate from actin cytoskeletons but not from cell membranes. Therefore, the apparent contact angle was merely used as an indicator for the significance of cell deformation on hydrogel substrates. Fig. 3c exemplified how the order parameter <S> could be calculated. The image analysis with a series of elongated Laplace of Gaussian filters yields a pixel orientation map in which the color code coincides with the orientational distribution of actin filaments with respect to a major axis of a cell (θ = 0°, indicated by an arrow). Here, the order parameter can be determined from the pixel numbers multiplied by the corresponding pixel fluorescence intensities, reflecting the amount of actin filaments at each orientation. As indicated in both panels in Fig. 3c, a significant reduction in order parameter was observed from <S40 kPa> = 0.42 to <S2 kPa> = −0.07 by dynamic softening of the hydrogel substrate.
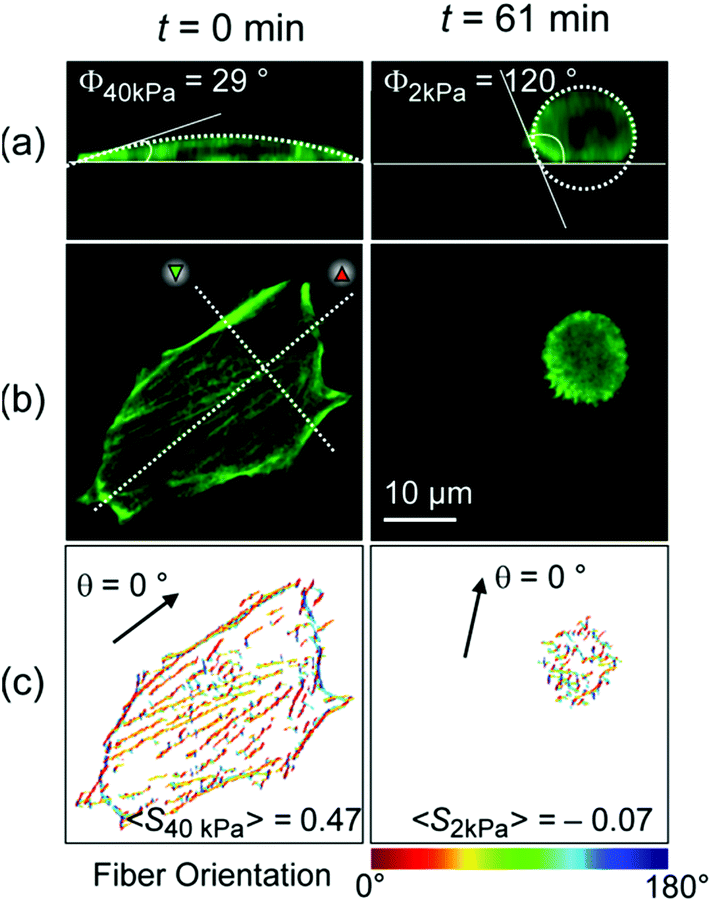 | ||
| Fig. 3 (a) Side and (b) top views of the reconstructed three-dimensional profiles of a live C2C12 cell before (t = 0 min, left panels) and after (t = 61 min, right panels) the morphological transition. (c) Pixel orientation maps used for the calculation of order parameter <S>. | ||
Fig. 4 represents the temporal changes in the projected area A, aspect ratio AR, contact angle Φ, and order parameter <S>. Green and red symbols in Fig. 4c correspond to the contact angle taken from red and green cross-sections in Fig. 3b, respectively. It is noteworthy that the changes in A, AR, and Φ commenced at approximately the same time point (t = 30 min). However, the changes in AR, A, and Φ happened within 4 min. This finding suggests that the cell loses tension and quickly reaches its steady state as soon as the focal adhesions (pinning centres) are detached. Remarkably, the orientational order parameter of actin filaments <S> exhibited a distinct delay (approx. 10 min) before it decreased rapidly. The combination of live cell imaging of cytoskeletons and dynamic mechanical cues given by stimulus-responsive hydrogels enables one to detect the kinetics of cytoskeletal remodelling in live cells for the first time. Such a discontinuous change in the order parameter of actin cytoskeletons observed here can be generalized as a symmetry break.
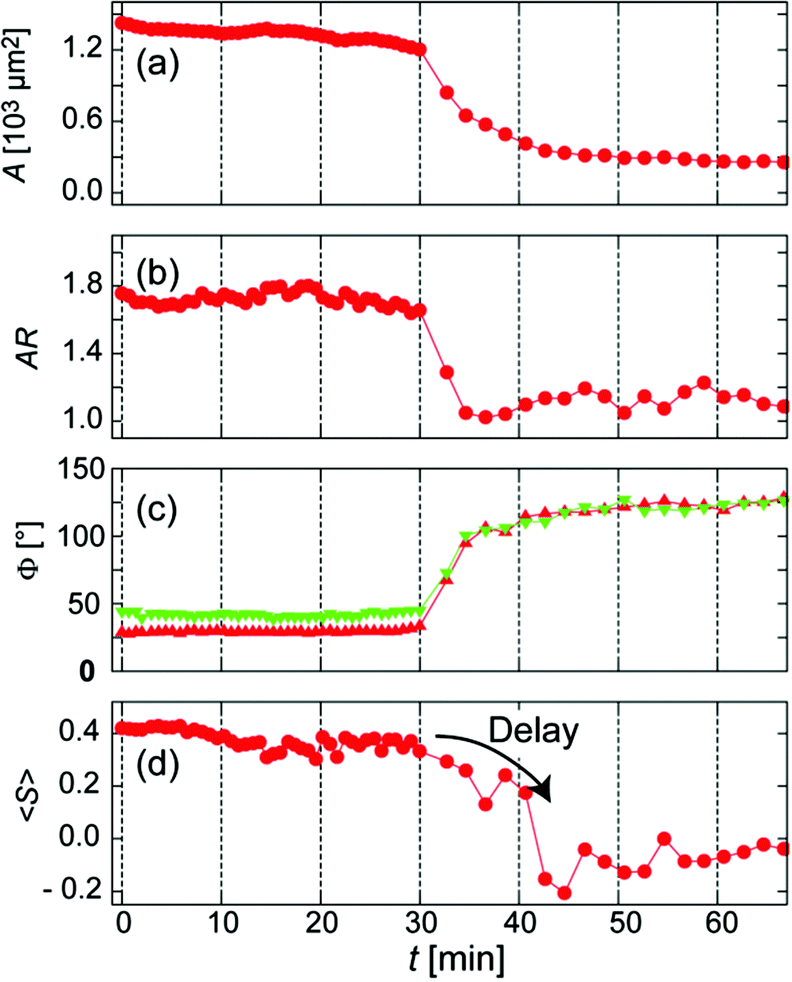 | ||
| Fig. 4 Temporal changes in (a) projected area A, (b) aspect ratio AR, (c) contact angle Φ, and (d) orientational order parameter of actin filaments <S>. Note that the change in <S> exhibited a clear delay (approx. 10 min). | ||
To further understand the relationship between loss of adhesion points and dynamic actin remodeling, we applied blebbistatin, which blocks the head group of myosin II.28,29Fig. 5 shows the temporal changes in cell morphology caused by addition of 20 μM blebbistatin. When a C2C12 cell placed on a stiff gel with E = 40 kPa (Fig. 5a) and was exposed to blebbistatin, we observed a drastic change in cell shape due to the loss of actomyosin complex (Fig. 5b). After t = 50 min, the cell attained its equilibrium shape. However, several pinning centres (indicated by arrows) could be identified even after the cell body changed to a rounded shape (Fig. 5b). In the presence of blebbistatin, the softening of the substrate does not lead to any detectable change due to the inhibition of binding of myosin II to actin filaments (Fig. 5c). The depletion of blebbistatin resulted in a tense, spindle-like shape, suggesting the recovery of actomyosin complexes (Fig. 5d). The clearly different cell morphology to the one presented in Fig. 2 can be attributed to the fact that the focal contacts (indicated by arrows) remained over time due to the abrupt disruption of mechano-induced signalling pathways between integrin and cytoskeleton. Since the binding between integrin and fibronectin was sustained, the re-polymerization of actin cytoskeletons occurred from the former contact points. Such a “memory” of initial contact points on stiff substrates causes a hysteresis in the morphological changes. Indeed, once the blebbistatin was added again (Fig. 5e), the cells take almost identical shape as Fig. 5c, suggesting that the effect of addition/depletion of blebbistatin is reproducible. Indeed, we increased the substrate elasticity to 40 kPa in the absence of blebbistatin and found a pronounced cell spreading, which confirms that blebbistatin does not cause any irreversible damage to cells (Fig. 5f).
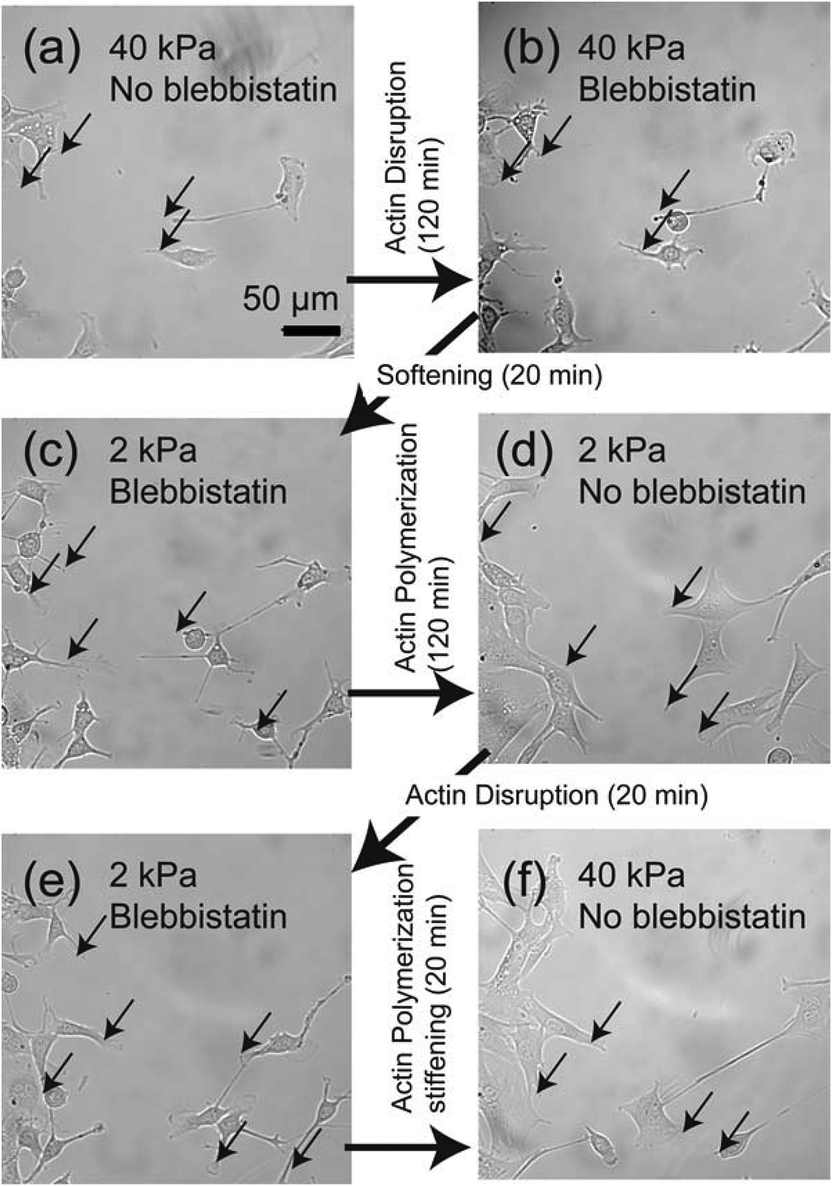 | ||
| Fig. 5 (a) Bright field image of C2C12 cells cultured on stiff gel (E = 40 kPa). (b) The cell underwent a clear morphological transition on the same substrate (E = 40 kPa) after incubation with myosin blocker (blebbistatin, 20 μM). (c) Softening of the substrate (E = 2 kPa) in the presence of blebbistatin did not result in a major morphological change. The reproducibility of the morphological response was confirmed by (d) the depletion of blebbistatin and (e) re-incubation with blebbistatin on the soft substrate (E = 2 kPa). (f) A significant spreading of cells could be observed upon the depletion of blebbistatin and increase in the substrate elasticity to E = 40 kPa. Black arrows indicate the remaining contact points (pinning centres) that causes a remarkable hysteresis in morphological changes. | ||
In fact, we found a larger number of more concentrated focal adhesion contacts in the cells cultured on stiff substrates (Fig. 6a), compared to those on soft substrates (Fig. 6b). The clear difference in the density of focal adhesion contacts support our hypothesis that the softening of substrates destabilizes and reduces pinning centres, as reported by ex situ experiments on chemically cross-linked polyacrylamide gels.9 This leads to a release of adhesion-induced tension that quickly alters the contact area A (Fig. 4a), the projected cell shape from top (AR, Fig. 4b) and side views (Φ, Fig. 4c). This may explain the distinct phase delay of the orientational order parameter of actin filaments <S>, as the depolymerisation of actin filaments begins only after cancellation of focal adhesion. In contrast, the abrupt disruption of acto-myosin complexes with blebbistatin did not cancel the focal contacts established on stiff substrates (Fig. 5b, indicated by arrows), which results in a distinct hysteresis in morphological dynamics (Fig. 5d).
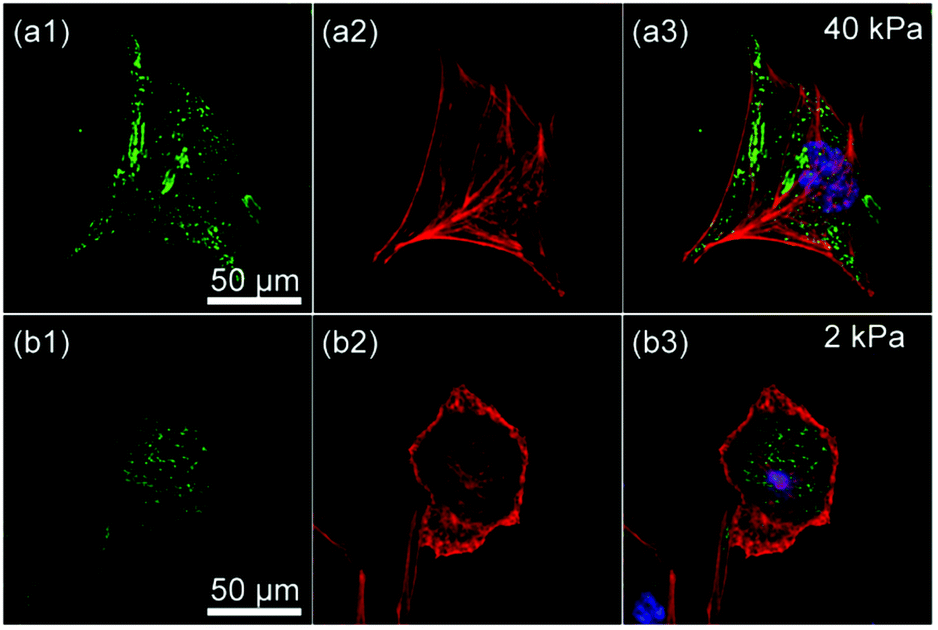 | ||
| Fig. 6 Confocal fluorescence images of C2C12 on substrates with (a) E = 40 kPa and (b) E = 2 kPa showing (1) integrin β1 (green), (2) actin (red), and (3) overlay with nuclei (blue). | ||
Conclusions
The combination of stimulus-responsive copolymer hydrogels and the transient transfection with LifeAct-GFP enabled us to monitor the dynamic response of myoblast cells experiencing an abrupt change in micromechanical environments. With aid of multi-parameter tracking of live cells and the combination with chemical myosin inhibitor (blebbistatin), we could characterize various dynamic processes, such as release of tension by cancellation of focal adhesions (AR), a reduction in the area of cell-substrate contacts (A), and changes in the global height profile (Φ). The live cell image analysis of actin cytoskeletons enabled us to identify the discontinuous change in orientational order parameter <S> (symmetry break) for the first time. Such dynamically tunable copolymer hydrogels can be used as a new material for in vitro cellular micro-environments that can exert time-dependent mechanical commands to the target cells.Acknowledgements
The authors thank H. Umeshima, M. Kengaku, and R. Medda for helpful suggestions, and C. Ackermann (Nikon Imaging Centre Heidelberg) for technical support. M.T. thanks the German Science Foundation (DFG CRC873 B07), Kyoto University SPRITS Program, Sumitomo Foundation, EU FP7 “ActiveSoft”, MEXT (No. 26103521), and JSPS (No. 26247070). S.P.A. thanks EPSRC for a Platform grant EP/J007846/1. M.T. is a member of German Excellence Cluster “CellNetwork”. iCeMS is supported by World Premier International Research Centre Initiative (WPI), MEXT, Japan. The authors declare that they have no competing financial interests.References
- V. Vogel and M. Sheetz, Nat. Rev. Mol. Cell Biol., 2006, 7, 265–275 CrossRef CAS PubMed.
- D. E. Discher, P. Janmey and Y. L. Wang, Science, 2005, 310, 1139–1143 CrossRef CAS PubMed.
- A. Engler, L. Bacakova, C. Newman, A. Hategan, M. Griffin and D. Discher, Biophys. J., 2004, 86, 617–628 CrossRef CAS.
- S. Kidoaki and T. Matsuda, J. Biotechnol., 2008, 133, 225–230 CrossRef CAS PubMed.
- J. H. Wen, L. G. Vincent, A. Fuhrmann, Y. S. Choi, K. C. Hribar, H. Taylor-Weiner, S. Chen and A. J. Engler, Nat. Mater., 2014, 13, 979–987 CrossRef CAS PubMed.
- P. C. Georges, W. J. Miller, D. F. Meaney, E. S. Sawyer and P. A. Janmey, Biophys. J., 2006, 90, 3012–3018 CrossRef CAS PubMed.
- C. M. Lo, H. B. Wang, M. Dembo and Y. L. Wang, Biophys. J., 2000, 79, 144–152 CrossRef CAS.
- D. S. Gray, J. Tien and C. S. Chen, J. Biomed. Mater. Res., Part A, 2003, 66A, 605–614 CrossRef CAS PubMed.
- R. J. Pelham and Y.-L. Wang, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 13661–13665 CrossRef CAS.
- J. P. Winer, P. A. Janmey, M. E. McCormick and M. Funaki, Tissue Eng., Part A, 2008, 15, 147–154 CrossRef PubMed.
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126, 677–689 CrossRef CAS PubMed.
- C. A. Simmons, E. Alsberg, S. Hsiong, W. J. Kim and D. J. Mooney, Bone, 2004, 35, 562–569 CrossRef CAS PubMed.
- K. Wolf, I. Mazo, H. Leung, K. Engelke, U. H. von Andrian, E. I. Deryugina, A. Y. Strongin, E. B. Brocker and P. Friedl, J. Cell Biol., 2003, 160, 267–277 CrossRef CAS PubMed.
- J. A. Burdick and W. L. Murphy, Nat. Commun., 2012, 3, 1269 CrossRef PubMed.
- X. Z. Shu, Y. C. Liu, Y. Luo, M. C. Roberts and G. D. Prestwich, Biomacromolecules, 2002, 3, 1304–1311 CrossRef CAS PubMed.
- M. Hörning, S. Kidoaki, T. Kawano and K. Yoshikawa, Biophys. J., 2012, 102, 379–387 CrossRef PubMed.
- X. Z. Shu, S. Ahmad, Y. C. Liu and G. D. Prestwich, J. Biomed. Mater. Res., Part A, 2006, 79A, 902–912 CrossRef CAS PubMed.
- H. Y. Yoshikawa, F. F. Rossetti, S. Kaufmann, T. Kaindl, J. Madsen, U. Engel, A. L. Lewis, S. P. Armes and M. Tanaka, J. Am. Chem. Soc., 2011, 133, 1367–1374 CrossRef CAS PubMed.
- A. Zemel, F. Rehfeldt, A. E. X. Brown, D. E. Discher and S. A. Safran, Nat. Phys., 2010, 6, 468–473 CrossRef CAS PubMed.
- H. Y. Yoshikawa, T. Kawano, T. Matsuda, S. Kidoaki and M. Tanaka, J. Phys. Chem. B, 2013, 117, 4081–4088 CrossRef CAS PubMed.
- Y. H. Ma, Y. Q. Tang, N. C. Billingham, S. P. Armes and A. L. Lewis, Biomacromolecules, 2003, 4, 864–868 CrossRef CAS PubMed.
- J. Madsen, S. P. Armes, K. Bertal, H. Lomas, S. MacNeil and A. L. Lewis, Biomacromolecules, 2008, 9, 2265–2275 CrossRef CAS PubMed.
- A. W. Adamson, Physical Chemistry of Surfaces, Wiley, 1990 Search PubMed.
- N. Otsu, IEEE Trans. Syst. Man Cybern., 1979, 9, 62–66 CrossRef.
- R. Simson, E. Wallraff, J. Faix, J. Niewöhner, G. Gerisch and E. Sackmann, Biophys. J., 1998, 74, 514–522 CrossRef CAS.
- R. Bruinsma, in Proceedings of the NATO Advanced Institute of Physics and Biomaterials, NATO Advanced Study Institute Series B: Physics, Kluwer, London, 1995 Search PubMed.
- S. Gönnenwein, M. Tanaka, B. Hu, L. Moroder and E. Sackmann, Biophys. J., 2003, 48, 646–655 CrossRef.
- M. Kovács, J. Tóth, C. Hetényi, A. Málnási-Csizmadia and J. R. Sellers, J. Biol. Chem., 2004, 279, 35557–35563 CrossRef PubMed.
- J. S. Allingham, R. Smith and I. Rayment, Nat. Struct. Mol. Biol., 2005, 12, 378–379 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5bm00205b |
| ‡ Present Address: Global R&D, Kao Corporation, Wakayama 640-8580, Japan. |
| § These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |