
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Systemic in vivo delivery of siRNA to tumours using combination of polyethyleneimine and transferrin–polyethyleneimine conjugates†‡
Anna M.
Grabowska
*a,
Ralf
Kircheis
b,
Rajendra
Kumari
c,
Philip
Clarke
a,
Andrew
McKenzie
c,
Jaime
Hughes
a,
Cerys
Mayne
a,
Arpan
Desai
d,
Luana
Sasso
d,
Susan A.
Watson
a and
Cameron
Alexander
*d
aCancer Biology, Division of Cancer and Stem Cells, University of Nottingham, NG7 2UH, UK. E-mail: anna.grabowska@nottingham.ac.uk; Fax: +44 (0)1158 231137; Tel: +44 (0)1158 231135
bVirologik GmbH, Erlangen, Germany
cPRECOS Ltd, Loughborough, UK
dSchool of Pharmacy, University of Nottingham, NG7 2RD, UK. E-mail: cameron.alexander@nottingham.ac.uk
First published on 9th September 2015
Abstract
Materials for delivery of oligonucleotides need to be simple to produce yet effective in vivo to be considered for clinical applications. Formulations of biomaterials based on combinations of existing demonstrated polymeric gene carriers with targeted derivatives are potential candidates for rapid translation but have not been fully explored for siRNA applications. Here we investigated formulations based on derivatised PEI for delivery of siRNA to gastrointestinal cancer cells. siRNA was complexed with linear PEI alone or with a mixture of linear PEI and transferrin-conjugated branched PEI (TfPEI), and knockdown of reporter genes was investigated. Overall, the in vitro use of complexes containing TfPEI resulted in up to 93% knockdown at 72 h post-transfection. Sustained knockdown was also achieved in a bioluminescent xenograft model. When complexes were delivered intratumorally, a 43% reduction in luminescence was achieved in the treated group compared with the control group 48 h after treatment. For systemic administration, only the intraperitoneal route, and not the intravenous route was effective, with 49% knockdown achieved at 72 h and sustained up to 144 h (44%) after a single administration of TfPEI-complexed siRNA. No toxicity or induction of the interferon response was observed. These findings demonstrate that simple formulations of transferrin-conjugated PEI with a ‘parent’ polymer such as linear PEI have potential as a method for therapeutic delivery of siRNA when administered either intratumorally or systemically.
Introduction
Small interfering RNAs (siRNAs) are a powerful tool for down-regulation of gene expression in mammalian cells.1 Oligonucleotides of this type can be designed for selective targeting of specific mRNAs, leading to their cleavage and degradation, by the RNA-induced silencing complex (RISC). Cellular recycling of the RISC enables targeting of further mRNAs, making the overall gene silencing process highly efficient. These factors, together with the high sequence specificity of siRNAs, means they have potential as therapeutics,2 and in particular, as anti-cancer agents for downregulation of oncogenes.3Whilst in vitro delivery of siRNA can be readily achieved, in vivo delivery has proved more difficult.4 For treatment of cancer, systemic delivery of siRNA is required to target distant metastases as well as primary tumours but potential losses via the kidney and liver or through degradation must be avoided. Delivery of uncomplexed siRNA has been achieved through the use of hydrodynamic injection or attachment of cell-targeting ligands5,6 but neither provides a complete solution.7 Alternatively, siRNAs can be complexed with macromolecules to protect them from degradative enzymes and increase cellular uptake,8 but liposomes, for example, activate the innate immune response and result in toxicity when used systemically.9
The polycation polyethyleneimine (PEI) has been successfully used for DNA delivery10,11 and protects siRNA from serum-associated enzymes. Multiple protonatable amine groups make PEI efficient in condensing DNA by electrostatic interactions and separation of nitrogen atoms by a 2 carbon spacer along the polymer backbone modulates the overall basicity. The resulting strong pH buffering capacity of linear and branched PEI has been suggested to enhance endosomal escape leading to an efficient release of DNA complexes into the cytoplasm.12 Linear and branched PEI can be synthesised and different molar mass forms are readily available; linear and low molecular weight PEI have both been associated with lower toxicity13,14 and higher efficiency of DNA delivery.15,16 In addition, PEI can be readily functionalised,17,18 as the presence of primary amines enables standard bioconjugation chemistries to be used to introduce targeting ligands, self-assembly inducers and steric shielding groups. These functionalities can enhance circulation times and specific tissue accumulation in vivo, although there is an inevitable trade-off between introduction of a targeting functionality at PEI-amine groups and loss of binding affinity and buffering capacity. In addition, for a practical pharmaceutical application, there is a need to make use of expensive or delicate functional affinity ligands to the minimum level associated with effective targeting. Therefore, despite the many potential advantages of PEI as an oligonucleotide delivery system, the inherent lack of targeting of the unsubstituted parent polycation and the balances of the introduced functionality, oligonucleotide binding ability, buffering capacity and toxicity have resulted in rather limited in vivo studies of this polymer as a carrier of siRNA.19–21
Accordingly, for this study we aimed at evaluating if a ‘minimal-functionalized’ PEI formulation could be made to enable cancer cell targeting without systemic toxicity, while retaining strong RNA binding during transit in vivo to maintain the therapeutic efficacy. Here we describe the use of a simple linear PEI co-formulated with a ligand-conjugated branched PEI for delivery of siRNA to gastrointestinal (GI) cancer cells in vitro and in vivo. For these studies we utilised an siRNA sequence for knockdown of luciferase in bioluminescent xenograft models, and transferrin as the ligand for cancer cell targeting. The receptor for transferrin is expressed on proliferating cells22 and has been used for delivery of a range of therapeutic agents23 to cancer cells, including siRNA.24 Incorporation of transferrin-conjugated PEI into complexes has also been shown to improve DNA delivery to tumour cells;25,26 the mechanism may involve shielding the positive charge on PEI–DNA complexes in vivo, reducing accumulation in ‘first pass’ organs and enabling them to reach the target tissues including tumours.27 However, while there have been several in vitro and in vivo studies of Tf-conjugated PEI alone with siRNA,20,21 the combination of the linear 22k PEI with branched PEI conjugated to Tf had not been evaluated prior to this study. Therefore, as a first step towards a low cost targeted siRNA formulation, a mixture of free, and transferrin-conjugated PEI, was assessed for systemic delivery of siRNA to cancer cells in vivo.
Methods
Cells
Bioluminescent HCT116 (colorectal, ECACC ref. no. 91091005) and MGLVA1 (gastric, ascitic variant of MKN 45G28) cells, stably transfected with firefly luciferase (pORF-LucSh-CpG, InvivoGen, Toulouse, France) were routinely cultured in RPMI1640 culture medium (Gibco, Paisley, UK) containing 10% (v/v) heat inactivated foetal bovine serum (FBS, Sigma, Poole, UK) at 37 °C in 5% CO2 and under humidified conditions.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio.25 Linear PEI (22 kDa) was obtained from MBI-Fermentas (St. Leon-Rot, Germany); this form of PEI was chosen based on the better in vivo properties observed when PEI mixed with TfPEI was used for DNA delivery previously.27
:P ratio are given in each figure for each of the individual conditions tested. The amounts used for the in vivo work are in the Materials and methods section ‘In vivo siRNA delivery’. The appropriate quantity of siRNA was diluted to 50 μl in Opti-MEM1 (Gibco, Paisley, UK). The required amount of linear PEI and transferrin-conjugated PEI (TfPEI), when used, was thawed, vortexed, mixed in the required ratio, diluted to 50 μl in Opti-MEM1, added to the nucleic acid and immediately mixed by repeated pipetting. Following incubation at room temperature for 20 min, the complexes were added to cells and cells were returned to the incubator. Each experiment was repeated on at least two occasions and the representative data are shown in the figures. For competitive experiments, free transferrin (Sigma) was added to the cells immediately prior to addition of the transfection mix in a range of concentrations between 500 and 10 μg ml−1.
:1 molar ratio.25 Linear PEI (22 kDa) was obtained from MBI-Fermentas (St. Leon-Rot, Germany); this form of PEI was chosen based on the better in vivo properties observed when PEI mixed with TfPEI was used for DNA delivery previously.27
:P ratio are given in each figure for each of the individual conditions tested. The amounts used for the in vivo work are in the Materials and methods section ‘In vivo siRNA delivery’. The appropriate quantity of siRNA was diluted to 50 μl in Opti-MEM1 (Gibco, Paisley, UK). The required amount of linear PEI and transferrin-conjugated PEI (TfPEI), when used, was thawed, vortexed, mixed in the required ratio, diluted to 50 μl in Opti-MEM1, added to the nucleic acid and immediately mixed by repeated pipetting. Following incubation at room temperature for 20 min, the complexes were added to cells and cells were returned to the incubator. Each experiment was repeated on at least two occasions and the representative data are shown in the figures. For competitive experiments, free transferrin (Sigma) was added to the cells immediately prior to addition of the transfection mix in a range of concentrations between 500 and 10 μg ml−1.
Mice were divided into groups, to be injected with a luciferase or control siRNA. For the intratumoral route of injection, 20 μg of the appropriate siRNA was complexed with a 1:4 mixture of TfPEI and PEI as described above but glucose was added to a final concentration of 3%, in a total volume of 50 μl and the complexed siRNA was injected into two sites within each tumour. For systemic delivery, single intraperitoneal injections of 50 μg of siRNA in a final volume of 200 μl were used. The mice were imaged again for bioluminescence at timepoints between 24 and 144 h after administration as indicated; bioluminescence post-treatment was expressed as a percentage of bioluminescence prior to siRNA injection.
:biotin blocking kit (Vector Labs, UK) using a concentration matched rat IgG2aκ (BD Pharmingen, UK) as a control. Primary labelling was visualised with biotinylated goat anti-rat secondary antibody (BD Pharmingen, UK), followed by a streptavidin binding complex (Vector Labs Ltd, UK) and diaminobenzidine tetrahydrochloride chromogen (Dakocytomation, UK). Following counterstaining with Mayer's Haemalum, sections were coverslipped using DPX. The microvessel density was analysed by assessment of vessel hotspots across the surface area of the section at 10× magnification.
Results
Characterisation of complexes
The biological activity of polymer/oligonucleotide polyelectrolyte complexes is strongly dependent on the particle size charge and method of preparation.32 Previous studies on Tf–PEI complexes with DNA prepared at a Tf:PEI25 molar ratio of 1:1 had shown greater transfection efficiency for particles of diameters of <200 nm and positive zeta potentials,27 thus we utilised Tf:PEI at a 1:1 molar ratio for the targeted siRNA delivery systems. Characterisation of siRNA complexes prepared using mixtures of TfPEI:PEI (1:4) and TfPEI:PEI (1:15) at N/P ratios of 8 and 12 was thus carried out using dynamic light scattering in comparison with PEI complexes (Table 1). The particle size distributions of PEI complexes were well-defined with hydrodynamic diameters of 20 or 45 nm depending on the N:P ratio, based on the number distributions of particles sizes calculated from scattered light intensities. Inspection of correlation functions and calculated particle sizes from intensity distribution data revealed the presence of weakly-scattering species in the PEI complexes with hydrodynamic diameters of <10 nm. Features in this size range were observed in the absence of siRNA, suggesting that these signals most likely corresponded to free PEI. Use of TfPEI:PEI (1:4) also resulted in particles in the 20–40 nm diameter range, with essentially all of the particulate content being of these sizes by number distribution, although the presence of larger aggregates was apparent in the scattered light intensity distributions. These variations in the apparent size were expected for DLS intensity distributions of heterogeneous particle populations, owing to the sixth power dependency of scattered light with the particle diameter. There was no detectable free polymer–protein conjugate (i.e. TfPEI in the presence of the polyplexes) and nor were signals corresponding to free PEI observed. For non-PEI complexes, increased N/P ratios decreased the observed hydrodynamic diameters, implying better condensation of the oligonucleotide with increased polycation. By contrast, for TfPEI–siRNA complexes at TfPEI:PEI of 1:4, increased N/P ratios resulted in an apparent increase in the sizes of the most abundant particles, which may have reflected the increased content of Tf (which has an isoelectric point of 5.2–5.5 dependent on pH) and the resulting potential for interparticle association between polycation and negative domains on the Tf surface. However, the differences in the sizes of PEI and TfPEI:PEI (1:4) complexes over this N/P range (i.e. ∼10 nm for both N/P 8 and 12) were considered unlikely to result in any major changes in biological behaviour, for example in cell uptake pathways, based on these size variations alone. The TfPEI:PEI–siRNA complexes at N:P 8 and N:P 12 were of similar size distributions to the PEI:siRNA complexes, indicating the likely predominance of the unconjugated PEI in the physicochemical behaviour of these species.
| Polycation | N/P | D H (number distribution ± SD)/nm | D H (intensity distribution ± SD)/nm |
|---|---|---|---|
| Figures in parentheses are the percentage of the sample population. | |||
| PEI | 8 | 45 ± 2 (100) | 48 ± 2 (6), 204 ± 23 (89), >1 μm (5) |
| PEI | 12 | 24 ± 1 (100) | 22 ± 1 (1), 270 ± 27 (87), >1 μm (12) |
| TfPEI:PEI (1:4) |
8 | 20 ± 1 (100) | 20 ± 1 (2), 300 ± 31 (43), 462 ± 88 (27), >1 μm (28) |
| TfPEI:PEI (1:4) |
12 | 38 ± 2 (100) | 44 ± 3 (6), 276 ± 40 (88), >1 μm (6) |
| TfPEI:PEI (1:15) |
8 | 23 ± 9 (100) | 30 ± 14 (29), 136 ± 60 (68), >1 μm (2) |
| TfPEI:PEI (1:15) |
12 | 21 ± 12 | 36 ± 9 (63), 274 ± 92 (35), aggregates (2) |
Complexes obtained with an N/P ratio of 8 were also characterised in terms of their surface charge (Table 2). PEI complexes exhibited significantly higher mean zeta potentials (24 mV) compared to TfPEI:PEI (1:4) complexes at the same N/P ratio (zeta potential of 17 mV, p < 0.01). These data were in accord with those reported previously for Tf–PEI/DNA complexes,33 and suggested a partial shielding of the high surface charge of PEI in the polyelectrolyte complexes, even at high N:P ratios, by the attached transferrin (molar mass 80 kDa, pI = 5.5–5.8, dependent on the Fe content).
Uptake of TfPEI![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :PEI-conjugated siRNA
:PEI-conjugated siRNA
Initial in vitro experiments investigated the effects of different amounts of transferrin within the PEI–siRNA complexes on the uptake of fluorescently-labelled siRNA by HCT116 cells, chosen as a model cell line well-characterised for expression of the transferrin receptor, TfR2.34,35 Cells were transfected with complexes containing 1:15 mixtures of TfPEI:PEI, in a range of N/P ratios, and analysed by flow cytometry at 24 h. Transfection efficiencies, assessed as mean fluorescence, for complexes containing transferrin were similar to those obtained by using PEI alone. There was an increase in fluorescence at the N/P ratios of 8 and 16 compared with cells treated with no siRNA which was significant when 2 μg siRNA was used (Fig. 1a, p < 0.05 and p < 0.01 for PEI alone and TfPEI:PEI ratio of 1:15) and a decrease when the N/P ratio was further increased. Cells transfected with fluorescent siRNA were also examined microscopically 1, 4 and 24 h after transfection. In cells transfected using PEI only, at the earlier time-point, a positive signal was observed only in a proportion of the cells, and in positive cells, the fluorescence was localised to vesicles which were widely distributed in the cytoplasm. When TfPEI was included in the complex, almost all cells were fluorescent at 1 h after transfection. By 4 h, in cells treated with PEI alone, the fluorescence was clustered into a single area of the cell outside the nucleus, similar to the pattern in cells treated with complexes containing TfPEI. By 24 h, the fluorescence signals were reduced with all treatments, with the strongest reduction found when PEI alone was used. The representative images are shown in Fig. 1b. These data together show that despite the reduced protonatable nitrogen content of PEI following conjugation of transferrin, the overall uptake of PEI–siRNA complexes into this cell line was not diminished. In turn, this implied that the loss of non-specific charge-mediated uptake through Tf conjugation was at least partially offset by receptor-mediated endocytosis, even when the TfPEI:PEI ratio was as low as 1:15.
 | ||
| Fig. 1 Uptake of PEI-conjugated siRNA by gastrointestinal cancer cells. In (a) mean fluorescence is shown 24 h after transfection of HCT116 cells with 1 or 2 μg of TAMRA-labelled siRNA using PEI alone or transferrin-conjugated PEI (TfPEI) mixed with PEI in a ratio of 1:15 TfPEI:PEI. * and ** indicates a significant increase (p < 0.05 and p < 0.001 respectively) in fluorescence compared with cells treated with a transfection reagent alone (one way ANOVA with the Bonferroni multiple comparison post-test). In (b) fluorescence microscopy images are shown of cells at 1, 4 or 24 h after transfection with 2 μg TAMRA-labelled siRNA complexed with PEI alone, or TfPEI:PEI (1:4 or 1:15 mixture, at N/P of 8). | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :PEI-conjugated siRNA in gastrointestinal cancer cells.
We next investigated siRNA-mediated knockdown in vitro using TfPEI:PEI complexed siRNA. A luciferase siRNA, in complexes containing TfPEI (in a ratio of 1:15 or 1:4) in a range of N/P ratios, was transfected into HCT116 and MGLVA1 cells (chosen as an established gastric cancer cell line in vitro and in vivo)36 stably expressing luciferase and luciferase activity measured at d3. The initial experiments demonstrated that TfPEI:PEI complexes with siRNA were well-tolerated by both cell lines, with no significant loss in metabolic activity as measured by MTT assays in HCT-116 cells, and less than 20% loss in activity in MGLVA-1 cells after 24 h (Fig. S1, ESI†). Knockdown of approximately 60% was achieved in HCT116 cells at either TfPEI:PEI ratio, and knockdown was generally maintained at d3 (Fig. 2c). In MGLVA1 cells, there was delayed knockdown, especially at TfPEI:PEI (1:15) but knockdown of nearly 90% could be achieved by d3. Overall, maximal, sustained knockdown was achieved at TfPEI:PEI (1:4) using N/P 8–12.
:PEI-conjugated siRNA in gastrointestinal cancer cells.
We next investigated siRNA-mediated knockdown in vitro using TfPEI:PEI complexed siRNA. A luciferase siRNA, in complexes containing TfPEI (in a ratio of 1:15 or 1:4) in a range of N/P ratios, was transfected into HCT116 and MGLVA1 cells (chosen as an established gastric cancer cell line in vitro and in vivo)36 stably expressing luciferase and luciferase activity measured at d3. The initial experiments demonstrated that TfPEI:PEI complexes with siRNA were well-tolerated by both cell lines, with no significant loss in metabolic activity as measured by MTT assays in HCT-116 cells, and less than 20% loss in activity in MGLVA-1 cells after 24 h (Fig. S1, ESI†). Knockdown of approximately 60% was achieved in HCT116 cells at either TfPEI:PEI ratio, and knockdown was generally maintained at d3 (Fig. 2c). In MGLVA1 cells, there was delayed knockdown, especially at TfPEI:PEI (1:15) but knockdown of nearly 90% could be achieved by d3. Overall, maximal, sustained knockdown was achieved at TfPEI:PEI (1:4) using N/P 8–12.
 | ||
| Fig. 2 Knockdown of luciferase activity using TfPEI:PEI siRNA complexes. Percentage knockdown of luciferase activity achieved 24 h (a, b) and 72 h (c, d) after transfection of HCT116 (a, c) or MGLVA1 cells (b, d) using a luciferase-specific siRNA complexed with transferrin-conjugated PEI (TfPEI) mixed with PEI in a ratio of 1:4 or 1:15 TfPEI:PEI relative to a control siRNA. Inhibition of knockdown by free transferrin in MGLVA1 cells transfected with 2 μg 1:4 TfPEI:PEI and 2 μg of siRNA and transfection reagent (e). * indicates concentrations of transferrin that gave significant knockdown relative to ‘no transferrin’ control (p < 0.05). | ||
Under these conditions, we observed a small but significant inhibition of knockdown when free transferrin was added to the cells immediately before transfection at concentrations of 500 or 100 μg ml−1 suggesting that at least a part of the uptake is mediated via transferrin receptors (Fig. 2e).
In vivo delivery of siRNA using TfPEI–PEI complexes
:PEI complexed luciferase siRNA was investigated in an in vivo bioluminescent MGLVA1 subcutaneous tumour. These tumours showed good vascularisation and strong staining for transferrin receptors (Fig. S2†). A dose of 20 μg of luciferase or control siRNA complexed TfPEI:PEI (1:4) in an N/P ratio of 8, which had given sustained knockdown in MGLVA1 cells in vitro, was initially administered directly into each tumour and bioluminescence was monitored over 48 h. The representative images of tumours from both groups are shown in Fig. 3a. Bioluminescence at each time-point was expressed as a percentage of the initial bioluminescence (24 h prior to treatment, Fig. 3b). Over the whole experiment, taking all timepoints into consideration, there was a significant difference between the bioluminescence in the two groups (p = 0.005, two-way ANOVA). In the control siRNA group, luminescence increased over this period (+29%), reflecting continued tumour growth, while in the luciferase siRNA group, luminescence levelled out at 24 h post-injection, then decreased by 48 h (32% decrease), equating to 47.3% lower luminescence in the treated group compared with the control group at 48 h (p < 0.05, one-way ANOVA with Bonferroni correction).
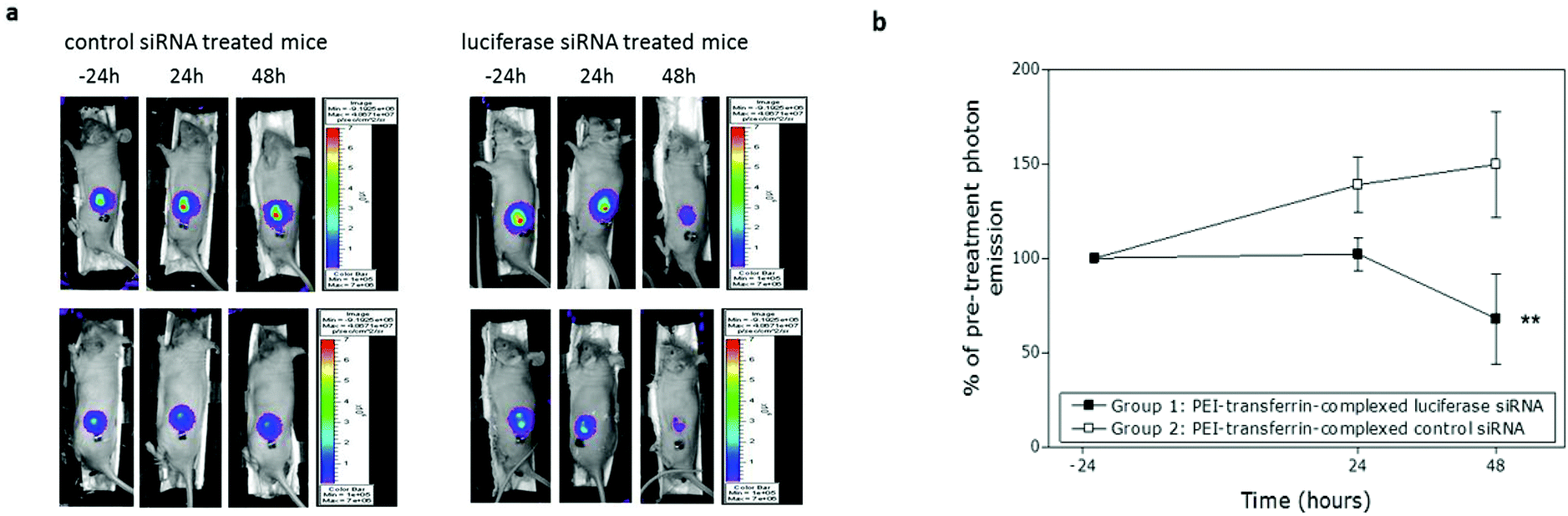 | ||
| Fig. 3 siRNA knock-down in vivo using the intratumoral route. MGLVA1 cells expressing luciferase were used to establish sub-cutaneous xenografts in nude mice and imaged 24 h before, and 24 and 48 h after intratumoral injection of a luciferase or control siRNA complexed with TfPEI:PEI in a ratio of 1:4 and an N/P ratio of 8. (a) Representative images of 2 mice treated with a luciferase or control siRNA taken 24 h before, 24 h after or 48 h after treatment. (b) Percentage change in bioluminescence at 24 and 48 h post-injection. A significant reduction in bioluminescence was observed in the luciferase siRNA-treated group (** indicates p = 0.005, 2-way ANOVA, n = 6) relative to the control siRNA-treated group (n = 7) over the whole course of the experiment, and a significant reduction in the luciferase siRNA compared with the control siRNA-treated group at 48 h (p < 0.05, one-way ANOVA with Bonferroni correction) but not at 24 h. | ||
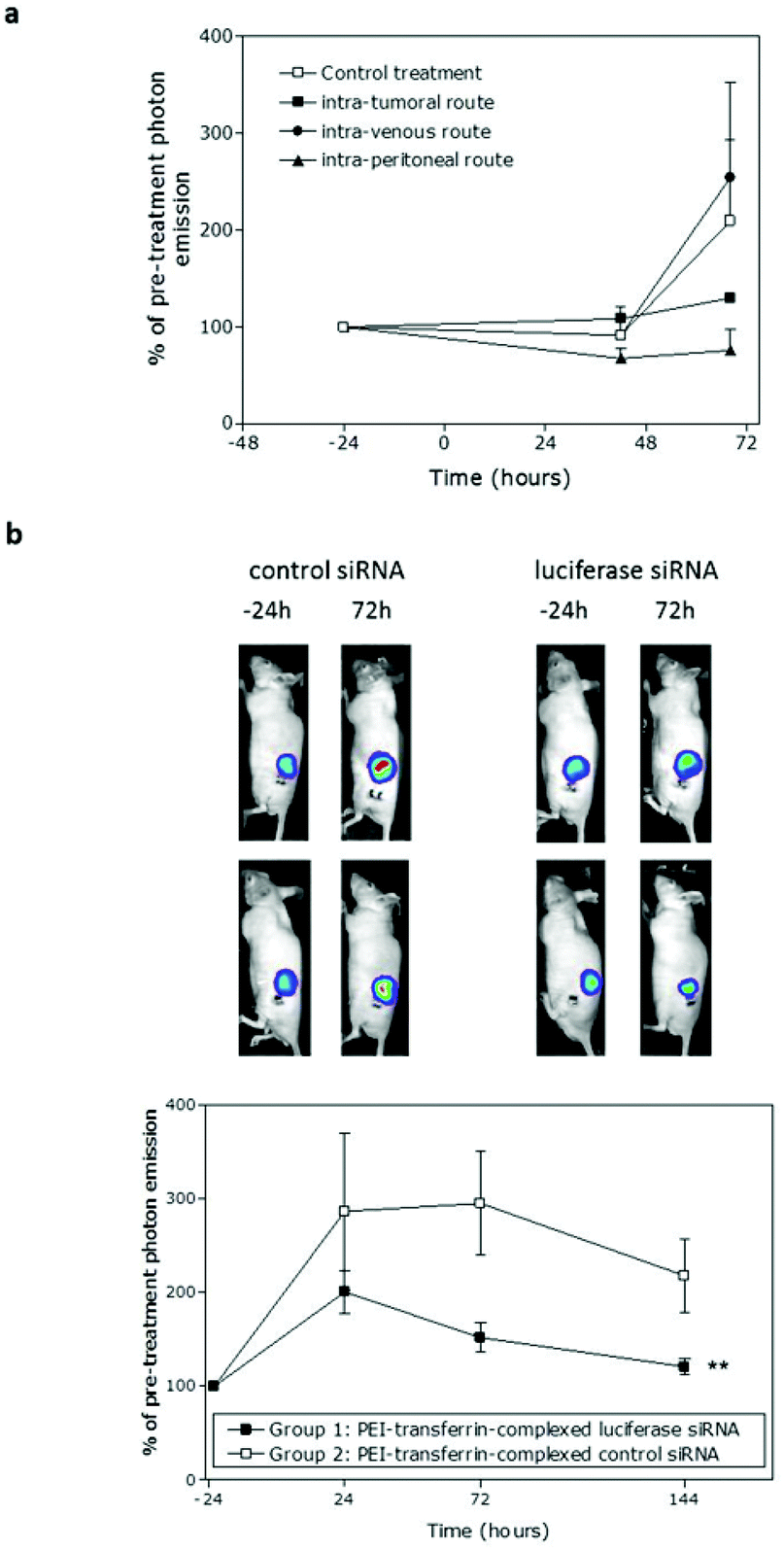 | ||
| Fig. 4 Systemic delivery of TfPEI-complexed siRNA in vivo. Bioluminescent sub-cutaneous MGLVA1 xenografts were established in nude mice. (a) TfPEI-complexed luciferase siRNA (TfPEI:PEI ratio of 1:4 and an N/P ratio of 8) was administered via intratumoral, intravenous or intraperitoneal routes (n = 4 mice per group) as a pilot study; a control siRNA was administered intravenously as control treatment. Bioluminescence at 42 and 68 h post-administration is shown relative to bioluminescence at 24 h pre-administration. Knockdown at 68 h was the greatest when the intratumoral or intraperitoneal routes of administration were used. (b) TfPEI-complexed luciferase or control siRNA (TfPEI:PEI ratio of 1:4 and an N/P ratio of 8) was administered via the intraperitoneal routes (n = 7 and 8 respectively) and imaged at 24 h before and 24, 72 and 144 h after injection. Representative images of 2 mice treated with control or luciferase siRNA taken 24 h before and 72 h after treatment are shown and the data for all mice are illustrated graphically. A significant reduction in bioluminescence was observed in the luciferase siRNA-treated group (** indicates p = 0.008, 2-way ANOVA) relative to the control siRNA-treated group. | ||
Next, a larger study was set up in which a complexed luciferase or control siRNA was administered intraperitoneally and bioluminescence was monitored at 24, 72 and 144 h after administration. As in the intratumoral study, whilst luminescence in both groups increased due to tumour growth, there was significant lower bioluminescence in the luciferase siRNA-treated group compared with the control group, taking into account all time-points (p = 0.008, two-way ANOVA). At 24 h, the reduction in bioluminescence (30%) was not significant, but by 72 h the reduction was significant (49%, p = 0.045, one-way ANOVA). Knockdown was sustained through to 144 h after siRNA administration (44% decrease, not sig.) and the lower bioluminescence in the luciferase siRNA group at the end-point compared with the control group was also reflected in a 39% reduction in the luciferase mRNA level (data not shown). General animal conditions were monitored daily and there was no apparent toxicity associated with delivery of siRNA to the animals with body weights maintained in both groups. There was also no significant difference in the growth of tumours in the two groups (Fig. S3a†) and no significant induction of the interferon response at the end-point (Fig. S3b and c†).
Discussion
The data together show that a relatively simple co-formulation of linear PEI with transferrin-conjugated PEI can be used for delivery of siRNA to colonic (HCT116) and gastric (MGLVA1) gastrointestinal cancer cells leading to specific knockdown of the target gene. The siRNA formulation which gave sustained knockdown in vitro was also effective in vivo at knocking down luciferase activity in MGLVA1 tumours growing subcutaneously in MF1 nude mice when delivered either intratumorally or systemically via the intraperitoneal route.The efficacy of the formulation derives from incorporation of transferrin into PEI/siRNA complexes. Although some inhibition of knockdown by free transferrin was observed, suggesting that at least some of the uptake is also via specific receptor binding, most probably the uptake is also due to a reduction in the polyelectrolyte complex surface charge by the large (80 kDa) and negatively charged conjugated transferrin, as shown by the reduced zeta potentials of the Tf–PEI siRNA complexes compared to those with PEI–siRNA only. Shielding of the positive charge of PEI in polyelectrolyte complexes has been hypothesised for PEGylated polymer/DNA complexes in vivo, through a reduction in plasma protein and red blood cell binding, leading to prolonged blood circulation and prevention of erythrocyte aggregation.37 However, there are significant differences in physical properties, particularly persistence length and compaction, of shorter oligonucleotides such as siRNAs compared to DNA plasmids.24 These in turn mean that it is not always possible to use polymers optimised for plasmid DNA therapeutics directly for oligonucleotide delivery. However, by simple mixing of linear PEI and branched PEI–Tf conjugates we were able to generate complexes of appropriate size and charge for siRNA delivery, and demonstrated the particle uptake in vitro in cancer cell lines. These data indicated that incorporation of transferrin and the resultant reduction in the positive charge was not detrimental to the cellular uptake, and were able to identify TfPEI:PEI and N/P ratios that were effective in promoting luciferase knockdown. The particles incorporating TfPEI were similar in size to those using PEI alone and while zeta potential measurements confirmed that incorporation of transferrin into the complexes reduced the positive charge of the particles for complexes prepared at N/P = 8, addition of transferrin did not generate polyplexes with an overall negative charge.
In this study we used 22 kDa linear PEI and 25 kDa branched PEI as similar molecular weight PEIs (<25 kDa) have previously been shown to be more effective than higher molecular weight forms (∼800 kDa) for delivery of DNA15,38 and were associated with lower toxicity.13,39 Low molecular weight linear PEI has been successfully used for intraperitoneal delivery of siRNA in mice, and it reduced the size of subcutaneously-grown tumours as well as achieving an ∼50% knockdown in target gene expression. However, in these studies, gene silencing effects were apparent after 11 days and multiple injections were required.40 The majority of studies investigating the use of PEI for siRNA delivery have used the branched form, either alone38 or conjugated to a peptide e.g. one targeted to integrin AvB3.41 In a study comparing branched and linear PEI for siRNA delivery, whilst binding of linear PEI to siRNA was similar to binding of branched PEI and the uptake by cells was also observed, the authors did not achieve knockdown of the target gene.13 This contrasts with our findings in which we achieved 80–90% knockdown in both the colonic and gastric cell-lines by day 1 and this was maintained up to day 3 of the assays. Differences in the efficacy of knockdown is likely due to differences in the characteristics of the particles used, including the size, charge and shielding which are dependent on particle formulation, or characteristics of the target cells. The formation of smaller complexes may lead to more efficient uptake due to the increased mobility of the complexes and increased interaction with negatively charged cell-surface proteoglycans, but at very high ratios, release of the nucleic acid into the cytoplasm may be impaired (i) by a very tight/strong (in extreme cases even non-reversible) condensation of nucleic acid and/or (ii) due to reduced ability of small-sized particles to act as a proton sponge.42
The effect of using complexes containing TfPEI has previously been shown to vary with different cell-types.15,25 In this study, in MGLVA1s there was a greater delay in knockdown which increased markedly between day 1 and day 3, using a number of the formulations, but in particular the TfPEI:PEI ratio of 1:15. This may mean that the route of uptake is different in the 2 cell-lines used or that release of the siRNA from the endosomes may be slower in the MGLVA1s.
Since we had identified the conditions that allowed efficient and prolonged siRNA delivery in vitro using complexes incorporating TfPEI, we investigated their function in an in vivo model which enabled real-time monitoring of the effect of siRNA administration. Both intratumoral delivery and systemic delivery via the peritoneum were effective at reducing luciferase activity in the xenografts whilst, in a pilot study, intravenous delivery was not effective. Biodistribution studies in which the intravenous delivery routes have been used previously showed that siRNA complexed with RGD-PEG-PEI complexes accumulates in the liver, and to a lesser extent in the lung, spleen, heart and kidney,43 while other studies with modified PEI complexes have shown accumulation in the lung predominantly,44 thus reducing the effectiveness of delivery. The intraperitoneal route has previously been successfully used for delivery of nucleic acids.40,45 In a study using PEI-complexed siRNA administered intraperitoneally and a subcutaneous ovarian cancer xenograft model, a HER-2 specific siRNA significantly reduced tumour growth over 14 days compared with a control siRNA.40 Further studies would be needed to verify the low efficiency of delivery by the intravenous route observed in our small pilot study, to determine the underlying mechanisms and to investigate whether intravenous delivery of TfPEI–siRNA complexes can be achieved; however, the current study provides the proof-of-principle that systemic delivery using TfPEI–siRNA complexes is feasible.
The downregulation of a luciferase gene as a result of siRNA administration in this study appears to be specific. The effects were observed only with an siRNA targeting the luciferase gene expressed by the cells and not when an siRNA, complexed in an identical manner and targeting a closely related luciferase gene, was used. The reduction in the bioluminescence signal observed in the luciferase siRNA group compared with the control group at the end-point following systemic administration (44%) was paralleled by a reduction in the luciferase mRNA of the same order of magnitude (39%). Interestingly, as in the in vitro studies, little knockdown was achieved at 24 h but there was an increase in knockdown at 72 h, which, in the systemic study, was maintained through to 144 h after administration of the siRNA. This may be a result of delayed release of the siRNA from the particles and subsequent gene knockdown following uptake of the siRNA/polycation particles.
In addition, there were no apparent adverse effects in the animals as a result of administration of the siRNA complexes; in the study where siRNA was administered systemically, no toxicity was observed in the animals over 6 days following injection and there was no effect on tumour growth, as anticipated since the siRNA used targets the luciferase gene and thus should not affect tumour cell proliferation. The absence of free PEI in the TfPEI-containing complexes in contrast with those containing PEI only, as observed from the light scattering studies, may have contributed to this low toxicity in addition to the reduced overall charge of the Tf–PEI complexes compared to PEI/siRNA complexes alone. Lastly, although we did not use modified siRNAs to prevent induction of the innate immune response,46,47 we saw only a small non-significant increase in the expression of OAS1 and downregulation of the STAT1 gene. These genes are both involved in the activation of the innate immune response,48 suggesting that the siRNAs used do not contain “trigger” sequences49 or that the formulation used protected them from uptake by immune cells. However, if delivery was shown to be mediated through the transferrin receptor rather than through the non-specific uptake as a result of transferrin-mediated shielding, further toxicity studies would be required to demonstrate that the absence of toxicity in this model was not a result of poor binding of transferrin to the mouse transferrin receptor.
This study provides the proof-of-principle that linear PEI (22 kDa) co-formulated with transferrin-conjugated branched PEI (25 kDa) can be used systemically for knockdown of tumour-expressed genes and therefore has potential as a delivery agent for treatment of GI cancer. Future studies will investigate the route of uptake, in vivo targeting50 and the relative efficacy of complexes containing a broader range of polymers. These include other derivatives of PEI, such as temperature and/or acid-responsive PEI conjugates,51 alternative cell-surface ligands41 and new polymer backbones and reporter groups52 which might further improve the efficacy and control of siRNA delivery and release in vivo.
Acknowledgements
This research was supported by a grant from Aphton Ltd and by the Engineering and Physical Sciences Research Council (EPSRC: Grants EP/H005625/1, EP/D501849/1). We also thank Ali Alazzo and Pam Collier for repeat DLS and MTT assays and Paul Cooling and Christine Grainger-Boultby for technical support.Notes and references
- S. M. Elbashir, J. Harborth, W. Lendeckel, A. Yalcin, K. Weber and T. Tuschl, Nature, 2001, 411, 494–498 CrossRef CAS PubMed.
- A. Gallas, C. Alexander, M. C. Davies, S. Puri and S. Allen, Chem. Soc. Rev., 2013, 42, 7983–7997 RSC.
- Y. Wang, J. Li, Y. Chen and D. Oupicky, Biomater. Sci., 2015, 3, 1114–1123 RSC.
- E. Wagner, Biomater. Sci., 2013, 1, 804–809 RSC.
- T. C. Chu, K. Y. Twu, A. D. Ellington and M. Levy, Nucleic Acids Res., 2006, 34, e73 CrossRef PubMed.
- J. Soutschek, A. Akinc, B. Bramlage, K. Charisse, R. Constien, M. Donoghue, S. Elbashir, A. Geick, P. Hadwiger, J. Harborth, M. John, V. Kesavan, G. Lavine, R. K. Pandey, T. Racie, K. G. Rajeev, I. Rohl, I. Toudjarska, G. Wang, S. Wuschko, D. Bumcrot, V. Koteliansky, S. Limmer, M. Manoharan and H. P. Vornlocher, Nature, 2004, 432, 173–178 CrossRef CAS PubMed.
- N. Kobayashi, M. Nishikawa and Y. Takakura, Adv. Drug Delivery Rev., 2005, 57, 713–731 CrossRef CAS PubMed.
- A. C. Holley, K. H. Parsons, W. Wan, D. F. Lyons, G. R. Bishop, J. J. Correia, F. Huang and C. L. McCormick, Polym. Chem., 2014, 5, 6967–6976 RSC.
- Z. Ma, J. Li, F. He, A. Wilson, B. Pitt and S. Li, Biochem. Biophys. Res. Commun., 2005, 330, 755–759 CrossRef CAS PubMed.
- E. Wagner, R. Kircheis and G. F. Walker, Biomed. Pharmacother., 2004, 58, 152–161 CrossRef CAS PubMed.
- Y. Nie, D. Schaffert, W. Rödl, M. Ogris, E. Wagner and M. Günther, J. Controlled Release, 2011, 152, 127–134 CrossRef CAS PubMed.
- O. Boussif, F. Lezoualc'h, M. A. Zanta, M. D. Mergny, D. Scherman, B. Demeneix and J. P. Behr, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7297–7301 CrossRef CAS.
- A. C. Grayson, A. M. Doody and D. Putnam, Pharm. Res., 2006, 23, 1868–1876 CrossRef PubMed.
- S. Choi and K. D. Lee, J. Controlled Release, 2008, 131, 70–76 CrossRef CAS PubMed.
- L. Wightman, R. Kircheis, V. Rossler, S. Carotta, R. Ruzicka, M. Kursa and E. Wagner, J. Gene Med., 2001, 3, 362–372 CrossRef CAS PubMed.
- P. Lampela, J. Raisanen, P. T. Mannisto, S. Yla-Herttuala and A. Raasmaja, J. Gene Med., 2002, 4, 205–214 CrossRef PubMed.
- Q. Peng, Z. Zhong and R. Zhuo, Bioconjugate Chem., 2008, 19, 499–506 CrossRef CAS PubMed.
- Y. Li, H. Tian, J. Ding, X. Dong, J. Chen and X. Chen, Polym. Chem., 2014, 5, 3598–3607 RSC.
- M. Günther, J. Lipka, A. Malek, D. Gutsch, W. Kreyling and A. Aigner, Eur. J. Pharm. Biopharm., 2011, 77, 438–449 CrossRef PubMed.
- S. Sajeesh, T. Y. Lee, S. W. Hong, P. Dua, J. Y. Choe, A. Kang, W. S. Yun, C. Song, S. H. Park, S. Kim, C. Li and D.-K. Lee, Mol. Pharm., 2014, 11, 872–884 CrossRef CAS PubMed.
- Y. Liu, J. Tao, Y. Li, J. Yang, Y. Yu, M. Wang, X. Xu, C. Huang, W. Huang, J. Dong, L. Li, J. Liu, G. Shen and Y. Tu, Mol. Ther., 2009, 17, 269–277 CrossRef CAS PubMed.
- P. T. Gomme, K. B. McCann and J. Bertolini, Drug Discovery Today, 2005, 10, 267–273 CrossRef CAS.
- T. R. Daniels, T. Delgado, G. Helguera and M. L. Penichet, Clin. Immunol., 2006, 121, 159–176 CrossRef CAS PubMed.
- M. E. Davis, Mol. Pharm., 2009, 6, 659–668 CrossRef CAS PubMed.
- R. Kircheis, A. Kichler, G. Wallner, M. Kursa, M. Ogris, T. Felzmann, M. Buchberger and E. Wagner, Gene Ther., 1997, 4, 409–418 CAS.
- Y. Liu, J. Tao, Y. Li, J. Yang, Y. Yu, M. Wang, X. Xu, C. Huang, W. Huang, J. Dong, L. Li, J. Liu, G. Shen and Y. Tu, Mol. Ther., 2009, 17, 269–277 CrossRef CAS PubMed.
- R. Kircheis, L. Wightman, A. Schreiber, B. Robitza, V. Rossler, M. Kursa and E. Wagner, Gene Ther., 2001, 8, 28–40 CrossRef CAS PubMed.
- S. A. Watson, L. G. Durrant and D. L. Morris, Int. J. Cancer, 1990, 45, 90–94 CrossRef CAS PubMed.
- A. M. Grabowska, J. Hughes and S. A. Watson, Br. J. Cancer, 2007, 96, 464–473 CrossRef CAS PubMed.
- United Kingdom Co-ordinating Committee on Cancer Research (UKCCCR), Guidelines for the Welfare of Animals in Experimental Neoplasia (Second Edition), Br. J. Cancer, 1998, 77, 1–10 Search PubMed.
- A. M. Grabowska, J. Hughes and S. A. Watson, 15th International Symposium on Regulatory Peptides, 2004.
- S. K. Cho, C. Dang, X. Wang, R. Ragan and Y. J. Kwon, Biomater. Sci., 2015, 3, 1124–1133 RSC.
- R. Kircheis, L. Wightman, A. Schreiber, B. Robitza, V. Rossler, M. Kursa and E. Wagner, Gene Ther., 2001, 8, 28–40 CrossRef CAS PubMed.
- A. Calzolari, I. Oliviero, S. Deaglio, G. Mariani, M. Biffoni, N. M. Sposi, F. Malavasi, C. Peschle and U. Testa, Blood Cells, Mol., Dis., 2007, 39, 82–91 CrossRef CAS PubMed.
- M. Soliman, R. Nasanit, S. R. Abulateefeh, S. Allen, M. C. Davies, S. S. Briggs, L. W. Seymour, J. A. Preece, A. M. Grabowska, S. A. Watson and C. Alexander, Mol. Pharm., 2012, 9, 1–13 CrossRef CAS PubMed.
- S. A. Watson, K. E. Robinson, D. McWilliams, D. Michaeli, A. M. Smith and G. Robinson, Int. J. Cancer, 2000, 87, 20–28 CrossRef CAS.
- M. Ogris, S. Brunner, S. Schuller, R. Kircheis and E. Wagner, Gene Ther., 1999, 6, 595–605 CrossRef CAS PubMed.
- S. Werth, B. Urban-Klein, L. Dai, S. Hobel, M. Grzelinski, U. Bakowsky, F. Czubayko and A. Aigner, J. Controlled Release, 2006, 112, 257–270 CrossRef CAS PubMed.
- T. Bieber and H. P. Elsasser, Biotechniques, 2001, 30 CAS , 74–77, 80–81.
- B. Urban-Klein, S. Werth, S. Abuharbeid, F. Czubayko and A. Aigner, Gene Ther., 2005, 12, 461–466 CrossRef CAS PubMed.
- R. M. Schiffelers, A. Ansari, J. Xu, Q. Zhou, Q. Tang, G. Storm, G. Molema, P. Y. Lu, P. V. Scaria and M. C. Woodle, Nucleic Acids Res., 2004, 32, e149 CrossRef PubMed.
- M. Ogris, P. Steinlein, M. Kursa, K. Mechtler, R. Kircheis and E. Wagner, Gene Ther., 1998, 5, 1425–1433 CAS.
- H. K. de Wolf, C. J. Snel, F. J. Verbaan, R. M. Schiffelers, W. E. Hennink and G. Storm, Int. J. Pharm., 2007, 331, 167–175 CrossRef CAS PubMed.
- M. Thomas, J. J. Lu, Q. Ge, C. Zhang, J. Chen and A. M. Klibanov, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 5679–5684 CrossRef CAS PubMed.
- M. H. Louis, S. Dutoit, Y. Denoux, P. Erbacher, E. Deslandes, J. P. Behr, P. Gauduchon and L. Poulain, Cancer Gene Ther., 2005, 13, 367–374 CrossRef PubMed.
- M. A. Behlke, Mol. Ther., 2006, 13, 644–670 CrossRef CAS PubMed.
- A. Reynolds, E. M. Anderson, A. Vermeulen, Y. Fedorov, K. Robinson, D. Leake, J. Karpilow, W. S. Marshall and A. Khvorova, RNA, 2006, 12, 988–993 CrossRef CAS PubMed.
- C. A. Sledz, M. Holko, M. J. de Veer, R. H. Silverman and B. R. Williams, Nat. Cell Biol., 2003, 5, 834–839 CrossRef CAS PubMed.
- S. Agrawal and E. R. Kandimalla, Nat. Biotechnol., 2004, 22, 1533–1537 CrossRef CAS PubMed.
- J. Eliezar, W. Scarano, N. R. B. Boase, K. J. Thurecht and M. H. Stenzel, Biomacromolecules, 2015, 16, 515–523 CrossRef CAS PubMed.
- F. Heath, A. O. Saeed, S. S. Pennadam, K. J. Thurecht and C. Alexander, Polym. Chem., 2010, 1, 1252–1262 RSC.
- A. V. Fuchs, A. C. Gemmell and K. J. Thurecht, Polym. Chem., 2015, 6, 868–880 RSC.
Footnotes |
| † This paper is dedicated to the memory of Professor Susan Watson. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c5bm00101c |
| This journal is © The Royal Society of Chemistry 2015 |