
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
MMP-9 triggered micelle-to-fibre transitions for slow release of doxorubicin†
Daniela
Kalafatovic
a,
Max
Nobis
b,
Nadeem
Javid
a,
Pim W. J. M.
Frederix
a,
Kurt I.
Anderson
b,
Brian R.
Saunders
c and
Rein V.
Ulijn
*ade
aWest CHEM, Department of Pure and Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow, G1 1XL, UK
bThe Beatson Institute for Cancer Research, Garscube Estate, Glasgow, G61 1BD, UK
cSchool of Materials, University of Manchester, Grosvenor Street, Manchester M1 7HS, UK
dAdvanced Science Research Centre (ASRC), City University of New York, 85 St Nicholas Terrace, New York, NY 10031, USA. E-mail: Rein.Ulijn@asrc.cuny.edu
eDepartment of Chemistry and Biochemistry, City University of New York – Hunter College, 695 Park Ave., New York, NY 10065, USA
First published on 28th October 2014
Abstract
Phenylacetyl-peptide amphiphiles were designed, which upon cleavage by a disease-associated enzyme reconfigure from micellar aggregates to fibres. Upon this morphological change, a doxorubicin payload could be retained in the fibres formed, which makes them valuable carriers for localised formation of nanofibre depots for slow release of hydrophobic anticancer drugs.
Peptide self-assembly is increasingly being investigated for a plethora of applications in biomedicine including drug release, tissue engineering, diagnostic studies and regenerative medicine.1 These nanostructures are of interest as they may contain bioactive peptide ligands, as well as structural components which enable access to a variety of nanoscale morphologies dictated by the amino acid sequence2 but also by the route of assembly.3 Enzymatic catalysis presents an attractive way to control molecular self-assembly.4 In this approach, non-assembling precursors that are “blocked” with enzyme cleavable groups are converted to self-assembling building blocks (including hydrogelators), enabling self-assembly on-demand under physiological conditions. The most frequently studied biocatalytic self-assembly systems are those based on aromatic peptide amphiphiles. They consist of short peptide sequences modified by aromatic groups such as phenyl, naphthyl, fluorenyl and others.5 Different enzymes such as phosphatases4b,6 and proteases (including matrix metalloproteinases (MMPs))7 have been used to trigger molecular self-assembly in vitro and in vivo.6c,8
Expression levels of enzymes dictate the difference between health and disease in many cases, including cancer. MMPs are a family of zinc dependent endopeptidases that are involved in the digestion and remodelling of the extracellular matrix.9 Some members of this family, such as MMP-9, have been reported to be overexpressed in breast, cervical, colon and other types of cancers.10 This makes them valuable triggers for responsive biomaterials and targeted self-assembly. Typically, MMP responsive peptide-based systems act via hydrolysis and dissociation of structures, i.e. using enzymatic cleavage to trigger dissociation of hydrogels (containing MMP sensitive crosslinks),11 supramolecular peptide filaments12 and polymer–peptide hybrids.13 The first example of the use of MMP-9 to form (rather than degrade) a peptide based supramolecular hydrogel was presented by the Xu group.7a A morphological change induced by MMP-7 was previously shown for an aliphatic, palmitylated peptide amphiphile system.14 A very recent study shows the development of a selective assay for MMP-9 via gelation.15
Based on this knowledge we set out to develop a peptide based enzyme-responsive system that is able to undergo a morphological change from micellar aggregates to fibres in response to cleavage by MMP-9 (Fig. 1) and use it for localised formation of a depot for slow release of hydrophobic drugs (e.g. doxorubicin) at the cancer site. There are three design requirements for such a system: (i) a biocompatible fibre forming self-assembly unit (phenylacetyl-FFAG) that also provides the hydrophobic binding region for drug candidates, (ii) the MMP-9 cleavable sequence and (iii) a hydrophilic unit (LDD) that modifies the amphiphilic balance of the precursor to favour micelle formation (Fig. 1). Thus, upon MMP-9 cleavage, the peptide micelles reconfigure into fibres, due to a change in the hydrophobic/hydrophilic balance of the sequence. In turn, this aspect may be used for hydrophobic drug entrapment into fibres, presenting a unique advantage for the development of drug delivery systems for prolonged release times after initial exposure.
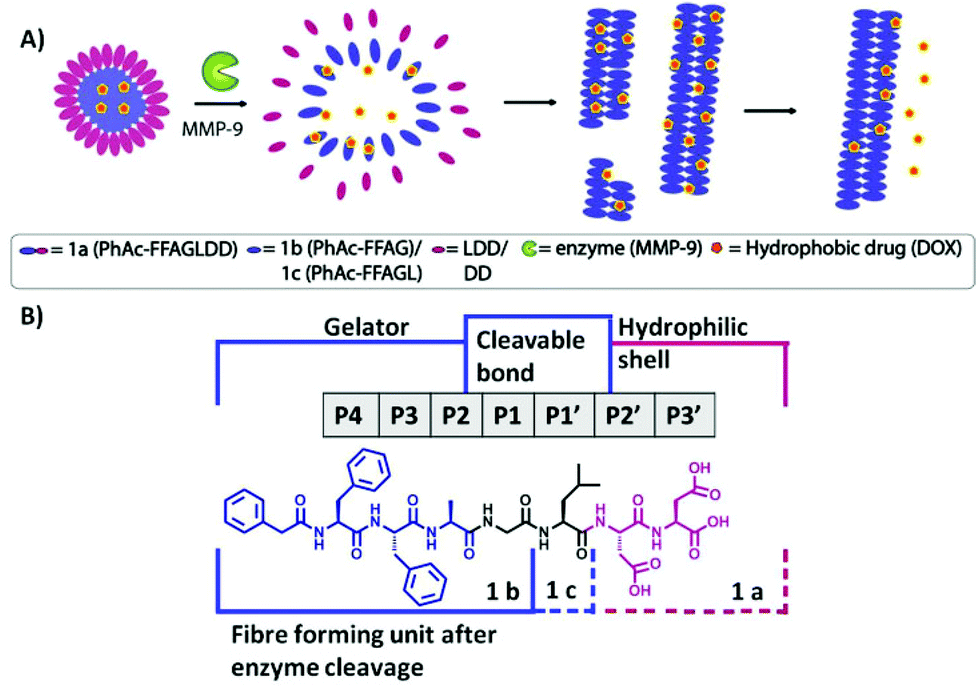 | ||
| Fig. 1 (A) Schematic representation of micelle to fibre transition induced by MMP-9 cleavage showing disassembly of micelles and the re-assembly into fibres after the removal of the hydrophilic group enabling prolonged drug release. (B) Chemical structure of the biocatalytic gelation system and its components. | ||
In order to design the enzyme cleavage site, the MEROPS16 database was used, which provides cleavage patterns for peptidases based on a collection of experimental data from the literature. This can be used for the design of substrates that simultaneously meet the three criteria mentioned above. MMP-9's specificity preference for P4–P3′ subsites (based on 367 cleavages reported in the literature) is based on the GPX1G↓LX2G sequence with G/L (P1–P1′) being the cleavable bond, X1 (P2) being preferentially alanine or leucine, and X2 (P2′) glycine as the preferred choice.16 MMP-9 requires longer substrates, of e.g. 7 residues (i.e. P4–P3′), in order to be able to recognise and efficiently cleave the G↓L bond,17 with GPLG↓LAG being an example.18 The length of the substrate and the presence of large substitutions (i.e. pyrene, naphthalene, etc.) at the N-terminus can lead to a shift in specificity of MMP-9.7b To fulfil the requirement of the gelator (fibre forming) unit in the P3 and P4 positions phenylalanine–phenylalanine19 was used (proline and glycine preferred, but phenylalanine known to be tolerated in P3 and P4). In P2 we chose alanine, while in P2′ and P3′ (referred to as amino acids flanking the scissile bond towards the C-terminus) aspartic acid–aspartic acid was used, as it is known to be tolerated in both positions, and will provide a negatively charged surface of the micellar aggregates.
First the PhAc-FFAGLDD (1a) and its expected product of enzyme cleavage PhAc-FFAG (1b) were synthesized and characterized by AFM, FTIR, DLS, rheology and fluorescence.
The peptide (1a) was directly dissolved in deionized water, the pH adjusted to 7.4 and its self-assembly behaviour investigated after a cycle of alternating sonication and vortexing. For the expected enzyme cleavage peptide fragment, i.e.1b the peptide was dissolved in DI water and the pH was increased (0.5 M NaOH) to solubilise 1b, followed by a slow decrease of pH achieved by addition of 0.5 M HCl, to a final pH of 6.5–7. This slightly acidic pH corresponds to that of the tumour microenvironment.20 Gelation was observed for the expected MMP-9 cleavage product 1b (Fig. 2A).
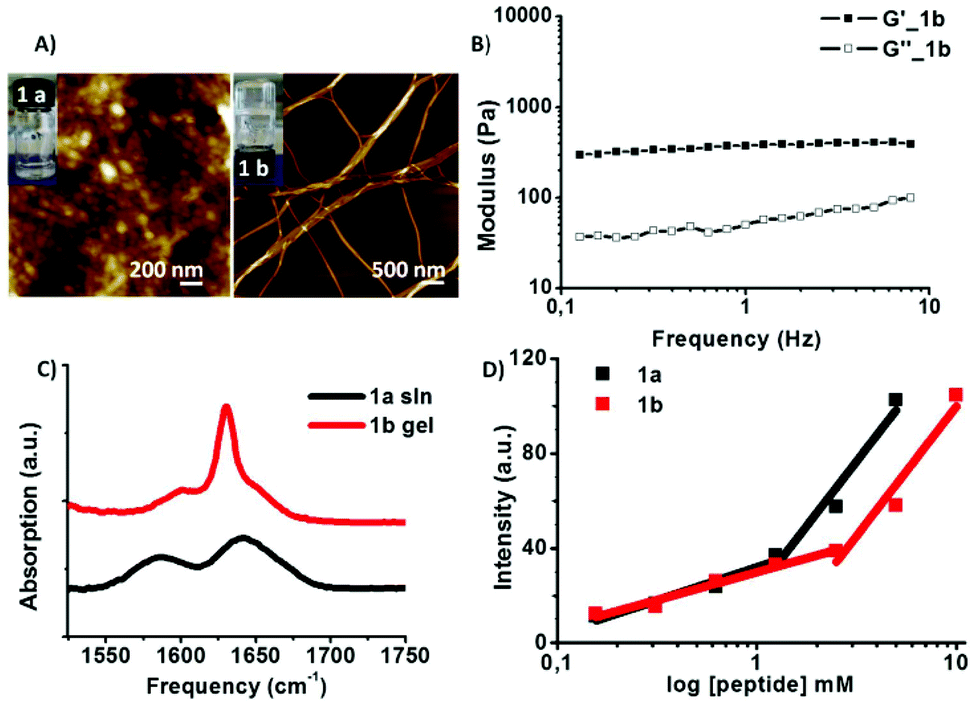 | ||
| Fig. 2 (A) AFM showing the micellar aggregates (solution) for 1a and fibres (hydrogels at 20 mM) for 1b. (B) Rheology data for 1b gel showing the plot of G′ (elastic modulus) and G′′ (viscous modulus) against frequency. (C) FTIR absorption spectrum in the amide I region (in D2O at pH 7): 1a (solution) and 1b (gel). (D) Critical aggregation concentration (CAC) of (1.32 mM (1a) and 2.88 mM (1b)). | ||
The AFM characterisation of the peptides revealed spherical aggregates for 1a (d = 43.6 ± 6.2 nm) while fibres were found for 1b having a micron scale length and 20–50 nm range diameter which corresponds to the size of peptide based fibres reported in the literature.2c Alternative supramolecular organisation was further supported by infrared (IR) spectroscopy data (Fig. 2C) suggesting the presence of ordered structures for the examined peptides due to aggregation via intramolecular hydrogen bonding.21 Peptide 1a shows a red shift and a broad peak at 1643 cm−1 compared to 1650–1655 cm−1 absorption values, typical for free peptides in solution. The 1570–1580 cm−1 absorption band is attributed to the aspartic acid side chain carboxylate group present in 1a.21 Extended structures are observed for 1b (transparent gel) characterised by a pronounced narrowing of the peak typical for short peptide β-sheets at 1630 cm−1, while the 1595 cm−1 characteristic of the C-terminus carboxylate group has a low intensity.21,22 Rheology measurements of the 1b hydrogel show the elastic modulus (G′) of 360 Pa, an order of magnitude higher than its viscous component (G′′ = 32 Pa) which is characteristic of entangled networks (Fig. 2B). Furthermore, DLS experiments (Fig. S3 and S4, ESI†) were performed for peptide solution samples 1a and 1b at various concentrations ranging from 0.625 mM to 5 mM. The diffusion coefficients (D) of samples 1a (micellar aggregates) and 1b (fibres) at 0.625 mM are 1.5 × 10−12 m2 s−1 and 6.8 × 10−13 m2 s−1 corresponding to RH values of 165 nm and 358 nm, respectively. The higher values of RH compared to AFM (dry samples) indicate that in the solution state the aggregates are bigger than the collapsed, dried ones.
In order to further investigate the self-assembly behaviour of peptide amphiphiles the critical aggregation concentration (CAC) in water was determined using the fluorescence intensity of the 8-anilino-1-naphthalenesulphonic acid (ANS probe) as a function of the peptide concentration (Fig. S5, ESI†). The determined CAC values were 1.32 mM for PhAc-FFAGLDD and 2.88 mM for PhAc-FFAG. Furthermore, the critical micelle concentration (CMC) for 1a was determined using pyrene as the fluorescent probe. The ratio of the fluorescence intensity of the first (λem = 372 nm) and the third peak (λem = 384 nm) was plotted as the function of the peptide concentration. The value calculated to be 1.25 mM (Fig. S6, ESI†) is in the same range of the CAC values for 1a.
After the designed peptides 1a and 1b were shown to be successful for controlling the morphology of the supramolecular aggregates based on the peptide length, i.e. hydrophobicity, the enzyme triggered micelle to fibre transition for peptide amphiphile 1a was explored. Peptide amphiphile 1a was treated with 50 ng mL−1 MMP-9 for 96 h and the morphological change was monitored by AFM, where fibre formation was observed (Fig. 3B). This enzyme concentration was chosen based on MMP-9 quantification of in vitro human cancer cell lines. Peptide 1a showed complete conversion after 96 h to the PhAc-FFAGL (1c) fragment (Fig. S7, ESI†) indicating the shift of the MMP-9 specificity for this heptapeptide to GL↓D instead of the expected G↓L in accordance with reported observations that cleavage sites of heptapeptides catalysed by MMP-9 differ from those of longer peptide sequences.7b According to MEROPS leucine and aspartic acid are tolerated in P1 and P1′ positions, respectively, but they do not seem to be preferentially recognised and reported as a cleavage site for MMP-9.
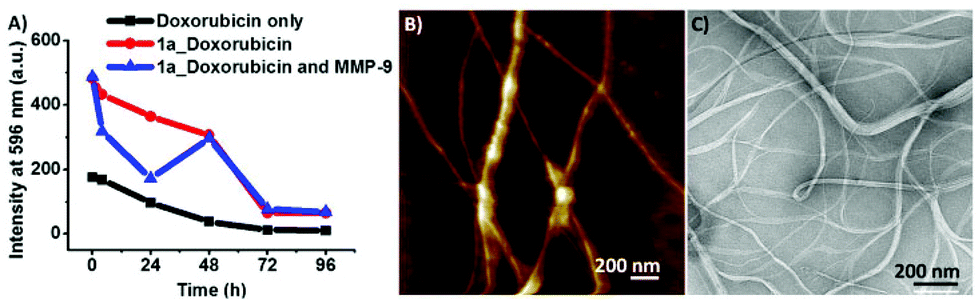 | ||
| Fig. 3 (A) Fluorescence intensities of doxorubicin monitored over time for doxorubicin only, doxorubicin loaded into precursor peptide (1a) micelles and MMP-9 treated precursor peptide (1a) micelles loaded with doxorubicin. (B) AFM images of MMP-9 induced fibre formation (PhAc-FFAGL) (1c) after 96 h. (C) TEM images of doxorubicin loaded samples treated with MMP-9 for 72 h showing that fibre formation was not disrupted by the presence of the drug. | ||
Following this it was investigated whether the micelles were capable of performing as mobile vehicles for encapsulation to immobilised fibre networks for the release of hydrophobic drugs. For this purpose, the release of an anticancer drug, doxorubicin, was studied. Doxorubicin was loaded into the micelles and its release by passive diffusion was monitored over time by fluorescence microscopy. The fluorescence intensity of doxorubicin at 596 nm which corresponds to the maximum intensity (λex = 480 nm) was monitored over 96 h when incorporated into the 1a peptide system. A control experiment with free doxorubicin shows a decrease in fluorescence intensity over time, probably due to aggregation. Due to its poor solubility in water, fluorescence quantification of doxorubicin is not reproducible, however, it becomes more stable when incorporated into peptides. The interaction of doxorubicin with the hydrophobic environment of 1c results in higher values of fluorescence intensity compared to free doxorubicin in water (doxorubicin emission in solvents of different polarities is shown in Fig. S8, ESI†). For 1a doxorubicin fluorescence intensity drops only slightly over 48 h suggesting that the payload stays incorporated into micelles over that time period showing only low release, followed by a significant decrease after 72 h. When treated with MMP-9 there is a release from micelles (a significant drop in fluorescence intensity upon exposure to water) followed by entrapment into fibres (resulting in an increase in fluorescence intensity). A similar discontinuous behaviour upon phosphatase triggered gelation was recently reported for a related system.23 Even if the overall release after 48 h is similar for the MMP-9 treated and untreated 1a the advantage of the enzymatic approach is that a localised effect is obtained with fibres expected to be significantly less mobile than the micelles.
After fibre formation, the increase of fluorescence intensity suggests that the doxorubicin re-enters a hydrophobic environment by becoming entrapped in the fibres, confirming the possibility of the system to temporarily retain the payload. TEM images were obtained on enzyme treated doxorubicin loaded peptides that confirm fibre formation and show that the presence of doxorubicin did not disrupt fibre formation (Fig. 3C).
In conclusion, an MMP-9 responsive peptide amphiphile is shown here that self-assembles into spherical aggregates. Enzyme triggered micelle to fibre transition shows that it is a substrate of MMP-9 and is capable of encapsulation and controlled release of doxorubicin. These observations suggest the use as a mobile carrier for the anticancer drug that in turn is expected to be selectively delivered to tumour tissues, where it assembles to form a localised fibre-based depot by exploiting local MMP-9 overexpression. Furthermore, the assembled fibres provide a scaffold for localised drug delivery due to the partial entrapment of the drug and the intrinsic biodegradable nature of peptide carriers themselves. These systems are now studied in animal models. It should be noted that when used in vivo the system may also respond to other MMPs and specifically to MMP-2 due to some overlapping in specificity profiles.24
Acknowledgements
We acknowledge Patricia Keating at the University of Strathclyde Mass Spectrometry facility for the help and support with analytical techniques and for valuable discussions and EPSRC for funding.Notes and references
- (a) S. Zhang, Nat. Biotechnol., 2003, 21, 1171 CrossRef CAS PubMed; (b) G. A. Silva, C. Czeisler, K. L. Niece, E. Beniash, D. A. Harrington, J. A. Kessler and S. I. Stupp, Science, 2004, 303, 1352 CrossRef CAS PubMed; (c) J. Naskar, G. Palui and A. Banerjee, J. Phys. Chem. B, 2009, 113, 11787 CrossRef CAS PubMed; (d) J. H. Collier, J. S. Rudra, J. Z. Gasiorowski and J. P. Jung, Chem. Soc. Rev., 2010, 39, 3413 RSC; (e) D. N. Woolfson and Z. N. Mahmoud, Chem. Soc. Rev., 2010, 39, 3464 RSC; (f) R. H. Zha, S. Sur and S. I. Stupp, Adv. Healthc. Mater., 2013, 2, 126 CrossRef CAS PubMed; (g) G. Fichman and E. Gazit, Acta Biomater., 2014, 10, 1671 CrossRef CAS PubMed.
- (a) H. Cui, M. J. Webber and S. I. Stupp, Pept. Sci., 2010, 94, 1 CrossRef CAS PubMed; (b) I. Hamley, Soft Matter, 2011, 7, 4122 RSC; (c) M. Hughes, L. S. Birchall, K. Zuberi, L. A. Aitken, S. Debnath, N. Javid and R. V. Ulijn, Soft Matter, 2012, 8, 11565 RSC.
- J. Raeburn, A. Z. Cardoso and D. J. Adams, Chem. Soc. Rev., 2013, 42, 5143 RSC.
- (a) R. J. Williams, R. J. Mart and R. V. Ulijn, Biopolymers, 2010, 94, 107 CrossRef CAS PubMed; (b) Z. Yang, G. Liang and B. Xu, Acc. Chem. Res., 2008, 41, 315 CrossRef CAS PubMed; (c) M. E. Hahn and N. C. Gianneschi, Chem. Commun., 2011, 47, 11814 RSC; (d) M. Zelzer, S. J. Todd, A. R. Hirst, T. O. McDonald and R. V. Ulijn, Biomater. Sci., 2013, 1, 11 RSC.
- S. Roy and R. V. Ulijn, in Enzymatic Polymerisation, ed. A. R. A. Palmans and A. Heise, Springer-Verlag, Berlin, 2010, pp. 127–143 Search PubMed.
- (a) Y. Gao, Z. M. Yang, Y. Kuang, M. L. Ma, J. Y. Li, F. Zhao and B. Xu, Biopolymers, 2010, 94, 19 CrossRef CAS PubMed; (b) Y. Gao, J. Shi, D. Yuan and B. Xu, Nat. Commun., 2012, 3, 1033 CrossRef PubMed; (c) Y. Gao, C. Berciu, Y. Kuang, J. Shi, D. Nicastro and B. Xu, ACS Nano, 2013, 7, 9055 CrossRef CAS PubMed; (d) J. W. Sadownik, J. Leckie and R. V. Ulijn, Chem. Commun., 2011, 47, 728 RSC.
- (a) Z. M. Yang, M. L. Ma and B. Xu, Soft Matter, 2009, 5, 2546 CAS; (b) Y. Huang, J. Shi, D. Yuan, N. Zhou and B. Xu, Biopolymers, 2013, 100, 790 CrossRef PubMed.
- M. P. Chien, M. P. Thompson, C. V. Barback, T. H. Ku, D. J. Hall and N. C. Gianneschi, Adv. Mater., 2013, 25, 3599 CrossRef CAS PubMed.
- Y. Itoh and H. Nagase, Essays Biochem., 2002, 38, 21 CAS.
- (a) C. E. Brinckerhoff, J. L. Rutter and U. Benbow, Clin. Cancer Res., 2000, 6, 4823 CAS; (b) M. Egeblad and Z. Werb, Nat. Rev. Cancer, 2002, 2, 161 CrossRef CAS PubMed; (c) M. W. Roomi, J. C. Monterrey, T. Kalinovsky, M. Rath and A. Niedzwiecki, Oncol. Rep., 2009, 21, 1323 CAS.
- (a) M. Lutolf, J. Lauer-Fields, H. Schmoekel, A. Metters, F. Weber, G. Fields and J. Hubbell, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5413 CrossRef CAS PubMed; (b) V. K. Garripelli, J. K. Kim, S. Son, W. J. Kim, M. A. Repka and S. Jo, Acta Biomater., 2011, 7, 1984 CrossRef CAS PubMed; (c) S. Sokic, M. C. Christenson, J. C. Larson, A. A. Appel, E. M. Brey and G. Papavasiliou, Biomater. Sci., 2014, 2, 1343 RSC.
- Y.-A. Lin, Y.-C. Ou, A. G. Cheetham and H. Cui, Biomacromolecules, 2014, 15, 1419 CrossRef CAS PubMed.
- D. Bacinello, E. Garanger, D. Taton, K. C. Tam and S. Lecommandoux, Biomacromolecules, 2014, 15, 1882 CrossRef CAS PubMed.
- D. Koda, T. Maruyama, N. Minakuchi, K. Nakashima and M. Goto, Chem. Commun., 2010, 46, 979 RSC.
- S. C. Bremmer, A. J. McNeil and M. B. Soellner, Chem. Commun., 2014, 50, 1691 RSC.
- N. D. Rawlings, F. R. Morton, C. Y. Kok, J. Kong and A. J. Barrett, Nucleic Acids Res., 2008, 36(Database issue), D320 CAS.
- (a) J. L. Seltzer, K. T. Akers, H. Weingarten, G. A. Grant, D. W. McCourt and A. Z. Eisen, J. Biol. Chem., 1990, 265, 20409 CAS; (b) S. Netzelarnett, Q. X. Sang, W. G. I. Moore, M. Navre, H. Birkedalhansen and H. E. Vanwart, Biochemistry, 1993, 32, 6427 CrossRef CAS.
- M. E. Hahn, L. M. Randolph, L. Adamiak, M. P. Thompson and N. C. Gianneschi, Chem. Commun., 2013, 49, 2873 RSC.
- M. Reches and E. Gazit, Science, 2003, 300, 625 CrossRef CAS PubMed.
- Y. Zhao, T. Ji, H. Wang, S. Li, Y. Zhao and G. Nie, J. Controlled Release, 2014, 177, 11 CrossRef CAS PubMed.
- S. Fleming, P. W. J. M. Frederix, I. Ramos Sasselli, N. T. Hunt, R. V. Ulijn and T. Tuttle, Langmuir, 2013, 29, 9510 CrossRef PubMed.
- A. Barth and C. Zscherp, Q. Rev. Biophys., 2002, 35, 369 CrossRef CAS.
- K. Thornton, Y. M. Abul-Haija, N. Hodson and R. V. Ulijn, Soft Matter, 2013, 9, 9430 RSC.
- E. I. Chen, S. J. Kridel, E. W. Howard, W. Li, A. Godzik and J. W. Smith, J. Biol. Chem., 2002, 277, 4485 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4bm00297k |
| This journal is © The Royal Society of Chemistry 2015 |