
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Comparison of different mass spectrometric techniques for the determination of polychlorinated biphenyls by isotope dilution using 37Cl-labelled analogues†
Lourdes
Somoano-Blanco
a,
Pablo
Rodríguez-González
*a,
Daniel
Pröfrock
*b,
Andreas
Prange
b and
J. Ignacio
García Alonso
a
aDepartment of Physical and Analytical Chemistry, Faculty of Chemistry, University of Oviedo, Julián Clavería 8, 33006 Oviedo, Spain. E-mail: rodriguezpablo@uniovi.es
bHelmholtz-Zentrum Geesthacht, Centre for Materials and Coastal Research, Institute of Coastal Research, Department for Marine Bioanalytical Chemistry, Max-Planck-Strasse 1, D-21502 Geesthacht, Germany. E-mail: daniel.proefrock@hzg.de
First published on 15th September 2015
Abstract
This work presents the comparison of four different mass spectrometric techniques coupled to gas chromatography (single quadrupole ICP-MS, triple quadrupole ICP-MS/MS, single quadrupole NCI-MS and triple quadrupole EI-MS/MS) for the detection of polychlorinated biphenyls (PCBs) in environmental samples and their determination by a new isotope dilution mass spectrometry (IDMS) approach. A mixture of twelve priority PCBs labelled with 37Cl was employed as the species-specific isotopically labelled internal standard. The 37Cl-labelled PCBs enable the use of both molecular and elemental ionization sources, such as ICP or NCI, as the isotopic label is in the heteroatom. First, the comparison was carried out by assessing the capabilities of all instruments to measure chlorine isotope ratios and calculating the isotopic enrichment of the labelled analogues. Finally, the analysis of the Certified Reference Material SRM 1941b (organics in marine sediment) containing PCBs in the low ng g−1 range was carried out for method comparison. Elemental ionization sources such as ICP and NCI combined with quadrupole mass spectrometry provided chlorine specific detection and high sensitivity for higher chlorinated compounds but suffered from high background signals from other chlorine containing, co-eluting compounds in the sample which prevented the accurate measurement of PCB-specific chlorine isotope ratios. On the other hand, the use of GC-MS/MS in the selected reaction monitoring mode (SRM) provided selective and accurate measurements but suffered from lower sensitivity for higher chlorinated compounds.
Introduction
PCBs are a group of 209 structurally related chemical compounds (congeners) that constitute a class of ubiquitous pollutants with aromatic structure, high chemical stability and extremely poor water solubility.1 Although their manufacture has stopped already in 1977,2 these toxic compounds are distributed at a global scale through atmospheric transport and can be found in almost every environmental compartment including air, water, sediments, fish and mammals as well as in every region of the world.3,4 For these reasons, the development of reliable methods for PCB determination in environmental samples is still highly important.5PCBs have been mainly determined in real samples using gas chromatography (GC) coupled to electron capture detection (ECD) or mass spectrometry (MS).6 GC coupled to ECD or negative chemical ionization (NCI-MS) provides high sensitivity but suffers from a lack of selectivity as all chlorinated compounds present in the sample matrix are detected.7–9 In contrast, the use of electron ionization (EI) coupled to MS using the selective ion monitoring mode (SIM) increases selectivity as only analyte-specific molecular fragments are detected. GC coupled to tandem mass spectrometry (GC-MS/MS) with EI using selected reaction monitoring (SRM) mode enhances even more the selectivity of PCB determination compared to SIM.10
The coupling of GC to inductively coupled plasma mass spectrometry (ICP-MS) has been established as a powerful hyphenated technique in trace element speciation analysis,11 particularly for the determination of volatile organometallic contaminants based on the element-specific determination of the heteroatom present in the molecule.12 However, the determination of relevant halogenated contaminants by GC-ICP-MS is very limited13–17 due to the high ionisation potential of halogen atoms, their low ionization efficiency in an argon plasma as well as different strong polyatomic interferents formed inside the plasma. For these reasons, GC-ICP-MS has never been applied to the determination of PCBs. Indeed, the only publication that can be found in the literature dealing with the determination of PCBs by ICP-MS is based on the use of electrothermal vaporization with the aim of differentiating “organic” chlorine and inorganic chlorine.18 The use of ICP-MS instruments equipped with collision cells has been demonstrated to be an efficient approach to remove spectral interference and decrease the detection limits of phosphorus, sulphur, bromine and chlorine or iodine containing compounds when analysed by GC-ICP-MS.13 The recent commercialisation of triple quadrupole systems with the ICP source (ICP-QqQ) allows the improved resolution of problematic spectral interference19,20 when operated in MS/MS mode but it has not been evaluated as a GC detector thus far.
Isotope dilution mass spectrometry (IDMS) provides accurate and precise determinations directly traceable to the International System of Units.21 The determination of PCBs by IDMS has been traditionally carried out using multiply 13C labelled analogues and methodological calibration graphs, where the labelled analogues are employed as internal standards.22 We have recently presented the synthesis of 37Cl-labelled PCBs and their application to the IDMS determination of PCBs in solid samples by GC-EI-MS/MS.23 The use of 37Cl-labelled analogues has been shown to provide analytical figures of merit comparable to those obtained using 13C-labelled compounds as internal standards. On the other hand, the use of 37Cl-labelled compounds allows for the selection of alternative ionization sources, both elemental and molecular. We present here for the first time the comparison of three different ionization sources (EI, NCI and ICP) and four different MS techniques (GC-MS, GC-MS/MS, GC-ICP-MS and GC-ICP-MS/MS) for the detection of PCBs in solid samples and the determination of selected PCBs by IDMS using their 37Cl-labelled analogues. For this purpose, we carried out the evaluation of different ion sources and mass spectrometry configurations for the characterization of a set of twelve 37Cl-labelled congeners in terms of isotopic measurements. Finally, the analysis of the certified reference material SRM 1941b (marine sediment) was performed to evaluate the capabilities of the different mass spectrometric techniques for the accurate determination of selected PCBs at low ng g−1 levels in real samples by IDMS.
Experimental section
Reagents and materials
A 10 μg g−1 solution of twelve PCBs (congeners 18, 28, 31, 44, 52, 101, 118, 138, 149, 153, 180, and 194) was purchased from Sigma-Aldrich (St Louis, MO, USA). The 37Cl-labelled PCB mixture was synthesised in our laboratory as reported elsewhere.23 Working standard solutions of labelled and unlabelled PCBs were prepared in isooctane (Sigma-Aldrich) and stored in the dark at 4 °C until use. Hexane and acetone were obtained from Sigma-Aldrich. Florisil was obtained from Supelco (Bellefonte, PA, USA). PTFE-coated magnetic stirring bars and 10 mL glass vessels employed for the microwave extractions were purchased from CEM Corporation (Matthews, NC, USA). Cu powder was purchased from Merck (Darmstadt, Germany). The standard reference material SRM 1941b (organics in marine sediment) was obtained from the National Institute of Standards and Technology (NIST, Gaithersburg, USA).Instrumentation
Gas chromatography was coupled in this work to four different mass spectrometric techniques. A single quadrupole Agilent 7700 (Agilent Technologies, Tokyo, Japan) was coupled to a GC model 6890 (Agilent Technologies, Waldbronn, Germany) equipped with a cold on-column injector and a DB-5MS column (15 m × 0.25 mm i.d. × 0.1 μm film thickness) from Agilent (Santa Clara, USA). The same gas chromatograph was also coupled to an Agilent 8800 triple quadrupole ICP-MS (Agilent Technologies). A single quadrupole GC-MS model QP2010 Plus (Shimadzu, Kyoto, Japan) equipped with a NCI source, a split/splitless injector and a DB-5MS capillary column (30 m × 0.25 mm i.d., 0.25 μm film thickness) was also employed. Finally a gas chromatograph model Agilent 7890A (Agilent Technologies) fitted with a split/splitless injector and a DB-5MS capillary column (30 m × 0.25 mm i.d. and 0.25 μm film thickness) was coupled to an Agilent 7000 triple quadrupole mass spectrometer equipped with an EI source. The operating conditions of all mass spectrometric techniques are summarized in Tables S1, S2 and S3 of the ESI.† The extraction of the PCBs from the solid sample was carried out by using a focused microwave Explorer-12-Hybrid system (CEM Corporation). The centrifugation of the samples was performed using a centrifuge model CENTRO-8 BL (JP Selecta, Barcelona, Spain). An analytical balance model AB204-S (Mettler Toledo, Zurich, Switzerland) was used for the gravimetric preparation of all solutions and standards, while a mini-vap evaporator/concentrator (Supelco, St. Louis, MO, USA) was used for the evaporation of the sample extracts to their final volume before injection.Procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of hexane and acetone. After microwave irradiation the extracts were centrifuged at 3000 rpm for 5 minutes. Then, 0.3 g of Cu powder was added to remove residual sulphur which is present in the sample. To assist the sulphur removal the sample was sonicated for 30 min. In addition, all the extracts were cleaned with a Florisil column. Finally the extracts were preconcentrated with a gentle nitrogen flow until a volume of approximately 50 μL. Please note that the accurate knowledge of the final volume is not required by IDMS.
:1 mixture of hexane and acetone. After microwave irradiation the extracts were centrifuged at 3000 rpm for 5 minutes. Then, 0.3 g of Cu powder was added to remove residual sulphur which is present in the sample. To assist the sulphur removal the sample was sonicated for 30 min. In addition, all the extracts were cleaned with a Florisil column. Finally the extracts were preconcentrated with a gentle nitrogen flow until a volume of approximately 50 μL. Please note that the accurate knowledge of the final volume is not required by IDMS.
Results and discussion
Measurement of elemental chlorine isotope ratios by GC-ICP-MS, GC-ICP-MS/MS and GC-NCI-MS
First, the precision and accuracy of the measurement of the compound-specific chlorine isotope ratios 37Cl/35Cl were evaluated by GC-ICP-MS, GC-ICP-MS/MS and GC-NCI-MS. For this purpose, five independent injections of a mixture of the naturally abundant PCBs were carried out with each technique. The concentration of the naturally abundant standard employed was 200 ng g−1 when using the ICP-MS instruments whereas three different concentration levels (100, 500 and 1000 ng g−1) were tested when using the NCI source. The detector dead time was corrected in all measurements and the final results obtained are given in Table 1. A significant mass discrimination effect was observed when using GC-ICP-MS and GC-ICP-MS/MS instruments. When comparing the experimental values with the theoretical 37Cl/35Cl isotope ratio, the relative error varied from 0.6% to 11.2% for GC-ICP-MS and from 2.8% to 11.9% for GC-ICP-MS/MS. The precision obtained, calculated as the relative standard deviation (RSD%) from three GC injections, ranged from 2.3% to 11.2% for GC-ICP-MS and from 2.7% to 10% for GC-ICP-MS/MS. A much better accuracy and precision were obtained when using the GC-NCI-MS system. The relative error obtained in the isotope ratio for all congeners ranged from 0.01% to 2.6% while the RSD values ranged from 0.1% to 0.7%. In addition, we did not observe any difference in the measured isotope ratios at the three levels of concentration tested, indicating that an adequate detector dead time correction was applied and the absence of other detector effects within the concentration range tested.| PCBs congeners | GC-ICP-MS 200 ng g—1 | GC-ICP-MS/MS 200 ng g—1 | GC-NCI-MS 100 ng g−1 | GC-NCI-MS 500 ng g−1 | GC-NCI-MS 1000 ng g−1 |
|---|---|---|---|---|---|
| PCB-18 | 0.343 ± 0.014 | 0.329 ± 0.020 | 0.3229 ± 0.0012 | 0.3236 ± 0.0015 | 0.3283 ± 0.0016 |
| PCB-28+31 | 0.337 ± 0.012 | 0.345 ± 0.015 | 0.3246 ± 0.0005 | 0.3191 ± 0.0017 | 0.3222 ± 0.0021 |
| PCB-52 | 0.341 ± 0.011 | 0.356 ± 0.036 | 0.3235 ± 0.0002 | 0.3222 ± 0.0015 | 0.3265 ± 0.0016 |
| PCB-44 | 0.322 ± 0.009 | 0.331 ± 0.018 | 0.3227 ± 0.0007 | 0.3208 ± 0.0017 | 0.3248 ± 0.0016 |
| PCB-101 | 0.330 ± 0.028 | 0.348 ± 0.026 | 0.3241 ± 0.0004 | 0.3222 ± 0.0016 | 0.3262 ± 0.0017 |
| PCB-118 | 0.349 ± 0.010 | 0.358 ± 0.025 | 0.3229 ± 0.0002 | 0.3208 ± 0.0017 | 0.3250 ± 0.0018 |
| PCB-149 | 0.317 ± 0.036 | 0.349 ± 0.020 | 0.3253 ± 0.0003 | 0.3236 ± 0.0017 | 0.3276 ± 0.0016 |
| PCB-153 | 0.338 ± 0.013 | 0.342 ± 0.014 | 0.3237 ± 0.0004 | 0.3220 ± 0.0018 | 0.3266 ± 0.0016 |
| PCB-138 | 0.350 ± 0.008 | 0.349 ± 0.014 | 0.3234 ± 0.0003 | 0.3202 ± 0.0018 | 0.3249 ± 0.0020 |
| PCB-180 | 0.359 ± 0.023 | 0.356 ± 0.010 | 0.3238 ± 0.0008 | 0.3216 ± 0.0019 | 0.3266 ± 0.0017 |
| PCB-194 | 0.341 ± 0.013 | 0.352 ± 0.018 | 0.3236 ± 0.0006 | 0.3222 ± 0.0015 | 0.3272 ± 0.0016 |
Determination of the isotope enrichment of 37Cl-labelled PCBs
The isotopic enrichment of 37Cl in the different 37Cl-labelled PCB congeners contained in the spike solution employed in this work was calculated on the basis of the results obtained by all the different techniques. The values obtained by GC-ICP-MS, GC-ICP-MS/MS and GC-NCI-MS were directly calculated from the detector dead time and mass bias corrected measurement of the compound-specific 37Cl/35Cl isotope ratio. Mass bias was corrected by external bracketing injecting a naturally abundant standard containing the twelve target PCB congeners every three samples. The isotopic enrichments obtained by GC-EI-MS/MS23 were calculated as described elsewhere.24 Briefly, the isotope distribution of each congener is compared with theoretically derived spectra calculated for different tentative isotope enrichments. The best isotope enrichment was that which provided the minimum in the square sum of residuals for the linear regression between the theoretical and experimental spectra. In all cases, the uncertainty of the values corresponds to the standard deviation of n = 5 independent GC injections. The results obtained are given in Table 2. As can be seen, most of the isotopic enrichments obtained agree well between all techniques. We only found significant differences when comparing the values obtained by GC-NCI-MS and GC-EI-MS/MS in four congeners, mainly due to the low standard deviation of the values in comparison with those obtained using GC-ICP-MS and GC-ICP-MS/MS.| PCBs congeners | GC-ICP-MS | GC-ICP-MS/MS | GC-NCI-MS | GC-EI-MS/MS23 |
|---|---|---|---|---|
| PCB-18 | 93.98 ± 0.68 | 93.50 ± 0.43 | 94.30 ± 0.11 | 94.14 ± 0.06 |
| PCB-28+31 | 94.06 ± 0.23 | 93.65 ± 0.41 | 93.83 ± 0.09 | 94.05 ± 0.04 |
| PCB-52 | 94.25 ± 0.25 | 94.59 ± 0.51 | 94.42 ± 0.02 | 94.16 ± 0.03 |
| PCB-44 | 94.38 ± 0.56 | 93.78 ± 1.99 | 94.36 ± 0.06 | 94.06 ± 0.03 |
| PCB-101 | 94.48 ± 0.23 | 94.16 ± 1.76 | 94.13 ± 0.30 | 94.06 ± 0.06 |
| PCB-118 | 94.21 ± 0.74 | 92.93 ± 1.81 | 93.93 ± 0.12 | 93.99 ± 0.03 |
| PCB-149 | 92.27 ± 0.61 | 91.95 ± 0.66 | 92.49 ± 0.07 | 92.51 ± 0.02 |
| PCB-153 | 93.57 ± 0.84 | 92.00 ± 1.64 | 92.70 ± 0.29 | 92.70 ± 0.03 |
| PCB-138 | 93.56 ± 0.38 | 92.54 ± 1.13 | 92.71 ± 0.04 | 92.70 ± 0.03 |
| PCB-180 | 92.13 ± 0.47 | 91.59 ± 0.67 | 91.91 ± 0.06 | 91.93 ± 0.03 |
| PCB-194 | 91.25 ± 0.46 | 91.34 ± 0.60 | 91.65 ± 0.08 | 91.45 ± 0.03 |
Determination of the 37Cl-labelled PCB concentration by reverse IDMS
The concentration of the 37Cl-labelled PCBs contained in the spike solution employed in this work was measured by reverse IDMS. For each of the instruments tested, three independent blends of the spike solution and a naturally abundant standard containing all target congeners were prepared and injected each in triplicate. The concentrations obtained by GC-ICP-MS, GC-ICP-MS/MS and GC-NCI-MS were calculated applying the classical IDMS equation using the compound specific 37Cl/35Cl isotope ratio. The concentrations obtained by GC-EI-MS/MS were calculated by multiple linear regression as described elsewhere.23Fig. 1 shows the comparison of the results obtained with the four techniques. As can be observed, significant differences between the different MS techniques were obtained for all congeners. As there are no reference values for this spike solution we cannot discuss the accuracy of the different measurements. Therefore, technique-specific concentrations of the labelled PCBs were employed for the subsequent analysis of the certified reference material 1941b.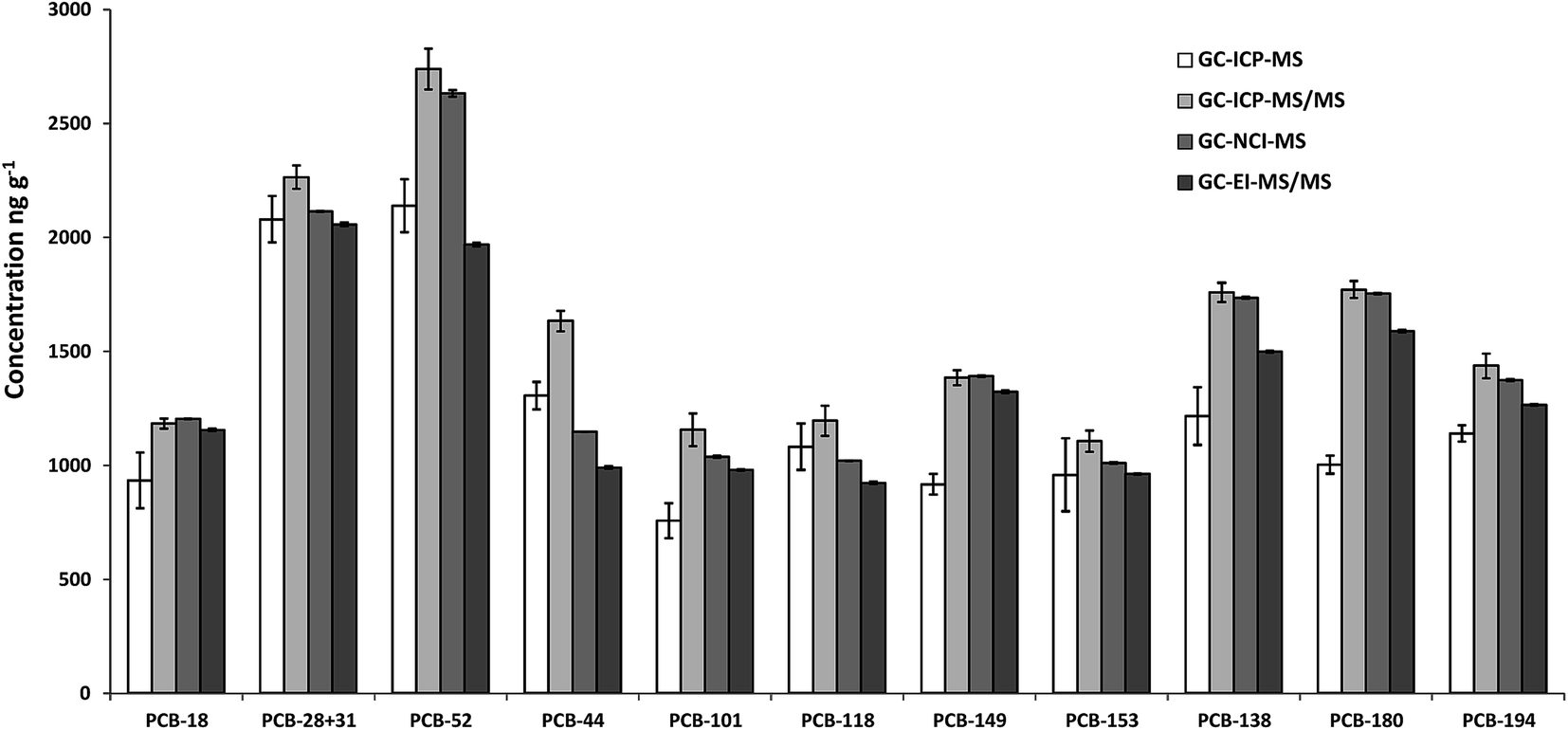 | ||
| Fig. 1 Concentration of the 37Cl-labelled PCBs contained in the spike solution employed in this work calculated by reverse IDMS using four different techniques (GC-ICP-MS, GC-ICP-MS/MS, GC-NCI-MS and GC-EI-MS/MS). Error bars correspond to the standard deviation of three independent blends of the spike solution and the naturally abundant standard containing all target congeners. | ||
Analysis of the certified reference material SRM 1941b (marine sediment)
We have previously demonstrated23 that the use of a 1:1 mixture of hexane and acetone and a focused microwave assisted extraction at 70 °C for 4 minutes provided quantitative recoveries for all target congeners present in the certified sediment SRM 1944 in a concentration range from 11 to 80 ng g−1. Therefore the same sample preparation procedure was employed here for the analysis of the sediment SRM 1941b. However, it is important to highlight that the concentration levels of the target PCB congeners in this material ranged from 1 to 8 ng g−1. It is also important to note that, besides the twelve target PCBs, this material also provides certified values for 30 additional PCB congeners as well as 7 chlorinated pesticides.
When using the GC-ICP-MS system (single quadrupole), helium was employed as the collision gas to remove spectral interference. However, preliminary analyses of this material showed that the sensitivity obtained was not enough to perform a reliable quantification of the target PCBs in the SRM 1941b concentration range. In addition to the low sensitivity, the coelution of other chlorinated compounds prevented the integration of most target congeners. Therefore, we could not provide concentration values for this material using GC-ICP-MS. It is worth stressing that the use of a 30 m column instead of a 15 m column might significantly improve the chromatographic resolution.
Fig. 2 shows the comparison of the certified values with the experimental values obtained in the analysis of SRM 1941b using GC-ICP-MS/MS, GC-NCI-MS and GC-EI-MS/MS. For GC-ICP-MS/MS analyses, H2 was employed as the reaction gas to remove spectral interference through the formation of 37Cl–H2 (m/z 39) and 35Cl–H2 (m/z 37).
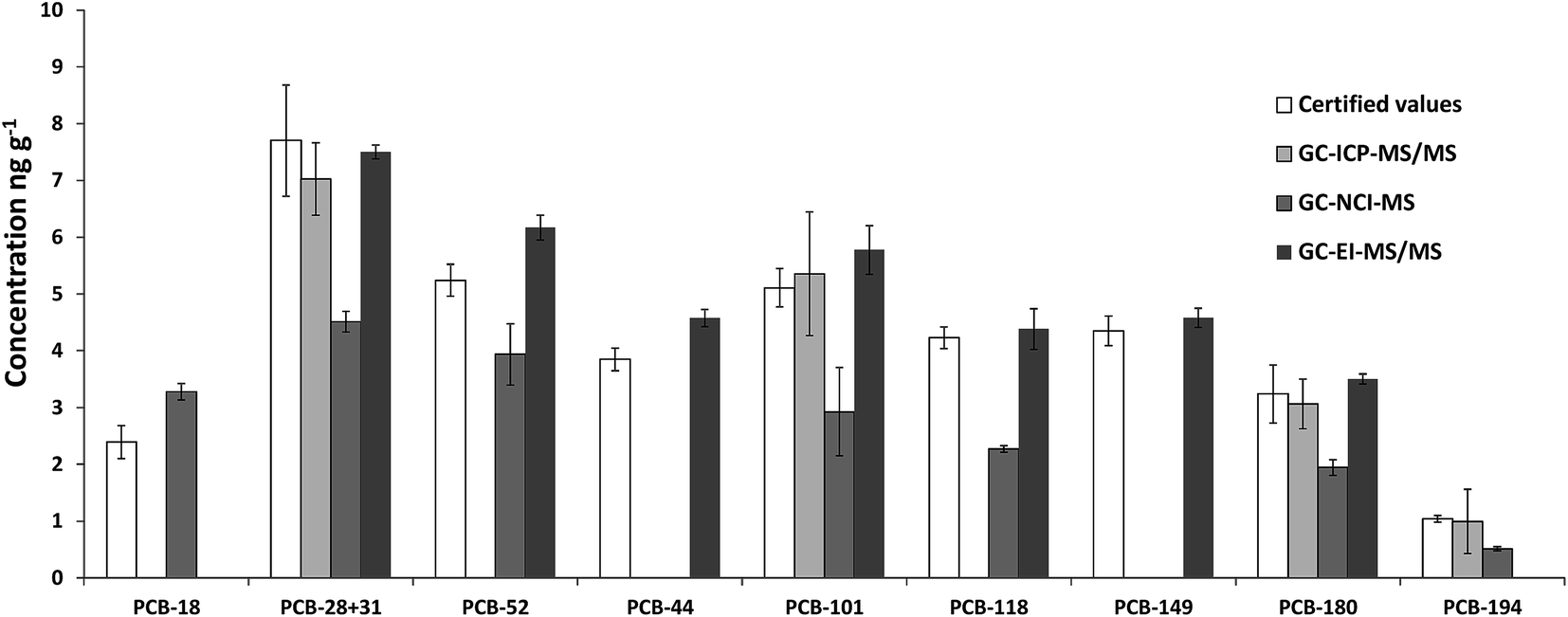 | ||
| Fig. 2 Comparison of the certified values and the experimental concentrations of the target PCBs obtained in the analysis of the SRM 1941b (organics in marine sediment) using three different techniques (GC-ICP-MS/MS, GC-NCI-MS and GC-EI-MS/MS). Error bars correspond to the standard deviations of n = 3 independent extractions. | ||
Fig. 3 shows a GC-ICP-MS/MS chromatogram of the SRM 1941b spiked with the mixture of 37Cl-labelled PCBs. The chromatogram for the transition 35Cl → 35Cl–H2 shows that the signal to noise ratio is not satisfactory and that the presence of other chlorinated compounds hampers the proper integration of many of the target congeners. In contrast, the chromatogram for the transition 37Cl → 37Cl–H2 shows a much better signal to noise ratio. Under these conditions, we could only determine experimental concentrations for PCB 28+31, 101, 180 and 194, as can be observed in Fig. 2, and they were in good agreement with the certified values. However, the RSD values obtained from three independent extractions ranged from 9 to 57%. Here the further improvement of the chromatographic separation conditions in terms of resolution (e.g. using a 30 m column length) might help to minimize possible coelution and therefore will improve the integration of the targeted compounds.
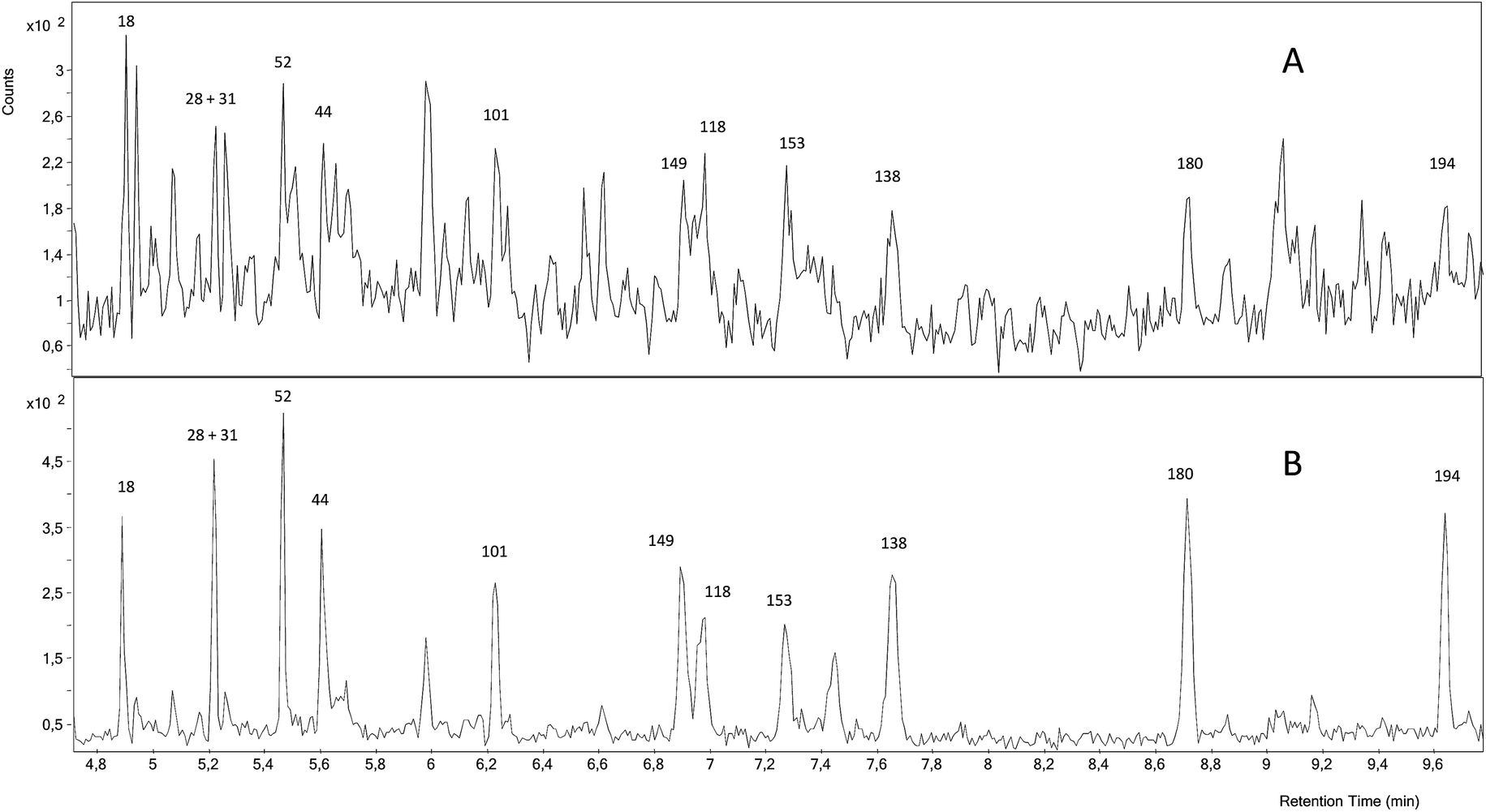 | ||
| Fig. 3 GC-ICP-MS/MS chromatogram of the certified reference sediment SRM 1941b spiked with the mixture of 37Cl-labelled PCBs. (A) chromatogram for the transition 35Cl → 35Cl–H2 and (B) chromatogram for the transition 37Cl → 37Cl–H2. | ||
Fig. 4 shows that the GC-NCI-MS system provides, among all the evaluated techniques, the highest sensitivity for PCB determination in SRM 1941b. However, the presence of other chlorinated compounds and an important background at m/z 35 hampered the proper integration of several congeners such as 138, 153 and 149. The results obtained using this technique for the rest of congeners are also shown in Fig. 2. As can be seen, we could not provide any concentration in agreement with the certified values of the material probably due to the high background at m/z 35. The RSD values obtained from three independent extractions ranged from 4 to 27%.
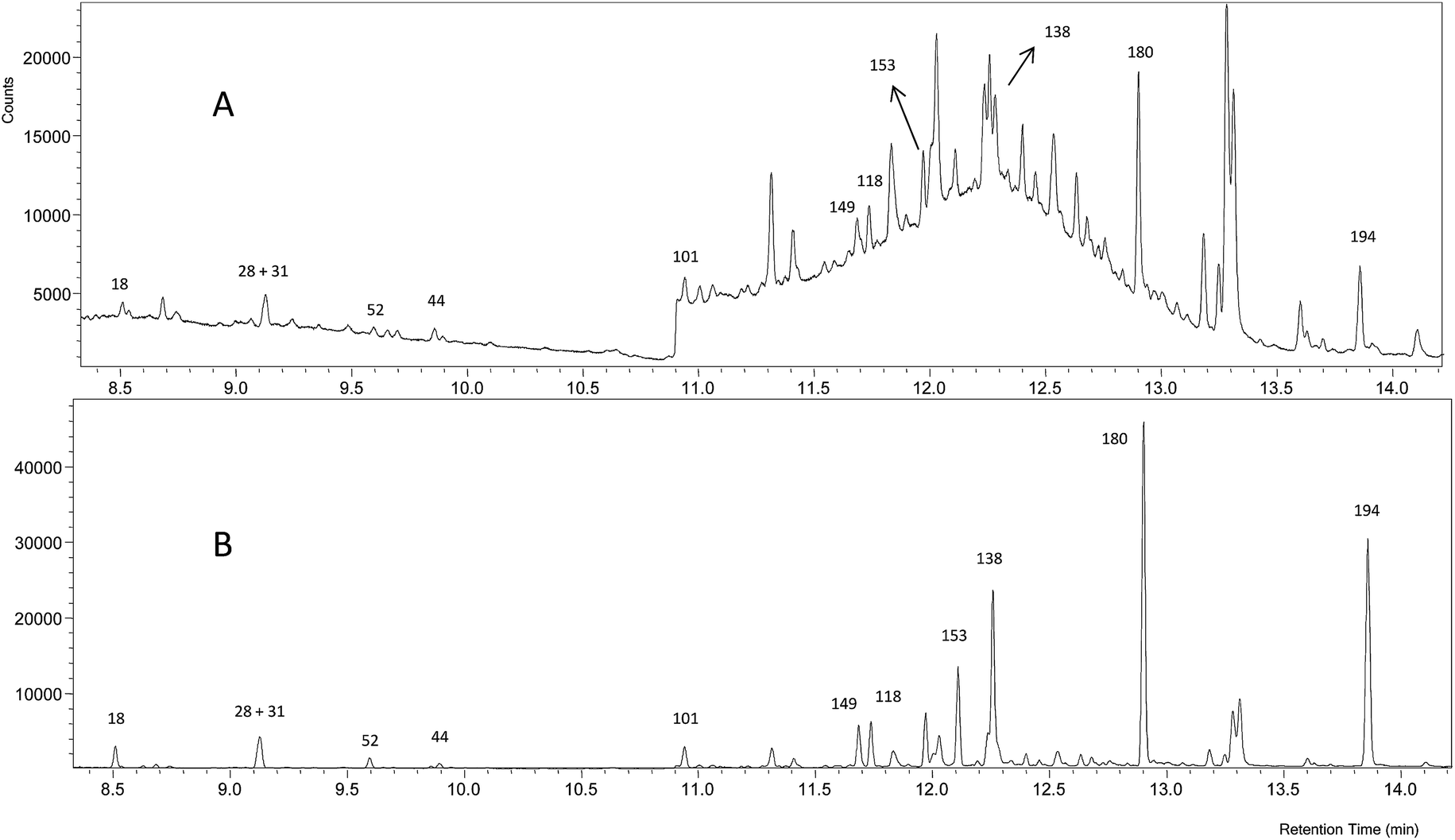 | ||
| Fig. 4 GC-NCI-MS chromatogram of the certified reference sediment SRM 1941b spiked with the mixture of 37Cl-labelled PCBs. (A) Chromatogram for 35Cl and (B) chromatogram for 37Cl. | ||
Finally, Fig. 5 shows that the GC-EI-MS/MS system provided the best signal to noise ratio and a very low background both for the naturally abundant PCBs and for the labelled analogues at the selected transitions. Using this technique, sequential detection windows based on the elution time and the PCB chlorination degree are programmed. In this way the only interference that may affect the analyte peak can only be due to a PCB congener with the same chlorination degree. As can be observed in Fig. 5, GC-EI-MS/MS allowed the accurate integration of all target congeners except for PCB 18, 153 and 138. Also, the concentration of PCB 194 was too low to provide an accurate integration. It is worth noting that none of the MS techniques employed in this work allowed us to obtain concentration values for PCB 153 and 138, indicating that the quantification problem for these congeners is due to an incomplete chromatographic separation rather than an instrumental limitation. Fig. 2 shows that GC-EI-MS/MS provided concentrations in good agreement with the certified values for the rest of the target PCBs with a precision ranging from 1.6 to 8.1% (RSD).
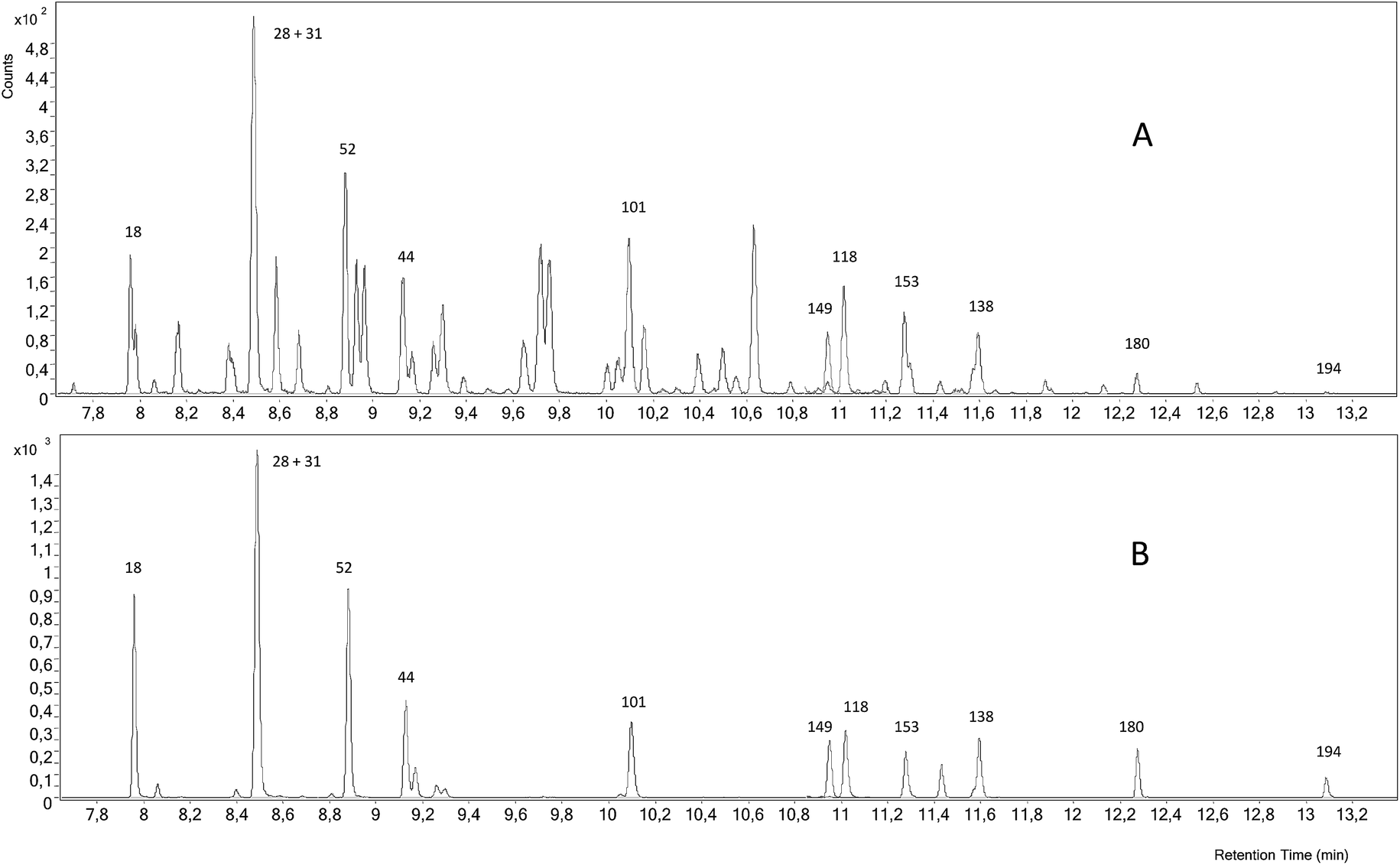 | ||
| Fig. 5 GC-EI-MS/MS chromatogram of the certified reference sediment SRM 1941b spiked with the mixture of 37Cl-labelled PCBs. (A) Chromatogram of the transitions for the naturally abundant PCBs and (B) chromatogram of the transitions for the 37Cl-labelled analogues. | ||
Conclusions
Using 37Cl-labelled PCBs it is possible to employ different ionization sources and mass analyzers to determine PCBs by IDMS. In this way, it is possible to compare the capabilities of different ion sources and mass analyzers coupled to GC. This work has shown that GC-EI-MS/MS is the technique that provides more accurate and precise results in the analysis of real samples as the SRM mode provides the most specific detection for each PCB congener. A higher instrumental sensitivity is obtained when using GC-NCI-MS, but the interference due to the presence of other co-eluting chlorinated compounds and the high background at m/z 35 prevented its application to real samples at low concentration levels. Nevertheless, it is worth commenting that accurate and precise compound specific isotope ratios were obtained by GC-NCI-MS. Finally, according to our results, the use of GC-ICP-MS and GC-ICP-MS/MS for the determination of PCBs offers no advantages over the more traditional GC-EI-MS/MS technique. Although we could obtain experimental concentrations in agreement with the certified values for 5 congeners in the analysis of SRM 1941b, the signal to noise ratio and the precision of the results were poor in comparison with GC-EI-MS/MS. In addition, the precision obtained in the measurement of the compound-specific chlorine isotope ratio in standards was not satisfactory, in comparison with that obtained by GC-NCI-MS. Here further instrumental improvements and optimizations might help to enhance the sensitivity and to further reduce the present interference. Also the optimization of the chromatographic conditions with respect to improving the resolution and therefore the separation between targeted PCBs and co-eluting as well as co-extracted chlorinated compounds will help to improve the performance of the different setups in the future.Acknowledgements
Financial support from the Spanish Ministry of Economy and Competitiveness through Project CTQ2012-36711 (cofounded by FEDER) is acknowledged. Additional funding for a research stay of Lourdes Somoano Blanco provided by the Helmholtz-Zentrum Geesthacht is also acknowledged. The UE is acknowledged for the provision of FEDER funds for the purchase of the GC-MS/MS instrument. P. Rodríguez-González acknowledges his research contract RYC-2010-06644 to the Spanish Ministry of Economy and Competitiveness through the Ramón y Cajal Program.References
- B. D. Rodan, D. W. Pennington, N. Eckley and R. Boethling, Environ. Sci. Technol., 1999, 33, 3482 CrossRef CAS.
- K. J. Kelly, E. Connelly, G. A. Reinhold, M. Byrne and D. J. Prezant, Arch. Environ. Health, 2002, 57, 282 CrossRef CAS PubMed.
- A. Beyer and M. Biziuk, Rev. Environ. Contam. Toxicol., 2009, 201, 137 CAS.
- K. Jaraczewska, J. Lulek, A. Covaci, S. Voorspoels, A. Kaluba-Skotarczak, K. Drews and P. Schepens, Sci. Total Environ., 2006, 372, 20 CrossRef CAS PubMed.
- H. P. Tang, TrAC, Trends Anal. Chem., 2013, 45, 48 CrossRef CAS PubMed.
- S. P. J. van Leeuwen and J. de Boer, J. Chromatogr. A, 2008, 1186, 161 CrossRef CAS PubMed.
- C. Danielsson, K. Wiberg, P. Korytár, S. Bergek, U. A. T. Brinkman and P. Haglund, J. Chromatogr. A, 2005, 1086, 61 CrossRef CAS PubMed.
- M. Ramil-Criado, D. Hernanz-Fernández, I. Rodríguez-Pereiro and R. Cela-Torrijos, J. Chromatogr. A, 2004, 1056, 187 CrossRef CAS PubMed.
- H. Kontsas and K. Pekari, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2003, 791, 117 CrossRef CAS.
- P. Plaza Bolaños, A. Garrido Frenich and J. L. Martinez Vidal, J. Chromatogr. A, 2007, 1167, 9 CrossRef PubMed.
- M. Popp, S. Hann and G. Koellensperger, Anal. Chim. Acta, 2010, 668, 114 CrossRef CAS PubMed.
- B. Bouyssiere, J. Szpunar and R. Lobinski, Spectrochim. Acta, Part B, 2002, 57, 805 CrossRef.
- D. Pröfrock, P. Leonhard, S. Wilbur and A. Prange, J. Anal. At. Spectrom., 2004, 19, 623 RSC.
- N. Fidalgo-Used, M. Montes-Bayon, E. Blanco-Gonzalez and A. Sanz-Medel, Talanta, 2008, 75, 710 CrossRef CAS PubMed.
- A. Gonzalez-Gago, J. M. Marchante-Gayon and J. I. Garcıa Alonso, J. Anal. At. Spectrom., 2007, 22, 1138 RSC.
- J. L. Gomez-Ariza, T. Garcıa-Barrera and F. Lorenzo, J. Anal. At. Spectrom., 2005, 20, 883 RSC.
- J. L. Gomez-Ariza and T. Garcıa-Barrera, J. Anal. At. Spectrom., 2006, 21, 884 RSC.
- P. Richner and S. Wunderli, J. Anal. At. Spectrom., 1993, 8, 45 RSC.
- S. Diez Fernández, N. Sugishama, J. Ruiz Encinar and A. Sanz-Medel, Anal. Chem., 2012, 84, 5851 CrossRef PubMed.
- L. Balcaen, G. Woods, M. Resano and F. Vanhaecke, J. Anal. At. Spectrom., 2013, 28, 33 RSC.
- J. I. García Alonso and P. Rodríguez González, in Isotope Dilution Mass Spectrometry, Royal Society of Chemistry, Cambridge, 2013 Search PubMed.
- T. Otake, Y. Aoyagi, T. Yarita and M. Numata, Anal. Bioanal. Chem., 2010, 397, 2569 CrossRef CAS PubMed.
- L. Somoano Blanco, P. Rodriguez-Gonzalez, S. García Fonseca and J. I. Garcia Alonso, Anal. Chem., 2015, 87, 7840 CrossRef CAS PubMed.
- A. González-Antuña, P. Rodríguez-González and J. I. García Alonso, J. Mass Spectrom., 2014, 49, 681 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ay01752a |
| This journal is © The Royal Society of Chemistry 2015 |