
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA flow-through enzymatic microreactor for the rapid conversion of triacylglycerols into fatty acid ethyl ester and fatty acid methyl ester derivatives for GC analysis
S. T.
Anuar
*ab,
S. M.
Mugo
c and
J. M.
Curtis
a
aLipid Chemistry Group (LCG), Department of Agricultural, Food and Nutritional Sciences (AFNS), University of Alberta, Edmonton, AB T6G 2P5, Canada
bSchool of Marine Science and Environment, Universiti Malaysia Terengganu, 21030 Terengganu, Malaysia. E-mail: sabiqahanuar@umt.edu.my
cDepartment of Physical Sciences (Chemistry), Grant MacEwan University, Edmonton, AB T5J 4S2, Canada
First published on 8th June 2015
Abstract
A flow-through enzymatic microreactor for the rapid conversion of triacylglycerols (TAG) into fatty acid ethyl ester (FAEE) or fatty acid methyl ester (FAME) derivatives was developed. The microreactor was a porous silica monolith fabricated within a 320 μm ID fused silica capillary with lipase from Candida antarctica immobilized onto the large surface area of the monolith. The microreactor was used for the room temperature ethanolysis of TAG from edible oils including canola, sesame, soybean and refined-bleached-deodorized palm oil. GC/MS-NCI and GC/FID were used to prove the identification of the FAEE and FAME products. The microreactor completely transformed the starting oils into FAEEs or FAMEs, without the use of any reagents other than alcohol, in quantities suitable for GC analysis. The prototype microreactors were reusable >5 times with ethanol and 2 times with methanol. The FAEE products obtained using the microreactor were similar to those produced using commercial Novozyme 435 enzyme beads as well as by catalysis with ethanolic H2SO4.
Introduction
Since at least 5 decades ago, gas chromatography (GC) has been the most widely used method to characterize the fatty acid composition of fats and oils as their methyl ester derivatives (FAME).1–6 During this period of time, there have been many developments in both GC technology and in derivatization techniques.4,7,8 The transesterification of animal or vegetable triacylglycerol (TAG) by methanol involves the use of either an alkaline or acid catalyst and the reaction conditions have been optimized for temperature, amount and time4–11 leading to the development of a number of widely used official methods. For instance, both the AOCS official methods Ce 2-66 and Ce 1b-89 include the use of a boron trifluoride/methanol (BF3/MeOH) solution to convert the lipid sample into FAMEs prior to GC analysis.11,12 In addition, there are several other standard methods for preparing FAMEs including AOCS Ce 1k-09 and AOAC 965.49.13,14 These methods have been shown to produce quantitative amounts of FAMEs but usually involve multiple steps of sample derivatization and work-up prior to obtaining the final solution for GC analysis. Another disadvantage is the time and cost for these procedures as well as the need for handling chemical reagents including BF3, strong acids or bases and various organic solvents. Hence, a move towards enzyme catalyzed transesterification reactions for analytical applications could be advantageous. It should be noted that although most methods use FAME derivatives, comparable separation of fatty acid ethyl esters (FAEEs) by GC is readily achieved. Thus, some official methods for the analysis of oils already in the FAEE form do not require conversion to FAMEs prior to GC analysis, such as Ph.Eur.2063 for omega-3-acid ethyl esters.15Recently, considerable advances have been made in industrial production of biodiesel and food grade ethyl esters using enzyme mediated approaches.16–20 The use of lipases is advantageous since it benefits from milder conditions, avoids the need for strong acids and bases and can result in high yields of fatty acid alkyl esters (FAAEs) with few side products.16–21 A challenge to the success of a biocatalytic process is the cost of the enzyme and ensuring its reusability under the reaction conditions. However, enzymes can be immobilized onto high surface area supports which can both facilitate the catalysis and aid in enzyme re-use. In this research, we propose to immobilize enzymes within a ‘microreactor’, a structure containing channels or networks in the μm range.22 This provides a high internal surface area for enzyme support and ultimately for efficient small-scale reactions. In the microreactor systems considered here, chemical reactions can take place under conditions of continuous flow. The substrate can be passed through the microreactor for as many cycles as the enzyme remains active, so that the microreactor is both versatile and reusable. Commercially, some types of microreactors for chemical synthesis are made by photolithographic and wet-etching techniques.23 However, in order to develop a process amenable to laboratory fabrication, other approaches are required. Here, we use a silica support, a platform with great potential for chemical modifications to allow the immobilization of catalysts including enzymes, organometallics or metals.23–28
Previously, we have reported the use of microreactors in which lipase was immobilized onto either a silica monolith (SM) within a fused-silica capillary or immobilized within a fused silica microstructured fiber (photonic fibre, MSF).29,30 These studies demonstrated the effectiveness of the prototype devices in transforming triolein into fatty acid ethyl esters29 and into 2-monoolein,30 depending on the experimental conditions. Lipase from Candida antarctica was chosen since it is a widely used and commercially produced enzyme for lipid transesterification.16–18,20,27–32 Others also have shown the use of enzymatic microreactors for chemical synthesis33,34 and as biosensor platforms.35,36
The present study investigates the possibility of using lipase microreactors for the transesterification of triacylglycerols into FAAE for GC analyses as an alternative to the widely used chemical procedures for derivatization to FAMEs.
Experimental procedures
Materials
Fused silica (ID: 320 μm) and microstructured fiber optic (MSF) (F-SM35, ID: 12–13 μm, outer diameter: 480 μm, 90 holes) capillaries were obtained from Polymicro Technologies (Pheonix, AZ, USA) and Newport Corp. (Irvine, CA, USA), respectively. A Harvard Model’11 Plus syringe pump was purchased from Harvard Apparatus (Holliston, MA, USA). All food grade canola (CanO), sesame seed (SSO) and soybean (SBO) oils were purchased from a local grocery store. Refined-bleached-deodorized palm olein (RBD-PO) was obtained from Malaysian Palm Oil Board. 3-(Aminopropyl)triethoxylane (APTES, 99%), sodium sulfate, sodium carbonate, sodium chloride, glutaraldehyde solution (BioChemika, ∼50% in H2O), sodium cyanoborohydride (reagent grade, 95%), Novozyme 435 lipase beads and lipase from C. antarctica (EC 3.1.1.3, BioChemika) were obtained from Sigma-Aldrich (Oakville, ON, Canada). Sulphuric acid and acetic acid (glacial, HPLC) were purchased from Fisher Scientific (Bridgewater, NJ, USA). Pure triolein (>99%) and all C16:0, C18:0, C18:1, C18:2 and C18:3 ethyl ester (FAEE) standards were purchased from Nu-Chek (Elysian, MN, USA). All organic solvents were of HPLC analytical grade which were obtained from Sigma-Aldrich (Oakville, ON, Canada).Fabrication of SM and MSF microreactors
The silica monolith (SM) was fabricated following the procedure described in detail earlier29 and is outlined in Fig. 1. The procedure for the immobilization of free lipase onto either the SM or within a silica microstructured fibre (MSF) involves the coupling of a linker molecule, aminopropyl ethoxysilane (APTES) onto the silica surface (Fig. 1). The lipase is then immobilized onto the active site by means of glutaraldehyde.27–30 Using these procedures, a number of identical SM and MSF microreactors were prepared for this study. | ||
| Fig. 1 Reaction scheme for the small-scale fabrication of silica monolith (SM) and microstructure fiber (MSF) capillary biocatalyst microreactors. | ||
Direct ethylation using the microreactors
Selected reaction mixtures of (i) 0.5 mg mL−1 canola oil (CanO), (ii) 0.5 mg mL−1 sesame seed oil (SSO) and (iii) 0.5 mg mL−1 soybean oil (SBO) were prepared in ethanol (EtOH) and vortexed vigorously to dissolve the lipids in ethanol. 0.5 mg mL−1 refined-bleached-deodorized palm olein (RBD-PO) was prepared in ethanol with excess of hexane (ethanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexane, 1:2, v/v). Each substrate solution was infused through the enzymatic microreactors at room temperature at 0.3 μL min−1 for 5 hours using a Harvard Model’11 Plus syringe pump. The eluent was collected and prepared in dichloromethane (DCM) prior to GC/FID and GC/MS injection.
:hexane, 1:2, v/v). Each substrate solution was infused through the enzymatic microreactors at room temperature at 0.3 μL min−1 for 5 hours using a Harvard Model’11 Plus syringe pump. The eluent was collected and prepared in dichloromethane (DCM) prior to GC/FID and GC/MS injection.
Batch ethylation using commercialized lipase beads
The same reaction mixtures of ethanolic CanO, SSO, SBO, and RBD-PO were each stirred at 80 rpm in the presence of 3 mg of commercialized lipase beads (Novozyme 435) in a small vial for 5 hours at room temperature. Aliquots of the ethylation reaction mixture (∼100 μL) were collected (i) after 10 min and (ii) after 5 h of the reaction. Samples were filtered through a syringe filter and diluted 5 times in DCM for GC/MS and GC/FID analyses.Acid-catalyzed ethylation using H2SO4
The ethylation of TAG in CanO, SSO, SBO and RBD-PO using an acid-catalyzed method followed a procedure described by Christie (1989).37 Briefly, 1 mg of each vegetable oil was mixed with 1% H2SO4 in 2 mL EtOH. 1 mL toluene was added to the reaction mixtures before they were left overnight at 50 °C. Then, the samples were washed with 5 mL of 5% NaCl in H2O before adding another 5 mL hexane to extract the ethyl esters. The extraction with hexane was performed twice to ensure that all of the FAEEs were extracted. The hexane layer was then washed with 2% NaHCO3 solution (2 mL) and then dried with 2 g of anhydrous sodium sulphate (Na2SO4). The solution was filtered and the lipid was concentrated under N2 gas. Prior to GC analysis, all samples were weighed and diluted with DCM.Reusability of the microreactor for selected alcohols
To test the robustness and reusability of the microreactor for performing alcoholysis, 2 identical SM microreactors were tested using (i) 0.2 mg mL−1 triolein in EtOH and (ii) 0.2 mg mL−1 triolein in methanol:toluene (1:2, v/v), both at 0.3 μL min−1 at room temperature. Each condition was repeated on the same microreactor 5–8 times, and the transesterification products that eluted from the microreactors were collected, identified and quantified by GC/MS-NCI and GC/FID. The uncorrected GC/FID peak areas were expressed as a percentage of the total peak area for FAEEs or FAMEs. The reusability of the microreactor was further tested using SSO, a natural oil sample. GC/FID peak areas of the resulting FAEE products were expressed as uncorrected area percentages. For each component, the overall precision was estimated by calculating the standard deviations over 5 runs.
GC/MS-NCI
An Agilent GC/MS system of GC/MS 5975C (Agilent Technologies, Santa Clara, CA, USA) operating in negative ion chemical ionization mode (NCI) was used with ammonia as reagent gas. All data were collected by Agilent Chemstation software (version G1701EA). The GC column used for separation was an HP-5MS 5% phenyl methyl siloxane (30 m × 0.25 mm × 0.25 μm) column (Agilent technologies; Santa Clara CA, USA). The make-up gas was He at a flow rate of 1 mL min−1, in split mode at 20:1. Reaction product samples were dissolved in DCM (1 mL) prior to injection. The injection volume was 2 μL and the split/splitless injector temperature was set at 275 °C. The oven program started at a temperature of 80 °C (0 min), and then increased at 15 °C min−1 until reaching 150 °C (0 min) before increasing again at 10 °C min−1 to 260 °C (15 min). A mass spectrometer scan range of m/z 35 to 600 was used.
GC/FID
An Agilent 7890 GC system (Agilent Technologies, Santa Clara, CA, USA) equipped with a flame ionization detector (FID), an autosampler and a split/splitless injector was used to run samples and standards on a BPX-70 column 110 m × 0.25 mm × 0.25 μm (Agilent Technologies; Santa Clara, CA, USA) and all data were collected by using Agilent Chemstation software (version G1701EA). The GC parameters were: 2 μL injection volume, split ratio 20:1, FID carrier gas: H2, flow 2 mL min−1, inlet temperature 250 °C, detector temperature 250 °C, make-up gas: He. Temperature program: 140 °C (hold 5 min); 8 °C min−1 to 180 °C (0 min); 4 °C min−1 to 210 °C (0 min); 20 °C min−1 to 270 °C (hold 7 min). All reaction product samples were dissolved in DCM prior to injection.
NARP-HPLC/ELSD
In order to confirm the disappearance of TAG from starting oil after the reaction, a non-aqueous reversed phased high-performance liquid chromatography (NARP-HPLC) analysis was performed using an Agilent 1200 HPLC system equipped with an evaporative light scattering detector (ELSD 1260 Infinity, Agilent Technologies, Santa Clara, CA, USA). Separations were achieved using an Agilent Zorbax HT C18 column (4.6 mm × 50 mm, 1.8 μm, Agilent Tech, Santa Clara, CA, USA) with a gradient of solvent A, acetonitrile:methanol (1:4, v/v) and solvent B, hexane:isopropanol (5:4, v/v). The starting condition was 0% B and held for 5 min, then increased to 70% B in 13.5 min, and held for another 5.5 min, before returning to 0% B at 19.1 min. This condition was held for 1 min (t = 20.1 min) in order to equilibrate the column. The ELSD temperature was set to 33 °C, with a N2 gas flow of 3 L min−1 at a pressure of 3.5 bar, and all data were collected by using Agilent Chemstation software (version G2180BA).
Results and discussion
A comparison of procedures for the ethanolysis of vegetable oils
Usually, the analysis of fatty acids by GC is preceded by their conversion into fatty acid alkyl esters. Previously, we have shown the potential use of microreactors for such simple lipid conversions.27–30 Here, we describe the conversion of triacylglycerol mixtures from edible oils into their FAEE derivatives using two types of laboratory prepared enzymatic capillary microreactors (SM and MSF) that were optimized for use at a low flow rate (0.3 μL min−1) and at room temperature. Under these conditions, the microreactors produce FAEEs in amounts that are suitable for GC analysis. The conversions of canola, sesame, soybean and RBD-palm oils into FAEEs were tested to evaluate the performance of the microreactors, as shown in Table 1. The results are expressed as uncorrected GC/FID peak area percentages.| FAEE compositione (%) | |||||
|---|---|---|---|---|---|
| C16:0 | C18:0 | C18:1 | C18:2 | C18:3 | |
| a Collected from the Silica Monolith Microreactor at 0.3 μL min−1 (constant continuous conversion over >5 h). b Collected from the Silica MSF Microreactor at 0.3 μL min−1 (constant continuous conversion over >5 h). c Collected by vortexed reactants with Novozyme 435 at 100 rpm after 5 h. d Collected from transesterification of the reactant using an acid catalyst after 12 h. e The FAEE compositions are uncorrected GC/FID peak area percentages. | |||||
| Canola oil (CO) | |||||
| SM microreactora | 4.2 | 1.1 | 69.7 | 18.2 | 5.9 |
| MSF microreactorb | 4.3 | 1.2 | 68.5 | 17.2 | 6.2 |
| Novozyme 435c | 4.2 | 1.1 | 69.9 | 17.0 | 5.7 |
| H2SO4 catalystd | 4.4 | 1.1 | 68.4 | 17.1 | 5.1 |
|
|||||
| Sesame seeds oil (SSO) | |||||
| SM microreactora | 9.3 | 5.3 | 40.2 | 43.2 | 1.2 |
| MSF microreactorb | 9.7 | 6.3 | 39.5 | 42.9 | 1.0 |
| Novozyme 435c | 9.2 | 5.6 | 40.4 | 43.1 | 1.2 |
| H2SO4 catalystd | 9.2 | 5.2 | 39.6 | 42.6 | 1.3 |
|
|||||
| Soybean oil (SYO) | |||||
| SM microreactora | 10.0 | 1.0 | 22.8 | 56.4 | 9.6 |
| MSF microreactorb | 9.7 | 1.0 | 22.5 | 56.6 | 9.2 |
| Novozyme 435c | 9.3 | 1.0 | 21.9 | 56.4 | 9.8 |
| H2SO4 catalystd | 8.9 | 1.2 | 21.1 | 53.8 | 8.9 |
|
|||||
| RBD palm olein (RBDPO) | |||||
| SM microreactora | 43.8 | 8.9 | 36.7 | 9.5 | 0.4 |
| MSF microreactorb | 43.6 | 8.9 | 36.6 | 9.2 | 0.4 |
| Novozyme 435c | 42.1 | 8.4 | 35.4 | 9.4 | 0.4 |
| H2SO4 catalystd | 40.8 | 8.5 | 33.2 | 9.3 | 0.4 |
The transesterification of CanO in ethanol using the SM and MSF microreactors resulted in virtually identical FAEE peak areas in the GC/FID trace, as indicated in Table 1. Furthermore, NARP-LC/ELSD analysis of the microreactor product indicated complete conversion from the TAG to the EE form of the lipid (data not shown). Quantitative conversion into FAEEs was expected based on a previous study in which a lipase immobilized silica monolith succeed in converting a pure standard of triolein (C18:1) completely into ethyl oleate when at room temperature and with flow rates through the microreactor of 0.2–0.5 μL min−1.29 In the case of conversion of RBD-PO, which has a high level of palmitic acid, the solubility in ethanol was very sparing and so hexane was added (1:2 v/v ethanol/hexane) in order to prepare a solution. However, under the same conditions of flow rate and temperature, the presence of hexane did not affect the conversion of TAG to FAEEs and complete conversion to ethyl esters was achieved.
Previous studies have also shown that the addition of modest amounts of organic solvents such as n-hexane, di-ethyl- and di-isopropyl-ether (DIPE) to enzymatic reactions in lipid transformations does not result in enzyme deactivation.20,38,39 The fatty acid distributions measured by GC/FID for the FAEE of each of the 4 oils following transesterification catalysed within the MSF or SM microreactors were all consistent with literature reports.29,30
The MSF and SM catalysed ethanolysis was then compared to ethanolysis of identical oils using commercial immobilized lipase beads (Novozyme 435, lipase immobilized on acrylic resin) as well as to a conventional acid catalysis using sulphuric acid.37 The FAEE compositions from both methods were consistent with the distribution of FAEEs that were produced from MSF and SM microreactors (Table 1), within experimental errors. Examples of GC/FID chromatograms for transesterification of canola oil in ethanol are shown in Fig. 2. Fig. 2B and C show the ethylation products that were eluted from the SM and MSF microreactors. These products had retention times that matched those of the corresponding FAEE standards (Fig. 2A). In addition, the FAEE distribution and retention times obtained using commercial immobilized lipase (Fig. 2D) and the acid catalyst (Fig. 2E) were consistent with results from MSF and SM microreactors.
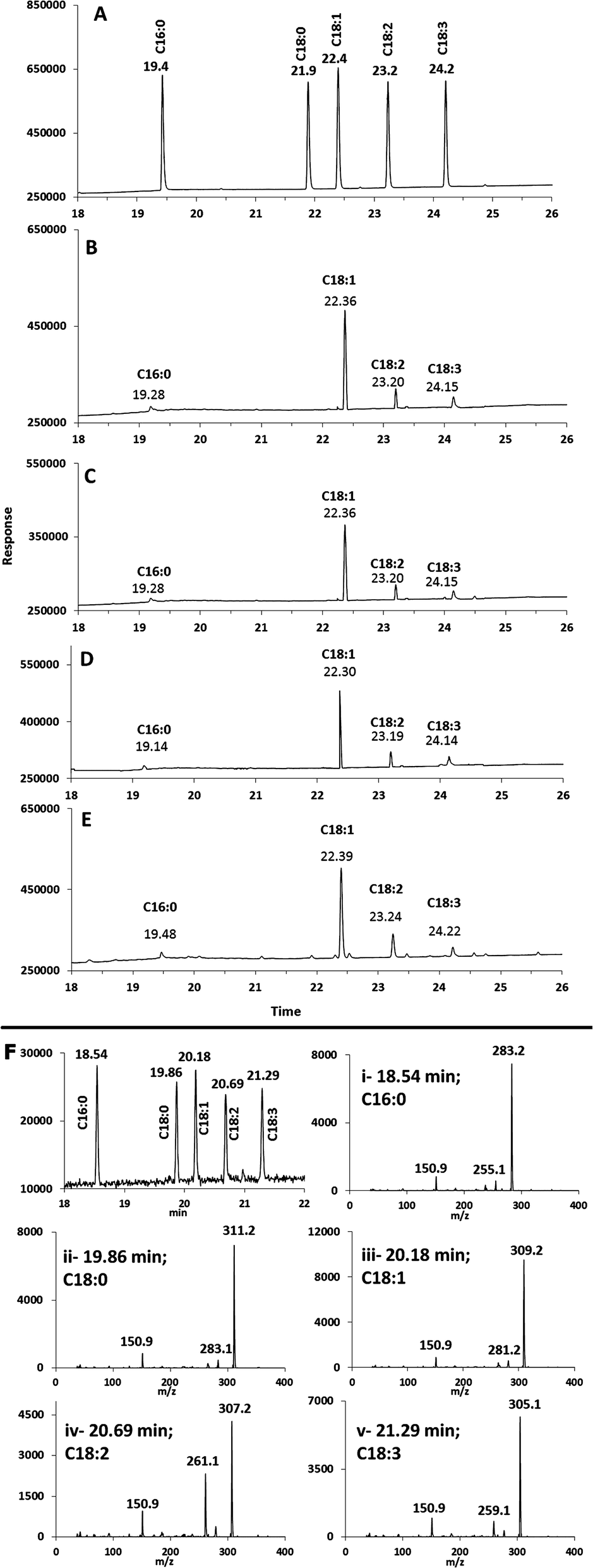 | ||
| Fig. 2 Comparison of GC/FID chromatograms for (A) ethyl ester FA standards and the products of canola oil (CanO) + EtOH transesterification using (B) SM microreactor at 0.3 μL min−1 flow-rate; (C) MSF microreactor at 0.3 μL min−1 flow-rate; (D) lipase beads after 5 h reaction time; (E) H2SO4 acid catalysis after 12 h reaction time; (F) GC/MS-NCI separation and spectra for ethyl ester FA standards. | ||
All of the reaction products were also identified by GC/MS using negative ion chemical ionization (GC/MS-NCI). In all four cases, the GC/MS-NCI traces, as exampled in Fig. 2F, closely resembled the GC/FID traces seen in Fig. 2A–E. Furthermore, the NCI mass spectra of the major peaks confirm the presence of FAEEs for both the MSF and SM; this is shown in Fig. 3 for the example of conversion of sesame seed oil in the SM. Specifically, the presence of ethyl oleate (C18:1) and ethyl linoleate (C18:2) was confirmed by their [M − H]− ions at m/z 309.1 and m/z 307.1 (Fig. 3C and D) as well as the characteristic fragment ions due to the loss of ethanol. Similarly, the [M − H]− ions for both FAEEs of palmitic and stearic acid GC peaks were observed (Fig. 3A and B). It is of importance to demonstrate the performance of the enzymatic microreactor in comparison to the use of the same lipase that is available commercially immobilized onto 0.3–1 mm sized beads since it might be possible to perform the transesterification using these, albeit with a considerably larger quantity of starting oil. Fig. 4 shows examples of the results for the transesterification of SBO in ethanol under several conditions: (A) SM microreactor at 0.3 μL min−1, (B) lipase beads (Novozyme 435, stirred for 10 min), and (C) lipase beads (Novozyme 435, stirred for 5 hours).
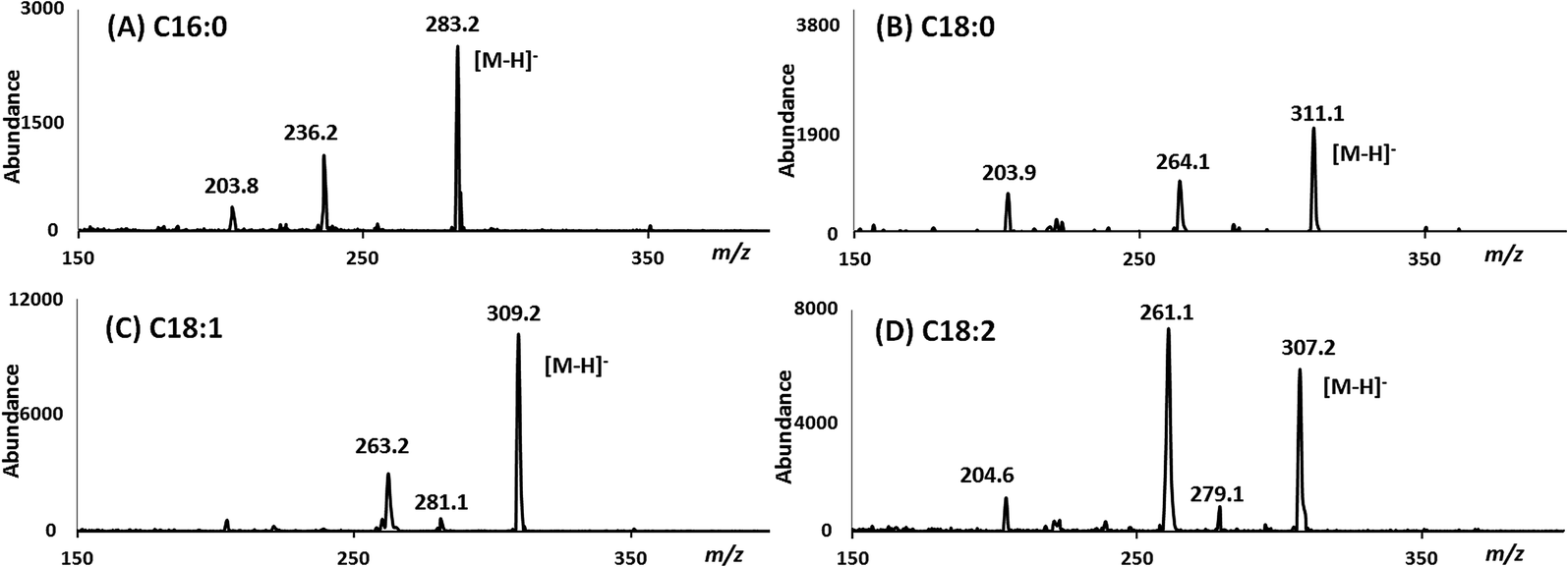 | ||
| Fig. 3 NCI-mass spectra from GC/MS separations of the products of sesame seed oil (SSO) and ethanol passing through the SM microreactor. Shown are examples of FAEEs identified including (A) C16:0 ethyl ester, (B) C18:0 ethyl ester, (C) C18:1 ethyl ester, and (D) C18:2 ethyl ester. | ||
 | ||
| Fig. 4 Comparison of the GC/MS-NCI total ion current (TIC) and NARP-LC/ELSD traces for the transesterification products of soybean oil (SBO) + EtOH using (i) (A and D) SM microreactor at 0.3 μL min−1, (ii) (B and E) Novozyme 435 lipase beads, 10 min reaction time, (iii) (C and F) Novozyme 435 lipase beads, 5 h reaction time and (iv) (G): NARP-LC/ELSD of SBO starting oil. | ||
The degree of conversion from TG to FAEEs was demonstrated by the NARP-LC/ELSD traces for the same experiments as shown in Fig. 4D–F, respectively (Fig. 4G is the starting material given for comparison). The ELSD was chosen for this purpose because of their versatility and availability. Both the unreacted oil and the product can be identified by this method. The result gives an indication of the relative reaction rates catalyzed by the same lipase either immobilized onto the monolithic microreactor (SM) support or onto beads. Comparing the manufacturer specified enzyme activity for the beads to the measured enzyme activity in the SM30 would predict a somewhat higher activity in the beads based on the conditions used here. However, what was found was that after 10 min reaction using lipase beads stirred in a vial (1 mL ethanol, 0.5 mg oil, 3 mg Novozyme 435 beads, see Fig. 4B and E), the starting SBO was only partially converted to FAEEs and the diacylglycerol intermediates were also present. After 5 hours of the stirred reaction at room temperature, there is complete conversion to FAEEs as seen in Fig. 4C and F.
In contrast, using the flow-through microreactor, rapid ethanolysis of SBO was achieved (Fig. 4A and D) resulting in complete conversion of TAG during the residence time of approximately 190 s. This greatly enhanced rate of ethanolysis (190 s vs. up to 5 h) can be partly explained by the vastly increased surface area available for the reaction as the solution passes through the monolithic structure (Fig. 1) which may be particularly important due to the formation of a biphasic system as glycerol and water are liberated. In addition, over time, the enzymatic action on complex lipid mixtures such as in SBO may also result in acyl migrations (i.e. interesterification reactions) that compete with ethanolysis.31 These processes, especially the much lower catalytic surface area, may be responsible for the much lower rate of ethanolysis with the lipase immobilized onto beads.
In summary, it was shown that at room temperature and low flow rates, the SM microreactor is a flow-through system that is able to achieve quantitative conversion of oils into FAEE derivatives. In order to demonstrate the reusability of SM microreactors, transesterification was performed by passing a 0.2 mg mL−1 solution of triolein in ethanol at a flow rate of 0.3 μL min−1 through a single microreactor at room temperature. 8 consecutive experiments were performed, separated by a sodium phosphate buffer (pH 7.23) flush between each run. It was found that 6 repeat ethanolysis reactions could be performed without any loss of conversion efficiency (see Fig. 5).
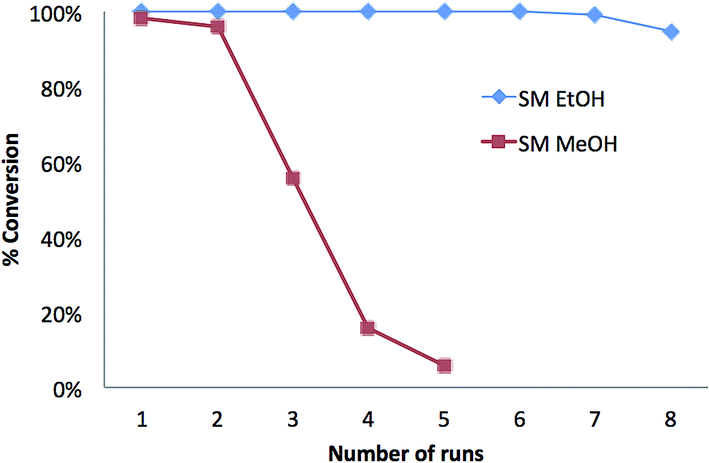 | ||
| Fig. 5 Percent conversion of triolein (TO) in ethanol to ethyl oleate (SM-EtOH) (◆) and in methanol to methyl oleate (SM-MeOH) (■) for 5–8 runs, using a single SM microreactor with a flow rate of 0.3 μL min−1 at room temperature. | ||
Over these 6 runs, the average GC/FID peak area of ethyl oleate, normalized to that of run 1, was 0.997 with an RSD of 0.19%. Complete conversion to ethyl oleate was also seen by NARP-LC/ELSD so that for runs 1 to 6 with no residual TAG peak observed. At runs 7 and 8, TAG was present at an estimated concentration of 0.6% and 2.5% of total lipid. The corresponding normalized GC/FID peak areas for triolein were 0.993 and 0.948, respectively, indicating <1% and <5% reduction in the FAEE yield for runs 7 and 8 compared to run 1. Hence, a single SM microreactor could be used 7 times whilst maintaining >99% conversion of triolein to FAEEs (Fig. 5). A similar result was also achieved for the conversion of SSO triacylglycerols into FAEEs using a single SM microreactor. It can be seen in Table 2 that the RSD values of the GC/FID peak areas for 5 runs were <1.5% for the 3 most abundant FAEEs; for less abundant FAEEs, the standard deviations were similar but resulting in RSDs of ∼7%. Hence, the SM microreactor was reused 5 times for the direct conversion of a natural vegetable oil to FAEEs, without the loss of efficiency.
| GC/FIDa (%) | |||||||
|---|---|---|---|---|---|---|---|
| FAEE | Run 1 | Run 2 | Run 3 | Run 4 | Run 5 | STDEV | % RSD |
| a The GC/FID was expressed by normalizing the individual peak area to the total peak area. b TAG was quantified using % NARP-LC/ELSD as described in materials and methods. Note that the normalized response factor of ELSD was higher for TAG (1) compared to that of FAEEs (0.5). c n/d not detectable. | |||||||
| C18:2 | 43.1 | 43.5 | 42.4 | 42.5 | 43.0 | 0.47 | 1.1 |
| C18:1 | 40.3 | 39.7 | 39.7 | 40.0 | 39.3 | 0.38 | 1.0 |
| C18:0 | 5.8 | 5.9 | 6.7 | 6.4 | 6.9 | 0.48 | 7.5 |
| C16:0 | 9.1 | 9.3 | 9.5 | 9.4 | 9.4 | 0.14 | 1.5 |
| C18:3 | 1.1 | 1.0 | 1.2 | 1.1 | 1.0 | 0.08 | 7.1 |
| TAGb,c | n/d | n/d | n/d | n/d | 0.17b | — | — |
It should be noted that in the above experiments, FAEEs were collected continuously over a period of 5 h and the samples of the collected fraction were used for GC/FID or LC/ELSD analysis. This long period of collection (>25 h total for the SSO data shown in Table 2) was chosen in order to demonstrate the longevity of the microreactor; in practice only a few minutes of collection time are required to produce a sample for GC analysis.
Hence, it is reasonable to estimate that if used in a flow-injection mode, even with only 1 sample per hour, the microreactor could be reused 25 times or more, if similar conditions are maintained.
The methanolysis of triolein using the SM microreactor
In oil derivatization for GC analysis using chemical catalysts, methanol is typically the alcohol used, producing FAMEs.4,40,41 However, concerns over the miscibility of the reaction mixture and product recovery may favour the use of longer chain alcohols such as ethanol and butanol.18,42–44 Furthermore, high concentrations of methanol can reduce the enzyme activity31,42,43 and therefore here we initially used ethanol in the alcoholysis reaction. Following this, experiments were performed in order to test the compatibility and reusability of the microreactor in a methanol environment (Fig. 5 and 6). Under the same conditions as described above for the formation of FAEEs, transesterification was performed on a 0.2 mg mL−1 solution of triolein in methanol. However, since methanol does not completely dissolve triolein, toluene was added into the system at 1:2 v/v (methanol:toluene). The conversion of triolein into C18:1 FAME was confirmed by GC/MS-NCI by the observation of the molecular ion at m/z 295.15 [M − H]− after the reaction (Fig. 6D). Complete methanolysis of triolein was achieved for 2 repeated runs as shown in Fig. 5 which compares the methanolysis to ethanolysis on a similar SM. Fig. 6A shows the chromatogram of methyl oleate (run 1) from a GC/MS-NCI experiment. However, on the third run, the conversion efficiency of the SM decreased to about 50% of the expected methyl oleate (Fig. 5 and 6B). In comparison, as described above, the formation of FAEEs remained quantitative for over 7 runs under the same reaction conditions (Fig. 5). After 5 runs, the SM microreactor used for methanolysis resulted in minimal conversion (∼5%) to FAMEs (Fig. 5 and 6C and D). This is likely due to partial denaturation of the lipase in the high methanol environment within the microreactor. Because of the low tolerance of lipase to methanol, previous studies have suggested that the stepwise addition of methanol into the system is preferable to obtain a high yield of FAMEs 45–48. It is also possible that the inclusion of toluene negatively affects the SM performance. However, the experiment has demonstrated the possibility of forming FAMEs using the SM, although further work is required to optimize conditions.
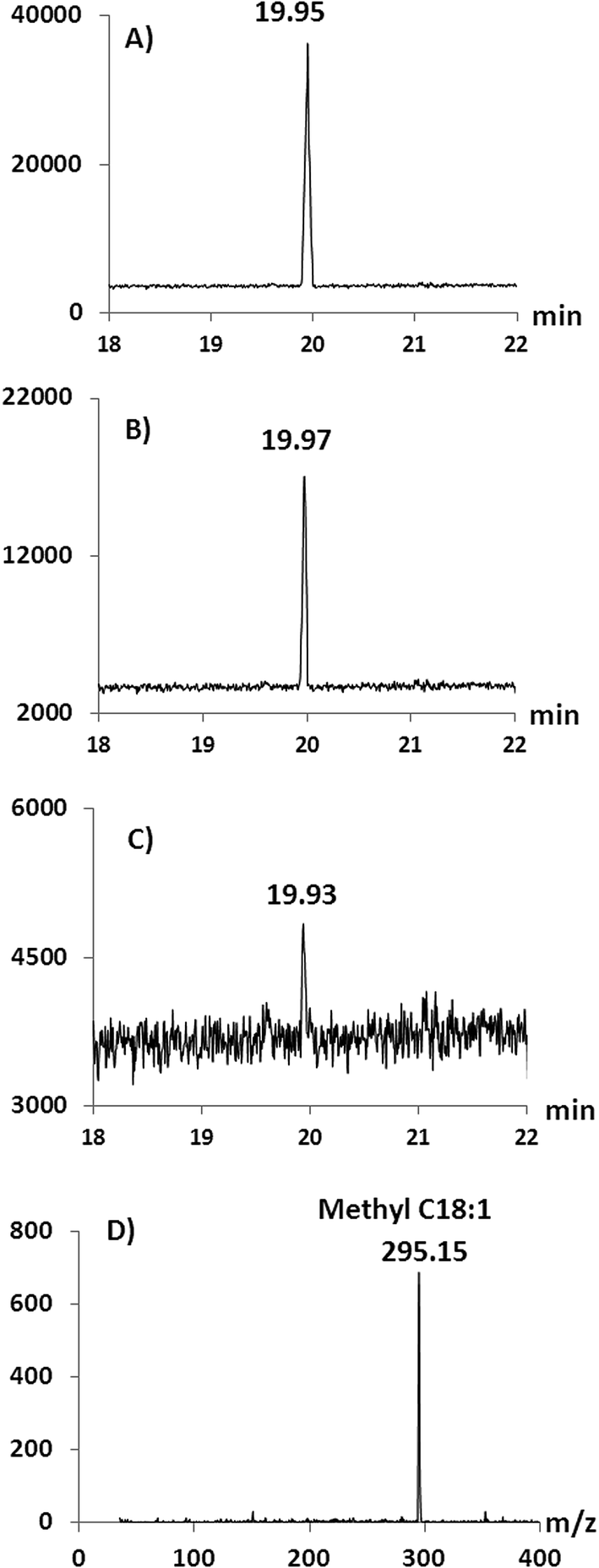 | ||
| Fig. 6 GC/MS traces for C18:1 FAME produced using a single SM microreactor: (A) run 1; (B) run 3; (C) run 5 and (D) NCI spectrum for run 5 extracted from the GC peak in (C) showing the presence of methyl oleate ([M − H]−m/z 295.15). | ||
Conclusion
In conclusion, the enzymatic microreactor technology employed in this study provides the ability to carry out the transesterification of TAG in a simple and rapid manner, which will be beneficial for the analysis of oils and fats. The products obtained from both the SM and MSF microreactors were consistently similar to the ethylation products obtained using both commercial immobilized lipase and conventional acid catalysts. This not only proves the success of the lipase immobilization within the microreactor but also demonstrates that plant oils can be directly converted to FAMEs or FAEEs using the prototype SM device with no prior sample preparation and only using alcohol in the reaction. The reusability of the microreactor provides an additional advantage that minimizes the cost of analysis and increases the potential for use in automation. The preliminary results presented here need to be followed by a quantitative validation of the fatty acid conversions achieved using similar microreactors. However, since the analysis of fatty acid composition by GC is so widely used, this approach to lipid derivatization could be of significant benefit.Acknowledgements
We gratefully acknowledge the NSERC Discovery grant 197273 to Dr J. M. Curtis and a Malaysian Ministry of Higher Education grant for a graduate student which supported this work. The authors would also like to thank Dr Yuan-Yuan Zhao and Mr Ereddad Kharraz of the University of Alberta Lipid Chemistry Group for the assistance in using the instruments.Notes and references
- F. D. Gunstone, The Chemistry of Oils and Fats: Sources, Composition, Properties and Uses, Blackwell Publishing Ltd, Oxford, 2004 Search PubMed.
- R. Aichholz and E. Lorbeer, J. High Resolut. Chromatogr., 1998, 21, 363 CrossRef CAS.
- K. Ichihara, A. Shibara, K. Yamamoto and T. Nakayama, Lipids, 1996, 31, 535 CrossRef CAS.
- W. Christie and X. Han, Lipid Analysis: Isolation, Separation, Identification and Lipidomic Analysis, 4th edn, Woodhead Publishing Ltd, Cambridge, 2012 Search PubMed.
- B. M. Craig and N. L. Murty, J. Am. Oil Chem. Soc., 1959, 36, 549 CrossRef.
- A. J. Sheppard and J. L. Iverson, J. Chromatogr. Sci., 1975, 13, 448 CrossRef CAS.
- A. I. Carrapiso and G. Carmen, Lipids, 2000, 35, 1167 CrossRef CAS PubMed.
- E. Indarti, M. I. S. A. Majid, R. Hashim and A. Chong, J. Food Compos. Anal., 2005, 18, 161 CrossRef CAS PubMed.
- M. Igarashi, T. Tsuzuki, T. Kambe and T. Miyazawa, J. Nutr. Sci. Vitaminol., 2004, 50, 121 CrossRef CAS.
- M. J. Goff, N. S. Bauer, S. Lopes, W. R. Sutterlin and G. J. Suppes, J. Am. Oil Chem. Soc., 2004, 81, 415 CrossRef CAS.
- AOCS, Official, Method Ce 2–66, in Official Methods and Recommended Practices of the American Oil Chemists' Society, ed. D. Firestone, 6th edn, 2nd printing, AOCS Urbana, Illinois, 2012 Search PubMed.
- AOCC, Official Method Ce lb-89, in Official Methods and Recommended Practices of the American Oil Chemists' Society, ed. D. Firestone, 6th edn, 2nd printing, AOCS Urbana, Illinois, 2012 Search PubMed.
- AOCS, Official Method Ce 1k–09, in Official Methods and Recommended Practices of the American Oil Chemists' Society, ed. D. Firestone, 6th edn, 2nd printing, AOCS Urbana, Illinois, 2012 Search PubMed.
- AOAC, Official Methods 965.49, in AOAC Official Methods for Fatty Acids in Oils and Fats, AOAC International, Maryland, 2000 Search PubMed.
- European Pharmacopoeia, Ph. Eur. Monograph number 2063, in Pharmacopoeia, 7th edn, Supplement 7.5, Pharmeuropa 22.4, European Pharmacopoeia Strasbourg, France, 2012 Search PubMed.
- D. Y. C. Leung, X. Wu and M. K. H. Leung, Appl. Energy, 2010, 87, 1083 CrossRef CAS PubMed.
- C. C. Akoh, J. Agric. Food Chem., 2007, 55, 8995 CrossRef CAS PubMed.
- O. S. Stamenković, A. V. Veličković and V. B. Veljković, Fuel, 2011, 90, 3141 CrossRef PubMed.
- M. Iso, B. Chen, M. Eguchi, T. Kudo and S. Shrestha, J. Mol. Catal. B: Enzym., 2001, 16, 53 CrossRef CAS.
- V. Dossat, D. Combes and A. Marty, Enzyme Microb. Technol., 2002, 30, 90 CrossRef CAS.
- H. Arroyo, F. J. Sánchez-Muniz, C. Cuesta, F. J. Burguillo and J. M. Sánchez-Montero, Lipids, 1996, 31, 1133 CrossRef.
- P. Watts and C. Wiles, Chem. Commun., 2007, 443 RSC.
- T. McCreedy, TrAC, Trends Anal. Chem., 2000, 19, 396 CrossRef CAS.
- J. Ma, Z. Liang, X. Qiao, Q. Deng, D. Tao, L. Zhang and Y. Zhang, Anal. Chem., 2008, 80, 2949 CrossRef CAS PubMed.
- C. Xie, M. Ye, X. Jiang, W. Jin and H. Zou, Mol. Cell. Proteomics, 2005, 5, 454 Search PubMed.
- N. T. S. Phan, D. H. Brown and P. Styring, Green Chem., 2004, 6, 526 RSC.
- S. M. Mugo and K. Ayton, J. Mol. Catal. B: Enzym., 2010, 67, 202 CrossRef CAS PubMed.
- S. M. Mugo and K. Ayton, J. Am. Oil Chem. Soc., 2013, 90, 65 CrossRef CAS PubMed.
- S. T. Anuar, Y.-Y. Zhao, S. M. Mugo and J. M. Curtis, J. Mol. Catal. B: Enzymol., 2013, 92, 67 CrossRef PubMed.
- S. T. Anuar, C. Villegas, S. M. Mugo and J. M. Curtis, Lipids, 2011, 46, 545 CrossRef PubMed.
- R. Irimescu, Y. Iwasaki and C. T. Hou, J. Am. Oil Chem. Soc., 2002, 79, 879 CrossRef CAS.
- Y. Shimada, Y. Watanabe, T. Samukawa, A. Sugihara, H. Noda, H. Fukuda and Y. Tominaga, J. Am. Oil Chem. Soc., 1999, 76, 789 CrossRef CAS.
- Y. Asanomi, H. Yamaguchi, M. Miyazaki and H. Maeda, Molecules, 2011, 16, 6041 CrossRef CAS PubMed.
- M. Brzozowski, J. A. Forni, G. Paul Savage and A. Polyzos, Chem. Commun., 2015, 51, 334 RSC.
- J. Liu, H. Guo, Q. Zhou, J. Wang, B. Lin, H. Zhang, Z. Gao, C. Xia and X. Zhou, J. Mol. Catal. B: Enzym., 2013, 96, 96 CrossRef CAS PubMed.
- S. K. Vashist, E. Lam, S. Hrapovic, K. B. Male and J. H. T. Luong, Chem. Rev., 2014, 114, 11083 CrossRef CAS PubMed.
- W. W. Christie, The Preparation of Derivatives of Fatty Acids. in Gas Chromatography and Lipids- a Practical Guide, 1st edn. The Oily Press, Scotland, 1989 Search PubMed.
- Y.-Y. Linko, M. Lämsä, X. Wu, E. Uosakainen, J. Seppälä and P. Linko, J. Biotechnol., 1998, 66, 41 CrossRef CAS.
- A. M. Fureby, L. Tian, P. Adlercreutz and B. Mattiasson, Enzyme Microb. Technol., 1997, 20, 198 CrossRef CAS.
- E. W. Hammond, Vegetable Oils- Types and Properties, in Encyclopedia of Food Sciences and Nutrition, ed. B. Caballero, L. C. Trugo and P. M. Finglas, Elsevier Science Ltd, Amsterdam, 2003, p. 5899 Search PubMed.
- D.-S. Lee, B.-S. Noh, S.-Y. Bae and K. Kim, Anal. Chim. Acta, 1998, 358, 163 CrossRef CAS.
- R. C. Rodeigues, G. Volpato, K. Wada and M. A. Z. Ayub, J. Am. Oil Chem. Soc., 2008, 85, 925 CrossRef PubMed.
- I. Kazanceva, V. Makarevičienė and K. Kazancev, Environ. Res. Eng. Manag., 2011, 56, 35 Search PubMed.
- M. Mittelbach, J. Am. Oil Chem. Soc., 1990, 67, 168 CrossRef CAS.
- L. Li, W. Du, D. Liu, L. Wang and Z. Li, J. Mol. Catal. B: Enzym., 2006, 43, 58 CrossRef CAS PubMed.
- A. Turkan and S. Kalay, J. Chromatogr. A, 2006, 1127, 34 CrossRef PubMed.
- Y. Watanabe, Y. Shimada, A. Sugihara, H. Noda, H. Fukuda and Y. Tominaga, J. Am. Oil Chem. Soc., 2000, 77, 355 CrossRef CAS PubMed.
- L. Deng, T. Tan, F. Wang and X. Xu, Eur. J. Lipid Sci. Technol., 2003, 105, 727 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |