
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Evaluation of uncertainty in the energy dispersive X-ray fluorescence determination of platinum in alumina
P. S.
Remya Devi
,
A. C.
Trupti
,
A.
Nicy
,
A. A.
Dalvi
,
K. K.
Swain
,
D. N.
Wagh
and
R.
Verma
*
Analytical Chemistry Division, Bhabha Atomic Research Centre, Trombay, Mumbai, India. E-mail: rverma@barc.gov.in; Fax: +91 22 25505331; Tel: +91 22 25595087
First published on 11th May 2015
Abstract
Evaluation of uncertainty in the Energy Dispersive X-ray Fluorescence (EDXRF) spectrometric determination of platinum in alumina catalysts is discussed. Pressed pellets of the platinum standard and a catalyst sample were prepared by using microcrystalline cellulose powder as the base material. A linear calibration of the X-ray fluorescence spectrometer was obtained in the range of 0.1–3 mg g−1 of platinum using pellets of matrix matched synthetic standards. The calibration function was obtained through bivariate least squares fitting, in conjunction with weighted regression of the residuals. The EDXRF results were compared with those obtained by instrumental neutron activation analysis and inductively coupled plasma optical emission spectrometry. Analysis of variance established the statistical parity of the results obtained by all the three techniques. A comprehensive evaluation of the various sources of uncertainty in the complete measurement process was carried out using a bottom-up approach. The main source of uncertainty was identified as the calibration of the EDXRF spectrometer, in which the major share was attributed to the intercept of the calibration function.
Introduction
Platinum group metals (PGMs) are widely used as catalysts in chemical processes.1 Most of the conventional oxidation catalysts are based on either platinum (Pt) or palladium (Pd) on an alumina support. Platinum based catalytic converters are used in automobiles.2 Platinum-alumina catalysts have been reported for the decomposition of sulphuric acid.3The efficiency and cost of the catalyst depend upon the concentration of PGMs and hence their accurate and precise determination is essential. Several solution sampling techniques viz. atomic absorption spectrometry (AAS), graphite furnace atomic absorption spectrometry (GF-AAS),4,5 inductively coupled plasma-mass spectrometry (ICPMS)6 and spectrophotometry7 have been reported for the determination of Pt in catalysts. These determinations put forward the inevitability of a validated analytical method with established precision, as dictated by economic considerations.8 Appropriate analytical techniques are chosen, depending on the concentration of Pt as well as the nature of the substrate material used in these catalysts.9 A method that requires the sample to be dissolved encounters certain difficulties during sample processing.10 The dissolution of alumina is difficult, due to its refractory nature. Hence, wet chemical routes are tedious and are not generally recommended for the analysis of alumina. In order to achieve complete dissolution of the alumina matrix, microwave assisted aqua-regia dissolution can be adopted.11 However, this process is tedious and time consuming. Cyanide leaching was reported for the beneficiation of the spent hydrogenation catalyst by Shams et al.12 This process is not practically adoptable since it generates hazardous cyanides as the by-products. Considering these practical difficulties, a purely instrumental method, which does not require sample dissolution, is desirable for the determination of Pt in the refractory alumina matrix. X-ray fluorescence (XRF)8,13,14 and neutron activation analysis (NAA)10,15 have been established as very efficient and versatile analytical techniques for the direct analysis of solids. Determination of Pt in catalysts by the NAA technique has been reported from our laboratory.16 Even though NAA is non-destructive, the availability of a nuclear reactor is indispensable for performing the analysis.
Energy dispersive X-ray fluorescence (EDXRF) spectrometry is a common solid sampling technique and is extensively used in industrial laboratories. The advantages of this technique include its non-destructive nature, simplicity, minimum sample preparation and fast operation. The chemical composition of the matrix severely affects the measured analyte line intensity during XRF measurements and hence matrix matched standards are required for accurate and precise determination. Thus, the EDXRF technique can be used when either matrix matched standard is available commercially or it is possible to prepare it synthetically. X-ray fluorescence methods have been reported for the determination of Pt in alumina catalysts using commercial and synthetic standards.8,13 The high energy-polarized beam-EDXRF technique was used for the determination of Pt, Pd and Rh in cordierite.14
The performance of a particular method is evaluated in terms of precision as well as trueness of the results. Recently, emphasis on measurement precision has greatly increased, as it is one of the most important parameters for assessing the quality of results. The precision of an analytical measurements is best represented in terms of measurement uncertainty, encompassing all probable sources along with their contribution, during the complete measurement process.17 For evaluating the uncertainty associated with the complete measurement process, either the bottom-up or the top-bottom approach can be adopted.18
Uncertainty evaluation during EDXRF measurements has been reported in the literature.19,20 The present report describes the evaluation of uncertainty during the EDXRF determination of Pt in alumina catalysts, adopting the bottom-up approach. Matrix-matched synthetic standards were used for calibration. Calibration, being one of the fundamental steps during the calculation of the concentration of the analyte, is discussed in detail in the present manuscript. Generally, ordinary least squares (OLS) fitting is adopted to arrive at the calibration function, which takes into account the error in the dependent variable only. However, there may be non-negligible errors associated with the preparation of calibration standards.21 In such cases, bivariate least squares (BLS) fitting can be used, which takes into account the errors in both axes.22 In the present work, the calibration function was derived, considering uncertainties in both the axes, along with heteroscedasticity in the instrumental response (i.e., unequal variances) at each point. Sources of uncertainty in the complete measurement process were identified and the combined uncertainty was evaluated systematically. In the absence of a suitable reference material, EDXRF results were validated using NAA and Inductively Coupled Plasma Optical Emission Spectrometry (ICPOES) techniques.
Experimental
Regents and chemicals
All reagents were of analytical reagent grade. Solutions were prepared using de-ionized water (conductivity = 0.05 μS cm−1). Microcrystalline cellulose powder (particle size ≤ 75 μm, Merck) and high purity alumina (Norton, USA) were used as the base materials for preparing pellets of calibration standards. Pt solution (1 mg mL−1) from Merck was used as the stock-standard for Pt.EDXRF determination of platinum in alumina
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 (w/w) in a Teflon dish. A known volume of Pt-standard solution was added to the above mixture (∼1 g), dried under an I.R. lamp and mixed thoroughly. Pellets were prepared using an automatic KBr press (AP-15, Technosearch Instruments), at a pressure of 10 tons. All pellets had identical dimensions (diameter = 2.5 cm and thickness = 0.2 cm).
:4 (w/w). Pellets having dimensions identical to those of the standard pellets were made in triplicate for each sample.
:4 (w/w) in a Teflon dish. A known volume of Pt-standard solution was added to the above mixture (∼1 g), dried under an I.R. lamp and mixed thoroughly. Pellets were prepared using an automatic KBr press (AP-15, Technosearch Instruments), at a pressure of 10 tons. All pellets had identical dimensions (diameter = 2.5 cm and thickness = 0.2 cm).
:4 (w/w). Pellets having dimensions identical to those of the standard pellets were made in triplicate for each sample.
EDXRF instrumentation and measurement procedure
XRF measurements were performed using an EDXRF spectrometer (EX-3600 M, Jordan Valley, Israel; resolution: 145 eV for the 5.9 keV Mn KLIII X-rays) and the experimental conditions are summarized in Table 1. Six replicate measurements were made on each standard pellet and the X-ray fluorescence intensities of Pt were obtained. Calibration was done by plotting the intensity of fluorescent X-rays against the concentration of Pt in the standards. Each sample pellet was measured twice (both sides). Analytical lines of Pt were the characteristic LIIIMV (9.439 keV) and LIIMIV (11.073 keV).23| Parameter | Value |
|---|---|
| Voltage (kV) | 35 |
| Current (μA) | 160 |
| Filter | Rh |
| Counting time (s) | 200 |
| Acquisition medium | Air |
NAA determination of platinum in alumina
Determination of Pt was carried out by NAA using two different nuclear reactors, independently.Neutron irradiation for 1 min duration was carried out in the Pneumatic Carrier Facility (PCF) of Dhruva reactor, Trombay, Mumbai, India24 and the neutron flux was ∼1013 cm−2 s−1. About 1–2 mg of the samples, Pt-standards (evaporated on filter paper) and alumina blank were heat-sealed in polyethylene separately and irradiated in a polypropylene capsule. Most of the matrix activity (28Al: t1/2 = 2.24 min) decayed within 15 min after irradiation. Gamma ray measurements were carried out, after 1 day of cooling, using a high purity germanium detector (45% relative efficiency, resolution: 1.9 keV at 1332 keV, Canberra) coupled to an 8k-channel analyzer. Characteristic gamma rays of 199Au (t1/2 = 3.13 d; 158.4 keV), the daughter of 199Pt (t1/2 = 30.8 min), were used for the quantification of Pt. The relative method of NAA was used for calculating the concentration.
The graphite reflector position of the Advanced Heavy Water Reactor Critical Facility (AHWR CF) reactor, Trombay, Mumbai, India25 was also utilized for neutron irradiation. About 500 mg of the alumina samples, along with Pt standards and blanks, were heat-sealed separately in polyethylene and irradiated for 4 h in a neutron flux of ∼108 cm−2 s−1. The pellets used for EDXRF measurements were also heat-sealed in polyethylene and irradiated along with the above samples. Gamma ray measurements were carried out as described above.
ICPOES determination of Pt in alumina
A microwave-assisted digestion procedure was adopted for bringing the alumina sample into solution. Approximately 0.2 g of accurately weighed sample was dissolved in 10 mL of aqua-regia in the microwave sample digestion system (ETHOS One, Milestone). The procedure was repeated twice, with fresh aqua regia each time, for complete dissolution of the sample. The solutions were evaporated nearly to dryness and made up to 50 mL, maintaining 5% acidity with respect to HNO3.These solutions were analyzed using an inductively coupled plasma optical emission spectrometer (JY 2000, Jobin YVON, Horiba Scientific). Calibration was carried out using Pt-standard solutions (5, 10 and 20 mg L−1), which were prepared by dilution of the Pt-stock solution (1 mg mL−1, Merck). The characteristic emission lines of Pt (214.120, 224.552 and 265.945 nm) were measured and the concentration of Pt in the samples was obtained using the calibration plot.
Results and discussion
A non-destructive EDXRF methodology was used, which obviates the need for sample dissolution, the most time consuming step. Pressed pellet and fusion bead methods were used for sample preparation in XRF measurements. Samples prepared by the fusion bead method have better homogeneity. However, there is a risk of loss/contamination from the platinum crucible which is used in the fusion bead method. Hence, the pressed pellet method was adopted for sample preparation, in the present work. Microcrystalline cellulose powder was used as the base material for preparing all pressed pellets, owing to its ease of preparation, mechanical strength and X-ray absorption/fluorescence characteristics. The optimized ratio between alumina and cellulose, during the present determinations, was 1:4 (w/w). Alumina as well as cellulose, being comprised of low-Z elements, is practically transparent to the excitation source (mass absorption coefficient, μ = 0.96 for 20.22 keV) and characteristic X-rays of Pt (μ = 6.55 for 9.44 keV). The sensitivity for the determination of Pt is high due to the high mass absorption coefficient of Pt for the source X-rays (μ = 75.74 for 20.22 keV)26 and the fluorescence yield (ωL = 0.35).23
The EDXRF spectrum of a typical fresh Pt-alumina catalyst (Fig. 1) shows that both the characteristic lines of Pt (i.e., LIIIMV and LIIMIV) have similar intensities and can be utilized for measurements.
 | ||
| Fig. 1 EDXRF spectrum of a fresh Pt–alumina sample. | ||
Calibration using platinum standard pellets
The quantification methodology in EDXRF analysis is usually different for thin, intermediate thickness and infinitely thick samples.19,27 All the pressed pellets (both sample and standard) used in this work could be categorized as intermediate thickness samples, since they satisfy the condition: mthin < m < mthick, where m is the mass per unit area of the sample and mthin and mthick are given by eqn (1) and (2).19,27 | (1) |
 | (2) |
| Composition of the pellet | Effective μ(E0)a [for E0 = 20.22 keV] | Effective μ(EPt)b [for EPt = 9.44 keV] | Ψ1 = Ψ2 = (radians) | m, mass per unit area of the pellets (g cm−2) | m thin (using eqn (2)) (g cm−2) | m thick (using eqn (3)) (g cm−2) |
|---|---|---|---|---|---|---|
| a Energy of the source X-rays was assumed = 20.22 keV, as reported in the literature.28 b Energy of the characteristic X-ray LIIIMV for Pt = 9.44 keV.23 | ||||||
| Alumina (Pt) + cellulose | 1.032 | 6.662 | 0.78 | 0.204 | 0.009 | 0.424 |
Calibration is the primary step in most of the instrumental analytical techniques.21 When the random uncertainties associated with each of the dependent variables, viz. the net counts, are not constant (designated as heteroscedasticity), the fitting should be done using the weighted regression method, instead of the most common ordinary regression.
Since, in the present calibration procedure, both the axes contribute to the final uncertainty, Bivariate Least Squares (BLS) fitting is the most appropriate regression method. Among all the regression techniques which consider uncertainty in both axes, the BLS technique more readily provides the regression coefficients as well as their associated variances.22 The BLS method calculates the coefficients of the straight line by taking into account the individual heteroscedastic random uncertainties in both the axes. Herein, the sum of the weighted residuals, S, is minimized as shown in eqn (3).
 | (3) |
![[N with combining circumflex]](https://www.rsc.org/images/entities/i_char_004e_0302.gif) j is the fitted value of Nj (net counts) and wj is the weighting factor that corresponds to the variance of the jth residual, represented by eqn (4).
j is the fitted value of Nj (net counts) and wj is the weighting factor that corresponds to the variance of the jth residual, represented by eqn (4).| wj = Sej2 = SNj2 + b12SCj2 − 2b1cov(Cj, Nj) | (4) |
 | (5) |
j and wj for the calibration standard pellets are listed in Table 3. The slope, b1 = 31633; intercept, b0 = 1707 and the RMS = 3.94 were obtained from the calibration.
| Pt in the standard pellets (mg g−1) | N j |
j
|
w j |
|---|---|---|---|
| 0.1411 | 5728 | 5286 | 33425 |
| 0.3547 | 13638 |
14348 |
29914 |
| 0.7077 | 24846 |
25595 |
37853 |
| 1.4153 | 44768 |
43056 |
73328 |
| 2.8414 | 91592 |
91592 |
181711 |
Least squares fitting could be applied to obtain the calibration function, since the variance in the instrument response (viz. the net counts for Pt) for each data point was much larger than the product of the slope and the variance in the concentration of Pt.21 Pearson's correlation coefficient (r = 0.9997) between the instrument response and the concentration of Pt in the pellets was greater than 0.995, further confirming the linear relationship between the two.30Fig. 2 depicts the linear calibration obtained for the Pt-standard pellets during EDXRF analysis. The linear calibration range of the instrument was 0.1–3 mg g−1 for Pt in the standard pellets.
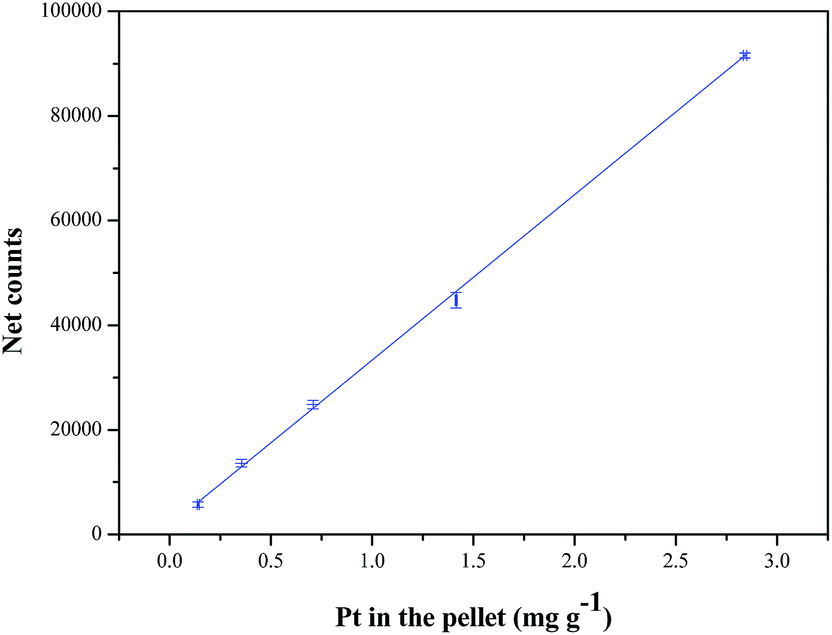 | ||
| Fig. 2 Calibration plot for Pt using an EDXRF spectrometer. | ||
The reliability of results depends on the extent of correlation between the measured X-ray fluorescence intensities of the samples and calibration standards, in EDXRF spectrometry. Errors are likely to arise when the matrices of samples and standards are not identical. During the present measurement, the matrix effect was overcome by maintaining identical matrices (1:4 w/w of alumina and cellulose) for all the samples and standards. The characteristic X-ray intensities in the samples were used for calculating the Pt concentrations by means of eqn (6), viz. the calibration equation.
 | (6) |
The advantage of using the calibration method during EDXRF includes a short analysis time (∼15–20 min for each sample; after the calibration has been performed) compared with the other methods, which require the tedious sample dissolution step. The limits of detection (LOD) and quantitation (LOQ) were 2 and 7 mg kg−1, respectively, calculated as per the guidelines given by IUPAC and ACS.31,32 However, the LOD and LOQ calculated using the method of error propagation33 were 10 and 35 mg kg−1, respectively. The method of error propagation considers the uncertainty in all the parameters and hence provides the practical limit of detection. The calibration standards, once prepared, can serve the purpose for all future determinations, provided that they are preserved appropriately.
Comparison of EDXRF results with those of NAA and ICPOES
The results obtained by EDXRF, NAA and ICPOES techniques are given in Table 4. All the results are rounded off according to the rules described in ASTM E29-13.34,35 The measurement repeatability of each method is given as one standard deviation of n numbers of replicate samples. Analysis of variance (ANOVA) was carried out to compare the results obtained by the three techniques. The calculated F values for fresh (F3,12,cal. = 1.79) and used (F3,14,cal. = 0.86) catalyst samples were less than the critical F values (F3,12,crit. = 3.49 and F3,14,crit. = 3.34),36 respectively, at the 95% confidence level. Thus, statistically, there is no difference, at the 95% confidence level, among the mean values obtained by EDXRF, NAA (both PCF and CF) and ICPOES techniques.Comparison of Pt concentration in fresh and used catalyst samples using ANOVA (F7,26,cal. = 0.97 and F7,26,crit. = 2.39)36 established that the fresh and used samples are statistically indistinguishable.
Evaluation of uncertainty in the EDXRF determination of platinum in alumina
Analytical results should always be expressed along with the corresponding uncertainty and the evaluation of uncertainty is an essential part of quantitative analysis.17 Top-down and bottom-up are the two methods adopted for arriving at the combined uncertainty.18 In the bottom-up approach of uncertainty evaluation, the analytical method is divided into sequential steps; various uncertainty sources are identified, quantified and combined appropriately.37 Basic equations of measurement uncertainty for linear calibrations in chemical analysis are presented and comprehensively discussed in the Eurachem-CITAC Guide, Appendix E.17 We have adopted the bottom-up approach of uncertainty evaluation.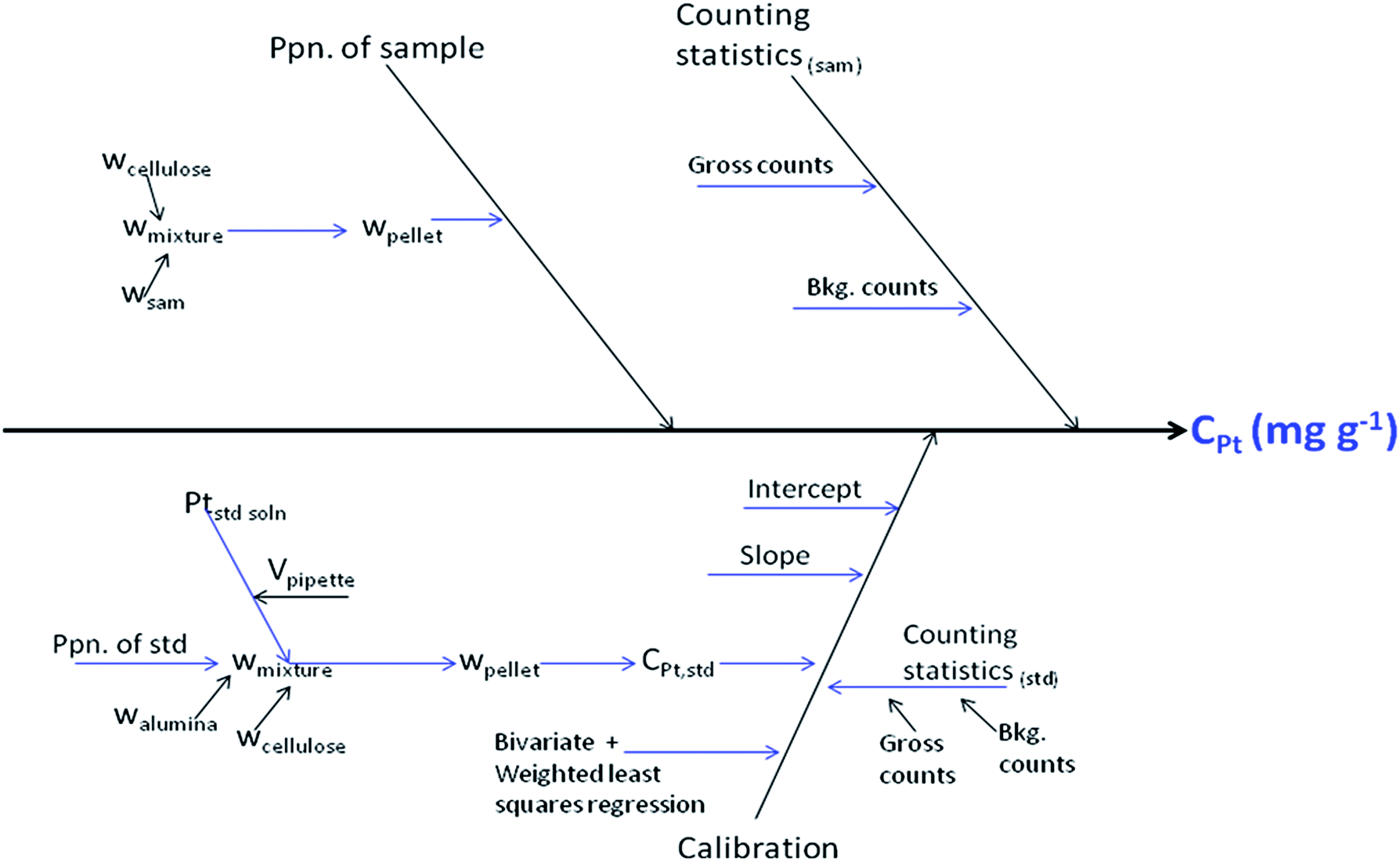 | ||
| Fig. 3 Cause and effect diagram: EDXRF determination of Pt. | ||
Uncertainty in the preparation of samples/standards arises mainly during weighing (i.e., the least count of the weighing balance, in the calculations) and pipetting of the Pt-standard solution. Contribution from counting statistics was considered in the usual manner as shown in eqn (7),38
| unet2 = ugross2 + ubackground2 | (7) |
The net counts were derived from the relationship,
| (net counts) = (gross counts) − (background counts) | (8) |
The uncertainty associated with counting is given by38
 | (9) |
Uncertainty contribution from the slope and intercept of the calibration plot was obtained on curve fitting, using origin software, through the standard procedure. The uncertainty from the linear least squares calibration was obtained using the RMS of the residuals19 as shown in eqn (10).
 | (10) |
The contribution of uncertainty from each source was evaluated, converted to the standard uncertainty and combined to get the final uncertainty in the determination of Pt using the EDXRF method. By applying the law of uncertainty propagation, the combined standard uncertainty was obtained as 0.090 mg g−1. Uncertainty contribution from each source for a fresh sample is illustrated in Fig. 4. It is evident from Fig. 4 that the calibration is the major contributor to uncertainty, which is manifested in terms of slope and intercept of the fitted line. The fractional contribution from counting statistics was found to be 0.032 mg g−1. The expanded uncertainty, calculated from the combined standard uncertainty using the coverage factor k = 2,17 was 0.18 mg g−1.
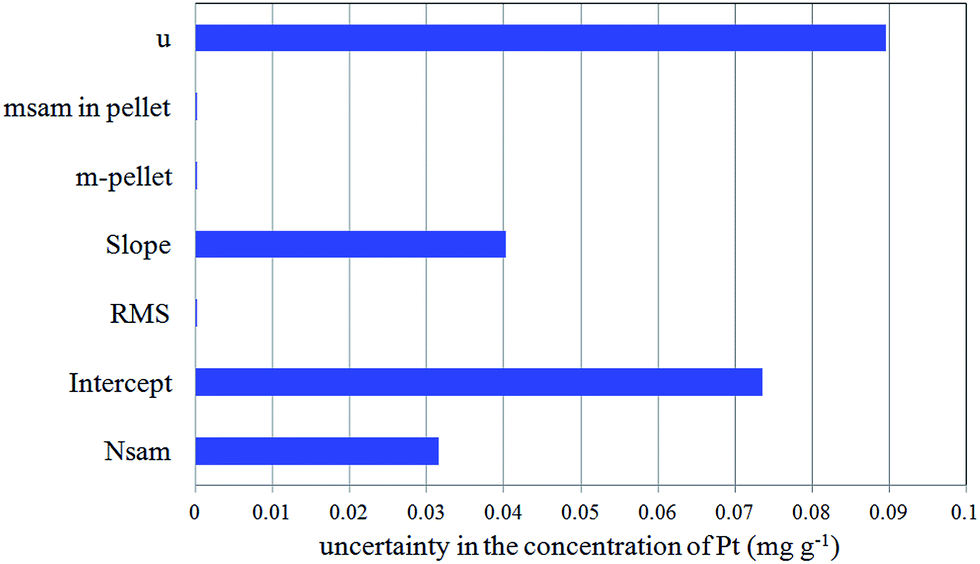 | ||
| Fig. 4 Uncertainty contribution from each source in the EDXRF determination of Pt. | ||
It was concluded that the sample was homogeneous at a sample size of 0.2 g as the standard deviation for the six replicate samples (s = 0.060 mg g−1) was less than the combined standard uncertainty (u = 0.090 mg g−1).39
Conclusion
Systematic evaluation of uncertainty during the EDXRF determination of Pt in alumina was carried out by way of the bottom-up approach. The calibration constants, viz. the slope and intercept, were found to bear the major share of uncertainty. By taking careful precautions in the above step, the overall uncertainty in the quantification procedure can be controlled to a large extent. The calibration function of the EDXRF spectrometer was derived through bivariate least squares fitting, in combination with weighted regression of the residuals. ANOVA revealed the statistical equivalence of the results obtained by EDXRF, NAA and ICPOES techniques. EDXRF is a fast, precise and accurate technique and hence can be used for quality control during the determination of Pt in alumina.Acknowledgements
The authors acknowledge Dr A. K. Tripathi and Dr A. M. Banerjee, Chemistry Division, BARC for providing the Pt-loaded alumina samples and Ms M. Venkatesh, Analytical Chemistry Division, BARC for the guidance during ICPOES analysis.Notes and references
- W. Keirn, Catalysis in C1 Chemistry, D. Reidel Publishing Co, 1983, p. 1 Search PubMed.
- T. Revathi Reddy, N. N. Meeravali and A. V. R. Reddy, Phase transfer catalyst assisted directly suspended droplet microextraction of platinum from geological and spent automobile converter samples prior to HR-CS AAS determination, Anal. Methods, 2013, 5, 2343–2351 RSC.
- D. M. Ginosar, L. M. Petkovic, A. W. Glenn and K. C. Burch, Stability of supported platinum sulphuric acid decomposition catalysts for use in thermochemical water splitting cycles, Int. J. Hydrogen Energy, 2007, 32(4), 482–488 CrossRef CAS PubMed.
- N. M. Potter, Determination of platinum and palladium in oxidation catalysts for automotive exhaust by atomic absorption spectrometry, Anal. Chem., 1976, 48(3), 531–534 CrossRef CAS.
- S. Scaccia and B. Goszczynska, Sequential determination of platinum, ruthenium, and molybdenum in carbon-supported Pt, Pt-Ru, and Pt-Mo catalysts by atomic absorption spectrometry, Talanta, 2004, 63(3), 791–796 CrossRef CAS PubMed.
- O. V. Borisov, D. M. Coleman, K. A. Oudesma and R. O. Carter, Determination of platinum, palladium, rhodium and titanium in automotive catalytic converters using inductively coupled plasma mass spectrometry with liquid nebulization, J. Anal. At. Spectrom., 1997, 12, 239–246 RSC.
- ASTM D4642-04, Standard test method for platinum in reforming catalysts by wet chemistry, ASTM International, USA, 2010 Search PubMed.
- R. V. C. Peddy, G. Kalpana, V. J. Koshy, N. V. R. Apparao, M. C. Jain and R. V. Patel, Determination of the platinum, rhenium and chlorine contents of alumina-based catalysts by X-ray fluorescence spectrometry, Analyst, 1991, 116, 847–850 RSC.
- C. R. M. Rao and G. S. Reddi, Platinum group metals (PGM); occurrence, use and recent trends in their determination, Trends Anal. Chem., 2009, 19(9), 565–586 CrossRef.
- R. H. Marsh and J. Butler, The determination of barium, platinum and ruthenium in experimental catalysts by neutron activation analysis, J. Radioanal. Chem., 1976, 29, 357–363 CrossRef CAS.
- D. Jafarifar, M. R. Daryanavard and S. Sheibani, Ultra fast microwave-assisted leaching for recovery of platinum from spent catalyst, Hydrometallurgy, 2005, 78(3–4), 166–171 CrossRef CAS PubMed.
- K. Shams, M. R. Beiggy and A. G. Shirazi, Platinum recovery from a spent industrial dehydrogenation catalyst using cyanide leaching followed by ion exchange, Appl. Catal., A, 2004, 258(2), 227–234 CrossRef CAS PubMed.
- E. L. Gunn, Determination of platinum in reforming catalyst by X-ray fluorescence, Anal. Chem, 1956, 28(9), 1433–1436 CrossRef CAS.
- K. V. Meel, A. Smekens, M. Behets, P. Kazandjian and R. V. Grieken, Determination of platinum, palladium, and rhodium in automotive catalysts using high-energy secondary target X-ray fluorescence spectrometry, Anal. Chem., 2007, 79, 6383–6389 CrossRef PubMed.
- A. Aksoy, F. Z. Khiari and A. Rahman, Study of Pt-Re/alumina reforming catalysts by neutron activation analysis, J. Radioanal. Nucl. Chem., 1998, 230(1–2), 75–78 CrossRef CAS.
- Utilization of Low Flux Nuclear Reactor for Neutron activation analysis, Nicy Ajith, Aditi A. Dalvi, K. K. Swain, Remya Devi P. S., M. Ghosh, R. Verma and A. V. R. Reddy, BARC Report (2014) E 018.
- S. L. R. Ellison and A. Williams, Quantifying uncertainty in analytical measurements, Eurachem/CITAC Guide CG 4, 3rd edn, QUAM, 2012 Search PubMed.
- www.measurementuncertainty.org, dated 22nd December 2014.
- IAEA TECDOC 1401, Quantifying Uncertainty in Nuclear Analytical Measurements, 2004, IAEA, Vienna.
- R. M. Rousseau, Detection limit and estimate of uncertainty of analytical XRF results, Rigaku J., 2001, 18(2), 33–47 CAS.
- J. Riu and F. X. Rius, Univariate regression models with errors in both axes, J. Chemom., 1995, 9, 343–362 CrossRef CAS PubMed.
- F. J. del Río, J. Riu and F. X. Rius, Linear regression taking into account errors in both axes in the presence of outliers, Anal. Lett., 2001, 34(14), 2547–2561 CrossRef.
- http://www.kayelaby.npl.co.uk/atomic_and_nuclear_physics/4_2/4_2_1.html, dated 30th December 2014.
- R. Acharya, K. K. Swain, A. D. Shinde, N. S. Bhamra, K. Chakrabarty, C. G. Karhadkar, T. Singh, Y. S. Rana, P. K. Pujari, D. K. Shukla and A. V. R. Reddy, Utilization of Pneumatic Carrier Facility of Dhruva Reactor for Trace Element Determination by Neutron Activation Analysis, J. Radioanal. Nucl. Chem., 2014, 302, 1525–1530 CrossRef CAS.
- K. K. Swain, N. Ajith, R. Acharya, R. Verma and A. V. R. Reddy, Large Sample Neutron Activation Analysis of Dross for Gold and Silver, J. Radioanal. Nucl. Chem., 2012, 294, 319–322 CrossRef CAS PubMed.
- http://physics.nist.gov/PhysRefData/XrayMassCoef/ElemTab/z13.html, dated 30th December 2014.
- A. Markowicz, An overview of quantification methods in energy-dispersive X-ray fluorescence analysis, Pramana, 2011, 76(2), 321–329 CrossRef.
- http://en.wikipedia.org/wiki/X%20ray%20fluorescence%20mediaviewer/File:%20TubeSpectrum.jpg, dated 30th December 2014.
- http://www.asdlib.org/onlineArticles/ecourseware/Palmer/ASDL%20Intro%20to%20XRF.pdf, (Slide No. 46 out of 79), dated 30th December 2014.
- R. J. N. B. da Silva and M. F. Camões, The quality of standards in least squares calibrations, Anal. Lett., 2010, 43, 1257–1266 CrossRef PubMed.
- IUPAC, Spectrochim. Acta, Part B, 1978, 33(6), 241–245 CrossRef.
- ACS committee on environmental improvement, Anal. Chem., 1980, 52(14), 2242–2249 CrossRef.
- G. L. Long and J. D. Winefordner, Limit of Detection: A Closer Look at the IUPAC Definition, Anal. Chem., 1983, 55(7), 712A–724A CAS.
- ASTM E29-13, Standard practice for using significant digits in test data to determine conformance with specifications, 2013, ASTM International, USA.
- M. Thompson, Precision in chemical analysis: a critical survey of uses and abuses, Anal. Methods, 2012, 4, 1598–1611 RSC.
- http://www.sussex.ac.uk/Users/grahamh/RM1web/F-ratio%20table%20M1web/F-ratio%20table%20202005.pdf, dated 30th December 2014.
- V. Pino, I. H. Martín, J. H. Ayala, V. González and A. M. Afonso, Evaluation of the uncertainty associated to the determination of heavy metals in seawater using graphite furnace atomic absorption spectrometry, Anal. Lett., 2007, 40, 3322–3342 CrossRef CAS PubMed.
- G. F. Knoll, Radiation Detection and Measurement, John Wiley and Sons Inc., USA, 3rd edn, 1999 Search PubMed.
- C. O. Ingamells and P. Switzer, A proposed sampling constant for use in geochemical analysis, Talanta, 1973, 20, 547–568 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |