
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring new dimensions in cadaveric decomposition odour analysis
P.-H.
Stefanuto†
*a,
K. A.
Perrault†
b,
R. M.
Lloyd
c,
B.
Stuart
b,
T.
Rai
d,
S. L.
Forbes
b and
J.-F.
Focant
a
aCART, Organic and Biological Chemistry Group, Chemistry Department, University of Liège, Allée de la chimie B6c, B-4000 Liège, Belgium. E-mail: phstefanuto@ulg.ac.be
bCentre for Forensic Science, University of Technology Sydney, PO Box 123 Broadway, NSW 2007, Australia
cDepartment of Chemistry, University of Leicester, University Road, Leicester LE1 7RH, United Kingdom
dSchool of Mathematical Sciences, University of Technology Sydney, PO Box 123 Broadway, NSW 2007, Australia
First published on 25th February 2015
Abstract
This study demonstrates the first documented use of comprehensive two-dimensional gas chromatography – high-resolution time-of-flight mass spectrometry (GC×GC-HRTOFMS) for volatile organic compound analysis in the forensic sciences. High-resolution mass spectral data provided higher confidence in analyte identification. GC×GC-HRTOFMS will be valuable for future studies of decomposition odour and other complex volatile matrices.
Gas chromatography (GC) coupled to mass spectrometry (MS) has long been used for the separation, detection and identification of volatile organic compounds (VOCs). Various gas chromatographic approaches have been used to analyse VOCs in a wide range of applications including environmental pollutant analysis, food and fragrance analysis, metabolomics and in the forensic sciences. In the forensic field, VOCs are relevant for understanding the alerts of detection dogs used by law enforcement to locate an array of contraband using olfactory mechanisms (e.g. currency, drugs, explosives, food, etc.). More specifically, human remains detection (HRD) canines can be trained to search for and locate deceased individuals by alerting to VOCs produced by decomposing remains. This is valuable for the search and recovery of human remains in outdoor environments in cases such as homicides and mass disasters. Chemical profiling of the human remains and other contraband items has provided information to detection dog trainers regarding the chemical composition of the target odours the canines are trained to detect. Because very little is known about how a positive canine alert is elicited, the identification of VOCs produced by decomposing remains (i.e. “decomposition VOCs”) is imperative to reach an improved understanding of potential analytes impacting canine olfaction.
The odour produced by decomposing remains is a complex mixture of various VOCs. Due to the complexity of the decomposition VOC profile, traditional one-dimensional gas chromatography (1D GC) often suffers from insufficient peak capacity to accurately separate the large number of analytes present, thus providing low mass spectral library matches and leading to the potential for compound misidentification. The decomposition VOC profile also exhibits a wide dynamic range whereby the presence of both high-level and low-level VOCs contribute to the complexity of the matrix. Background VOCs (of similar structure and behaviour to decomposition VOCs) are also present in the profile. For these reasons, decomposition VOC profiling benefits substantially from analysis by comprehensive two-dimensional gas chromatography (GC×GC). This comprehensive analysis allows two independent separation mechanisms to occur by attaching two GC columns together at a junction known as a modulator. Due to the narrow peaks that are produced by modulation between the columns, time-of-flight MS (TOFMS) is often coupled with GC×GC in order to provide the required acquisition rate (>50 Hz). Recently, GC×GC coupled to low-resolution (LR) TOFMS has been employed in this field leading to a more developed understanding of cadaveric decomposition odour.1–8
Recent developments in environmental monitoring have investigated the coupling of GC×GC with high-resolution time-of-flight mass spectrometry (GC×GC-HRTOFMS).9–13 Unit mass resolution using a low-resolution TOFMS may be insufficient for compound detection in complex matrices that exhibit interferences.14 GC×GC-HRTOFMS provides the potential to combine the high peak capacity of the GC×GC system with accurate mass spectral data. The mass measurement accuracy provides the ability to estimate the chemical formula of analytes, providing higher confidence in analyte identification.14 Differences in the mass measurement between analytes and interferences also affords improved deconvolution in complex matrices.14
In the non-targeted analysis of decomposition odour, criticism of compound identification is often met. There are various challenges encountered in decomposition odour profiling that make this concern challenging to address including: (1) the substantial costs associated with obtaining a large database of chemical reference standards; (2) the impossibility of obtaining chemical standards for every compound identified; (3) the lack of certified reference materials for the sample matrix; and (4) the limitation in replicating generated samples because of ethical, legal, and logistical challenges associated with obtaining human or animal remains. Thus, tools for increasing the confidence in mass spectral identifications are required in order to improve the accuracy of decomposition odour profiling.
In this study, the use of GC×GC-HRTOFMS was investigated for the analysis of decomposition VOCs in soil beneath carrion. Assessment of the instrumentation on a restricted data set was desired in order to determine the added value this instrumentation could have for the field of decomposition VOC analysis, specifically for future longitudinal studies involving large set-up and collection of field trial data. Determining whether absolute quantification would be possible from samples using the developed method was also of interest via the evaluation of the linear range of calibration curves for representative standards. As GC×GC-HRTOFMS is a novel and developing technique, quality control information of the mass accuracy data was required to ensure consistency and reliability of the results for future studies. The use of GC×GC-HRTOFMS for the VOC profiling of complex biological matrices presented herein may also be applied to other areas of forensic VOC monitoring as well as environmental, metabolomics and food science applications.
Experimental
Samples
Decomposition VOC samples were collected using sorbent tubes from the soil within the cadaver decomposition island (CDI) from four 70 kg pig carcasses (Sus scrofa domesticus L.) used as human analogues, which had undergone sufficient decomposition on the soil surface. This caused the soils to become loaded with decomposition by-products during a period of 3 months and represented late-stage decay, providing representative samples of many victim recovery scenarios. The soil gas was analysed in this study as it represents the biotic components of the soil community that also influence the decomposition VOC profile.15,16 Decomposition took place at an outdoor research facility in Sydney, Australia during January–March 2014 and VOCs were collected using previously documented methods5,6,8 from a VOC Mole™ soil probe onto Tenax TA/Carbograph 5TD sorbent tubes (Markes International Ltd., Lltantrisant, Wales, UK). This involved the collection of four experimental samples from the soil within the CDI and four control samples from soil where no remains were placed. VOC samples were collected in this manner in situ (i.e. directly from the ground at the field site). One of the experimental samples was lost to instrument malfunction which generated 3 replicate samples in the ‘experimental’ class and 4 replicate samples in the ‘control’ class. These class designations were used throughout the data processing and analysis.GC×GC-HRTOFMS analysis
Each sorbent tube was injected with an internal standard of 1 μL of 1.5 ppm GC-grade bromobenzene in HPLC grade methanol (Sigma Aldrich, Castle Hill, NSW, Australia) prior to analysis. Sorbent tubes were thermally desorbed for 4 minutes at 300 °C using a Markes Unity 2 Thermal Desorber (Markes International Ltd.). The desorbed sample was collected on a general purpose cold trap at −10 °C, and then secondary desorption was performed at 300 °C for 3 minutes under a 20 mL min−1 split flow. The instrument used was an Agilent 7890A gas chromatograph (Agilent Technologies, Palo Alto, CA, USA) coupled with an AccuTOF™ GCv 4 G high-resolution time-of-flight mass spectrometer (JEOL Ltd., Tokyo, Japan). A Rtx®-624Sil MS first dimension (1D) column (30 m × 0.250 mm ID, 1.40 μm film thickness, Restek Corporation, Bellefonte, PA, USA) and a Stabilwax® second dimension (2D) column (2 m × 0.250 mm ID, 0.50 μm film thickness, Restek Corporation) were used. Modulation between columns was performed using a ZX2 dual-stage thermal loop modulator (Zoex Corporation, Houston, TX, USA). The cold jets were cooled by the ZX2 system to −90 °C and hot jets were maintained at 200 °C by a thermal auxiliary. The 1D GC oven was ramped from 35 °C to 240 °C at a rate of 5 °C min−1 and held for an additional 5 minutes. No 2D oven was used. The modulation period (PM) was 4 seconds with a 600 millisecond hot pulse duration. The carrier gas (high purity ALPHAGAZ™, Air Liquide, Liège, Belgium) was held at a constant flow rate of 1.0 mL min−1.The HRTOFMS was operated in electron ionization (EI) mode with an ionizing voltage of 70 eV. An acquisition rate of 50 Hz was used with a mass range of 35–400 m/z and an acquisition delay of 1 minute. The plate voltage was 2150 V with a sampling interval of 0.25 μs. Data were acquired using MassCenter version 2.6.2b (JEOL Ltd.). Instrument tuning was performed using perfluorokerosene (PFK) (Tokyo Chemical Industry Co. Ltd., Tokyo, Japan) and the mass resolution was 7637 at m/z 293. Data was analysed in GC Image 2.5 HR (Zoex Corporation) using the GC Project and Image Investigator features. A cumulative image was created using all samples being analysed. A template was built using this image and removal of column bleed and other artefacts that were not specific to the analysis was performed. The feature template was then applied to the centroided sample files (.7rw). The configuration used to generate the cumulative image involved baseline correction, a 0.8 second baseline shift, and a minimum blob volume of 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. Compound identification was made by comparison to the 2011 NIST library with a manually-applied threshold match of 700.5,17 Linear retention indices from the 1D column were used to further verify analyte identifications along with mass measurement error from the raw profile data (.7rw). Compounds of interest were identified by comparing the Fisher ratio (FR) for the normalised volume of each compound to a critical F value (Fcrit). Where FR > Fcrit, the variance between the two classes was significant and the compound was considered to be decomposition-specific. This data analysis approach has been previously demonstrated using complex multivariate GC×GC data.18 Principal component analysis (PCA) was performed in The Unscrambler X version 10.3 (CAMO Software, Oslo, Norway) for visualisation based on scores (samples) and loadings (analytes).
000. Compound identification was made by comparison to the 2011 NIST library with a manually-applied threshold match of 700.5,17 Linear retention indices from the 1D column were used to further verify analyte identifications along with mass measurement error from the raw profile data (.7rw). Compounds of interest were identified by comparing the Fisher ratio (FR) for the normalised volume of each compound to a critical F value (Fcrit). Where FR > Fcrit, the variance between the two classes was significant and the compound was considered to be decomposition-specific. This data analysis approach has been previously demonstrated using complex multivariate GC×GC data.18 Principal component analysis (PCA) was performed in The Unscrambler X version 10.3 (CAMO Software, Oslo, Norway) for visualisation based on scores (samples) and loadings (analytes).
Results and discussion
Chromatographic considerations
The method described herein was adapted from previous work5 for use with the GC×GC-HRTOFMS instrument. It was important to assess method adaptation because GC×GC instrumentation may differ between laboratories and therefore parameters may not be directly transferrable when different hardware is used. Fig. 1 illustrates a contour plot of a typical soil sample using the new method with slices in the 1D and 2D. Similar chromatographic performance was observed using this instrumentation in comparison to previous work that used different commercial instrumentation (i.e. GC×GC-LRTOFMS).5,6,8 In comparison to the previous method, the PM for the adapted method was reduced from 5 seconds to 4 seconds without resulting in wrap-around. It is likely that this change was necessary given the different type of modulators used between the two systems (i.e. quad-jet cryogenic thermal modulator used in5,6,8vs. dual-jet cryogen-free loop type thermal modulator used in this study). The previous GC×GC-TOFMS study5 also used a 2D oven which was not present in the GC×GC-HRTOFMS system used herein. Despite these differences, the separation of the test mix on the chromatographic plane was comparable between the two studies.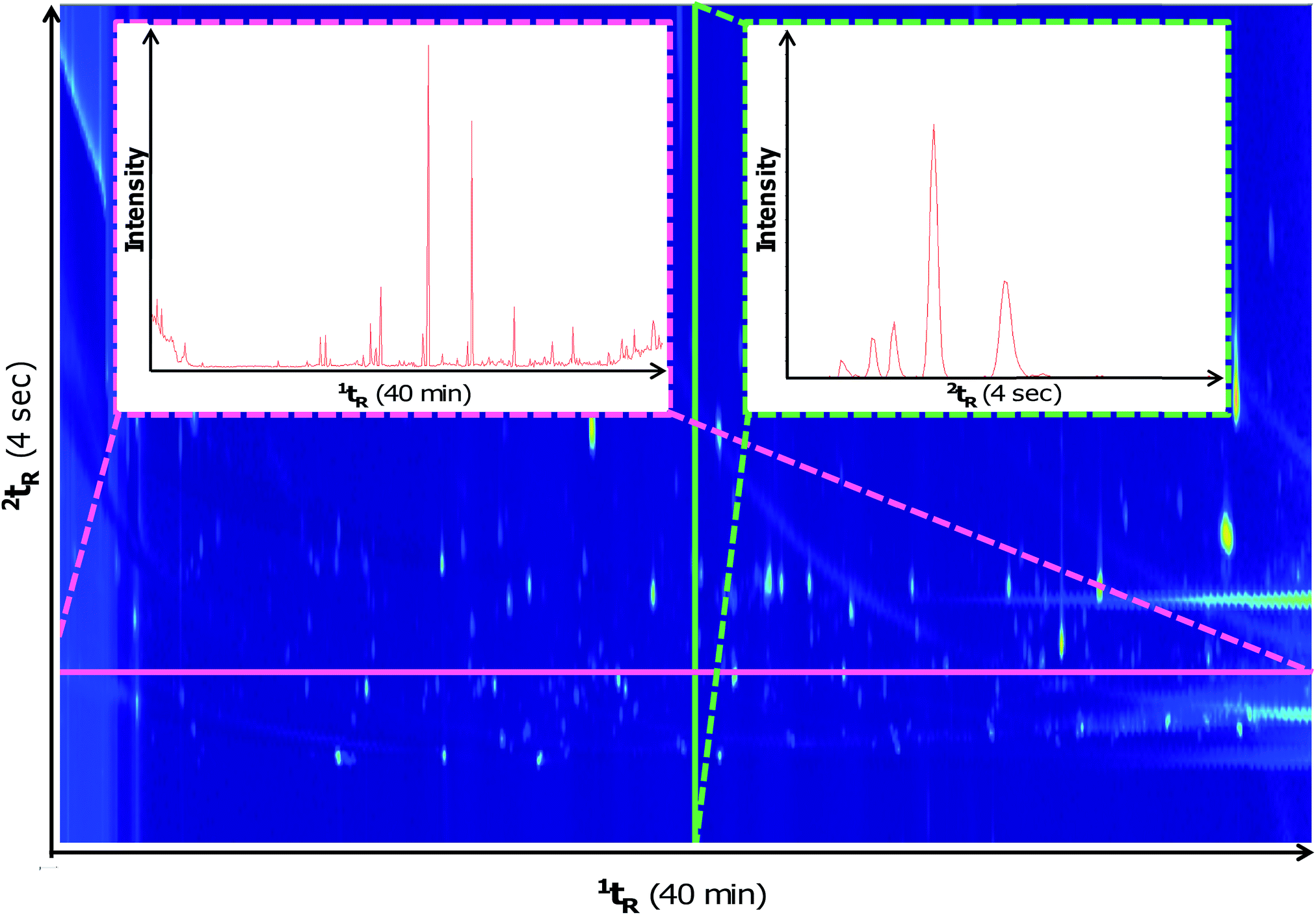 | ||
| Fig. 1 Total ion current (TIC) contour plot of a decomposition soil VOC sample. The x-axis represents first dimension retention time (1tR) and the y-axis represents second dimension retention time (2tR). Projected slices of the contour plot are displayed for the 1D (pink) and 2D (green). | ||
The GC×GC-HRTOFMS instrumentation exhibited dissatisfactory modulation of early-eluting analytes. This likely occurred due to their high volatility and the lack of cryogenic modulation. As a result, the oven method from previous work was adjusted by removing the 5 minute hold at 35 °C at the beginning of the sample run. Cryogen-free cold jet cooling has the potential to be problematic in profiling the entire decomposition odour profile. Cryogenic modulation would provide the best possible results due to the lower temperature reached in the cold jets (i.e. between −196 and −210 °C), allowing improved modulation of lighter compounds below C7. However, the use of a cryogen-free cooling system significantly reduces operational costs. Many low boiling point compounds detected in previous work still remained detectable using the method in this study (e.g. benzene, dimethyl disulphide, etc.).
Previous reports have demonstrated that HRTOFMS has a sufficient acquisition rate at 25 Hz to be coupled with GC×GC at no detriment to quantification.19 The maximum acquisition rate of the GC×GC-HRTOFMS was 50 Hz, which further expands its utility as a GC×GC detector. A slice of the 1D and 2D traces are represented in Fig. 1. Although the HRTOFMS detector exhibits a lower acquisition rate (i.e. maximum 50 Hz) than that of the LRTOFMS detector (i.e. typically 100 Hz or higher), its maximum acquisition rate was capable of providing a sufficient number of scans across the width of the narrow peaks generated by the modulator. For a typical peak eluting at the detector with a 200 millisecond width, 10 scans would be performed across the peak providing a high quality GC×GC chromatogram for performing analyte quantification and mass spectral deconvolution. Although past research demonstrated that HRTOFMS has a sufficient scan rate to be a good candidate for GC×GC detection,19 this confirms the assertion for the wide dynamic range experienced with decomposition odour samples.
Chromatogram alignment was necessary in order to ensure that the compounds reported in one sample were aligned with those from other samples. This alignment was conducted based on retention time (tR) and mass spectral matches between chromatograms. This was carried out based on the high-resolution mass spectral data. Therefore, alignment of high-resolution data provides a more robust alignment process, thus further increasing confidence in analyte identification across samples within a data set. In decomposition odour analysis, studies are often performed longitudinally and can last months or even years. Slight changes in response and conditions over time can result in difficulty comparing results over experimental days. High-resolution data alignment facilitates a robust comparison of analytes across an entire study, thereby providing more reliable results.
Decomposition VOCs
Table 1 displays the significant compounds for the most discriminating compounds obtained from the sample displayed in Fig. 1. Initially, there were 179 peaks identified within the data set. This was expected as it is well-accepted that decomposition odour is highly complex and exhibits a large number of compounds covering a broad range of chemical classes. In order to focus on the significant VOCs contributed by decomposition and ensure the removal of background VOCs, a pairwise FR comparison was used. The FR is a ratio between the within-class variance and the between-class variance. The Fcrit value was determined to be 6.608 based on the following criteria: (1) the data set exhibited n = 2 classes; (2) there were n = 4 and n = 3 replicates in the two respective classes; and (3) a significance level of 0.05 (i.e. 95% confidence level) was desired. Compounds with FRs exceeding Fcrit are displayed in Table 1 (arbitrarily sorted by decreasing FR values). This more focused approach, versus an exhaustive fingerprinting approach, was desired for reducing the complexity of the multivariate data based on compounds that were considered to be significant in the decomposition soil. Data reduction in decomposition odour profiling (and in other areas of VOC profiling from biological matrices) is often required in order to reduce the large number of background VOCs that fluctuate based on noise.5 The VOCs of interest for decomposition odour analysis are those which vary from the background matrix; therefore, the use of a FR threshold afforded more confidence for determining compounds of relevance for application purposes.| Compound name (previously referenced in) | Chemical formula | CAS # | Library forward match | Library reverse match | 1 t R (min) | 2 t R (s) | Mean signal-to-noise ratio | Fisher ratio (FR) | Linear retention index (LRI) | Exact m/z | Measured m/z | Mass error (ppm) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Heptadecane, 2,6,10,14-tetramethyl- | C21H44 | 18344-37-1 | 790 | 741 | 30.72 | 0.544 | 399.71 | 39.89 | 1403 | 198.2348 | 198.2315 | 16.2 |
| 2 | Benzonitrile3,5,6,8,15,16,20–22 | C7H5N | 100-47-0 | 915 | 878 | 20.35 | 1.980 | 299.14 | 31.80 | 1059 | 103.0422 | 103.0393 | 28.0 |
| 3 | Decane, 6-ethyl-2-methyl- | C13H28 | 62108-21-8 | 806 | 715 | 32.02 | 0.551 | 54.16 | 17.10 | 1450 | 140.1565 | 140.1553 | 8.45 |
| 4 | Thieno[2,3-c]pyridine | C7H5NS | 272-12-8 | 796 | 788 | 27.83 | 2.434 | 42.63 | 10.16 | 1298 | 135.0142 | 135.0105 | 27.7 |
| 5 | Naphthalene4,20,22–26 | C10H8 | 91-20-3 | 892 | 823 | 26.35 | 1.548 | 58.53 | 9.68 | 1248 | 128.0626 | 128.0587 | 30.8 |
| 6 | Benzenemethanol, α,α-dimethyl-22–24 | C9H12O | 617-94-7 | 745 | 743 | 23.43 | 2.043 | 60.02 | 8.81 | 1152 | 136.0888 | 136.0851 | 27.1 |
| 7 | Benzene, 2-methoxy-4-methyl-1-(1-methylethyl)- | C11H16O | 1076-56-8 | 742 | 648 | 27.20 | 0.926 | 26.45 | 8.23 | 1275 | 164.1201 | 164.1168 | 20.3 |
| 8 | Pentadecane6,15,16,20,26,27 | C15H32 | 629-62-9 | 838 | 806 | 33.35 | 0.560 | 713.76 | 8.23 | 1500 | 212.2504 | 212.2473 | 14.7 |
| 9 | Ethanol, 2-phenoxy-6 | C8H10O2 | 122-99-6 | 787 | 780 | 28.08 | 3.895 | 106.44 | 8.16 | 1307 | 138.0681 | 138.0643 | 27.5 |
| 10 | Nonane1,4,6,8,24–27 | C9H20 | 111-84-2 | 911 | 803 | 14.82 | 0.454 | 638.33 | 6.82 | 900 | 128.1565 | 128.1528 | 28.7 |
| 11 | Dimethyl trisulfide3,6,8,15,16,20,21,23 | C2H6S3 | 3658-80-8 | 782 | 742 | 18.77 | 1.129 | 51.61 | 6.74 | 1012 | 125.9632 | 125.9603 | 23.1 |
A low mass error (31 ppm or lower) was achieved for all compounds based on the m/z indicated in Table 1. The low mass error contributed to the overall confidence in analyte identification, which highlights one of the main benefits of using GC×GC-HRTOFMS. The main advantage of using this technique for VOC profiling was the ability to simultaneously monitor the library identification, accurate mass data, and linear retention index (LRI) for each compound. For example, in Table 1 the peak assignment for compound 8 (pentadecane) was originally identified with a high quality library match (>900) as 1-iodo-2-methylundecane, which has been previously reported as a decomposition VOC.4,8 However, based on the LRI of the analyte (i.e. 1500) and the lack of a molecular ion (m/z 296) present for 1-iodo-2-methylundecane in the analyte spectrum, it was possible to identify that this compound was, in fact, pentadecane. Pentadecane exhibited a lower match quality to the NIST library than 1-iodo-2-methylundecane and was one of the secondary peak hits. However, the low mass error obtained for pentadecane (14.7 ppm) and its LRI made it possible to confirm the correct identification (confirmed by standard injection). LRIs and mass measurement error further contributed to confirming other lower NIST library identifications with matches between 700 and 800. It is common in VOC profiling of complex matrices to obtain match factors between 700 and 800, and these compounds may not be reported in the interest of stringency. This may also occur when the analyte's reference mass spectrum within the NIST library database has been obtained by quadrupole mass spectrometry, since the fragmentation patterns can differ slightly to fragmentation by TOFMS. The combination of available information demonstrates the high confidence in analyte identification and the added value of obtaining high-resolution mass spectral data. The use of HRTOFMS in GC×GC analysis of complex VOC mixtures will provide confirmatory information regarding analyte identifications that can be useful for building reference databases for users to increase consistency in analyte reporting.
In Table 1, molecular ions were used for mass error measurement where possible. The aromatic VOCs (e.g. benzene, toluene, and styrene) exhibited strong molecular ion peaks based on the stability of their structures. However, for structures like alcohols, the molecular ion peak was only found in trace levels or was absent from the spectra. The formula calculator of the data processing software was used to determine the exact m/z of a selected fragment to facilitate computing of the mass error. However, in the future it may be useful to recollect a portion of the split flow of the desorption step onto a secondary tube that would be analysed at a lower ionization voltage. This would provide a higher chance of calculating the mass error based on the molecular ion of analytes with less stability that often displayed low or non-existent molecular ions in their spectra.
In decomposition odour analysis, there are often a large number of compounds identified that are present in the background VOC profile and fluctuate based on “noise”. PCA is often used in this field of research, yet generating PCA plots from a large multivariate data set can result in poor discrimination based on the principal components (PCs) even when differences exist between the groups. This is often managed by conducting PCA on the sum of compound classes or by reducing the number of compounds input into the analysis based on statistical thresholds. For this reason, only significant compounds from the pairwise FR analysis were used for PCA in this study. Visualisation of the remaining data structure after compound selection ensured that the selection of significant compounds of interest yielded differentiation between the control and experimental classes.
Fig. 2a demonstrates that it was possible to make a clear discrimination between the two classes along the first principal component (PC-1) axis. Fig. 2b demonstrates that the significant compounds chosen by the pairwise FR analysis were those that were indicative of the decomposition odour samples and not of the control samples, providing the distinction between the two compound classes in Fig. 2a. This is apparent because the compound loadings are located on the outer ring of the correlations loading plot (i.e. demonstrating their significance), and that they are on the right side of the plot where the experimental samples are located in the scores plot.
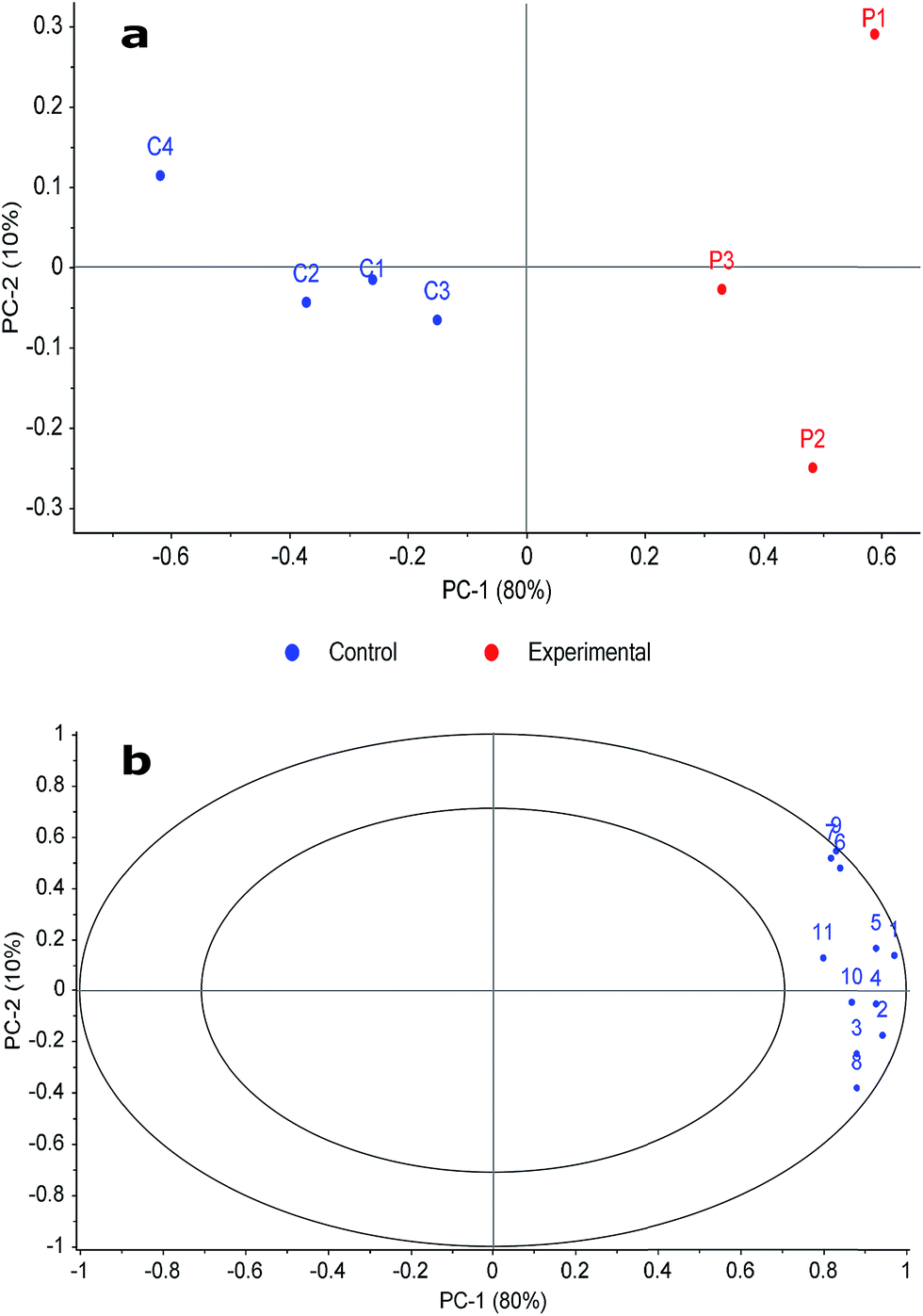 | ||
| Fig. 2 Principal component analysis (PCA) plots of (a) scores and (b) correlation loadings based on normalised peak volume of significant compounds. Compound loading labels are associated with the numbering in Table 1. | ||
Although data filtration considerably reduced the number of compounds evaluated from those detected during the original analysis, Fig. 2b shows that the significant compounds were those that were specific to decomposition. In addition, the number of compounds reported as being significant was expected to be low since these samples were collected three months post-mortem when the remains were skeletonised and a reduced odour was present. Extending this analysis to earlier stages of decomposition would be of interest in the future to compare the high-resolution identifications with previous work that characterised decomposition VOCs in early-stage decomposition using low-resolution TOFMS. Using a FR analysis of this nature will facilitate discrimination of the more complex, multivariate data expected under these circumstances.
Quality control study
Mass calibration was performed on manually assigned PFK peaks prior to the analysis of the samples. Calibration was also verified by running a test mix containing more than 100 compounds from various chemical classes, whose composition has been noted in previous work.5 Following the injection of samples, this test mix was injected once per day for an additional 14 days. Relevant compounds (one from each chemical class in the test mix) were chosen for monitoring the mass error over time and included: 1-heptanol, 2-decanol, 2-octanone, bromobenzene (internal standard), diethyl phthalate, naphthalene, nonane, pentylbenzene, p-xylene, pyridine and 1-chlorooctane. The mass error was tracked for each compound during this time. Tuning of the HRTOFMS was performed on each day for resolution and accuracy. This information was considered to be valuable when extending the use of GC×GC-HRTOFMS from this study to larger longitudinal data sets for decomposition odour profiling. The mass errors of the chosen compounds fluctuated over the two-week time period (Fig. 3). The mass error for select compounds did, on occasion, increase to high levels (>100 ppm) but would return to low error levels (<20 ppm) without mass calibration. In addition, all compounds appeared to follow the same trend and therefore it is likely that this fluctuation was associated with instrument response. For a longitudinal study, the mass error should be verified prior to each analysis using chemical standards in order to determine whether mass calibration is stable and sufficient to ensure reliability of the analysis. This is particularly important for thermal desorption introduction, as the sample cannot typically be re-analysed if the mass error within the sample is found to be high after injection.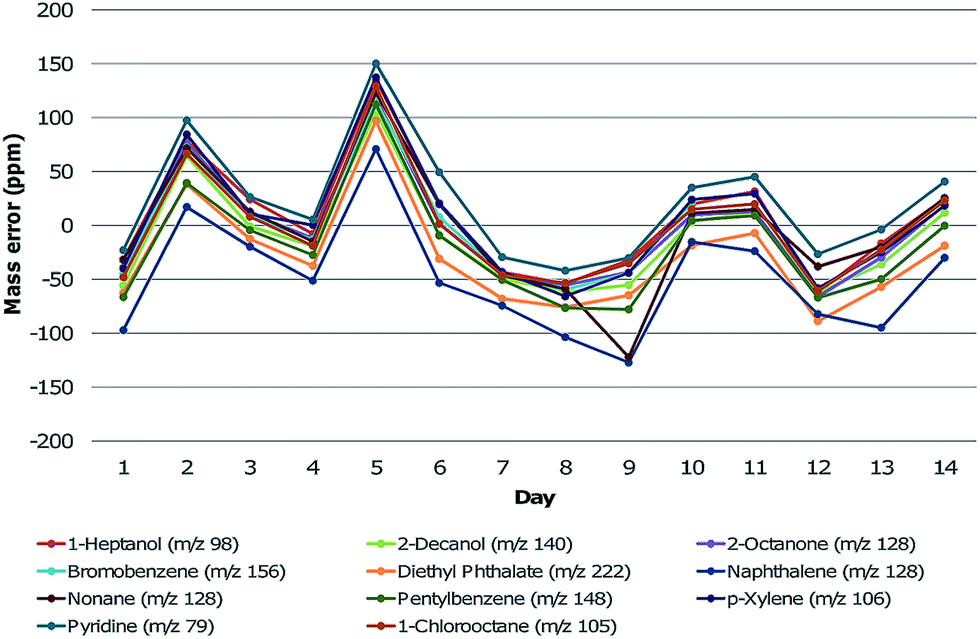 | ||
| Fig. 3 Mass errors (in ppm) of selected compounds based on the measured m/z in comparison to the accurate m/z over a two week period. | ||
Calibration curve study
Calibration curves were constructed for the target compounds selected from the test mix in order to investigate the ability to perform absolute quantification for decomposition studies using this instrument. Solutions of the test mix were prepared at concentrations of 0.1, 1, 2, 5 and 10 ppm in order to determine the linearity by linear regression. This range of concentrations was based on the typical range of VOCs expected within a sample of decomposition odour according to the dynamic range experienced within this matrix. Overall, acceptable R2 values were obtained for the compounds selected across the compound classes (R2 > 0.9) with the exceptions of nonane (R2 = 0.8920) and 1-chlorooctane (R2 = 0.8388). The linearity of some select compounds in decomposition odour analysis has been previously demonstrated for a narrow range of VOC concentrations using GC-LRMS27 and exhibited comparable R2 values to the study herein. The dynamic range exhibited by decomposition odour samples (i.e. presence of both high-level and low-level VOCs) requires that linearity be acceptable across a large range of concentrations, particularly because the ability to dilute and re-analyse samples does not exist. The linearity of calibration curves in this study demonstrated that adequate absolute concentration determination could be performed for most compounds within the test mix from a range of 0.1–10 ng mass loading on sorbent tubes, which represented the ranges of VOCs typically analysed in this study and in previous work by the authors. Altering the collection volume on the sorbent tubes between studies does indeed affect the mass loading of compounds on the sorbent tube and therefore, optimisation of collection parameters is essential prior to performing studies involving absolute quantification. Although most previous studies on decomposition odour have performed relative quantification of analytes based on an internal standard, moving forward with absolute quantification of analytes will be useful once a set of core decomposition VOC biomarkers has been developed in this area of research. Quantitative data of this nature will facilitate comparison of VOC concentration between studies that use different experimental methods, and may also have value for associating decomposition VOC response with post-mortem interval (PMI) estimation from autopsy specimens.Challenges
The use of GC×GC-HRTOFMS was valuable for providing additional confirmatory information about analyte identification in combination with the use of LRIs and NIST library identifications. One of the main challenges of this study was managing the elaborate size of the data produced by the samples (e.g. each file was approximately 40 GB). This challenge has also been noted by researchers using this instrumentation for environmental monitoring.9,11 Due to the longitudinal nature of decomposition odour studies, the number of samples analysed in a single study can be very large. This produces issues with data storage, computational resources, and processing time. With current software and computing capabilities, simply opening the raw profile data can significantly increase the throughput of sample analysis. In addition, access to GC×GC-HRTOFMS instrumentation may be limited for many researchers as it is still a novel technology.Due to these challenges, at this time it is suggested that the “added dimension” afforded by the HRTOFMS in this study would be most valuable to this area of research for generating reference databases of reported decomposition VOCs. This would provide a tool for researchers to refer to when conducting studies using GC or GC×GC with LRMS to provide a secondary (or tertiary) confirmation of analyte identification. Generating reference databases for decomposition odour analysis was first proposed in 2004 by Vass et al.22 and provided reference data for pioneering studies in decomposition odour analysis using GC-MS. Following on from this work, more information has now been made available about the decomposition VOC profile through implementation of GC×GC-TOFMS.1–8 It is now possible to develop decomposition VOC databases with even higher confidence using GC×GC-HRTOFMS. This provides a promising outlook for delivering an updated database of compounds based on the advances in analytical technology that have been employed in recent years.
Conclusions
GC×GC-HRTOFMS was used in this study to enhance confidence in analyte identification and data comparison of decomposition VOCs in soil. Quality control of the mass error showed that the instrumentation was robust and could be used for longitudinal studies. Furthermore, absolute quantification would be possible using this instrumentation based on the range of VOC concentrations exhibited in decomposition VOC studies. A reference database tool could be developed using GC×GC-HRTOFMS and would provide valuable complementary information for providing consistency and reliability of results across studies, especially when access to reference standards for certain compounds is not possible. GC×GC-HRTOFMS, in combination with the FR analysis, will improve the ability to target the relevant decomposition VOCs from this complex matrix, specifically for forensic applications that rely heavily on this information (i.e. HRD canines). Further studies involving the analysis of decomposition VOCs throughout all stages of decomposition and using human cadavers are required in order to develop a reference database of this nature. Providing reference tools by GC×GC-HRTOFMS can extend well beyond decomposition odour analysis. This instrumentation could also be used to improve analyte identification in research that profiles VOC from contraband that detection dogs are used to locate. Metabolomics also relies highly on the detection of compounds produced by complex biochemical processes and therefore it is also expected that GC×GC-HRTOFMS reference databases could be useful for confirming identifications. Providing information about the potential for compound misidentification using low-resolution instrumentation would be valuable for refining analyte reporting. With the developing commercial availability of GC×GC-HRTOFMS and associated software abilities, this tool will become an additional dimension for VOC profiling in many fields of research.Acknowledgements
This work was funded by the Australian Research Council (ARC), University of Technology Sydney (UTS), University of Liège and JEOL Ltd. Restek Corporation and SGE Analytical Science are gratefully acknowledged for contributing research supplies for this project.Notes and references
- S. Stadler, P.-H. Stefanuto, M. Brokl, S. L. Forbes and J.-F. Focant, Anal. Chem., 2013, 85, 998–1005 CrossRef CAS PubMed.
- P.-H. Stefanuto, K. Perrault, S. Stadler, R. Pesesse, M. Brokl, S. Forbes and J.-F. Focant, ChemPlusChem, 2014, 79, 786–789 CrossRef CAS.
- C. Brasseur, J. Dekeirsschieter, E. M. J. Schotsmans, S. de Koning, A. S. Wilson, E. Haubruge and J.-F. Focant, J. Chromatogr. A, 2012, 1255, 163–170 CrossRef CAS PubMed.
- J. Dekeirsschieter, P.-H. Stefanuto, C. Brasseur, E. Haubruge and J.-F. Focant, PLoS One, 2012, 7, e39005 CAS.
- K. A. Perrault, P.-H. Stefanuto, B. H. Stuart, T. Rai, J.-F. Focant and S. L. Forbes, J. Sep. Sci., 2014, 38, 73–80 CrossRef PubMed.
- S. L. Forbes, K. A. Perrault, P.-H. Stefanuto, K. D. Nizio and J.-F. Focant, PLoS One, 2014, 9, e113681 Search PubMed.
- J.-F. Focant, P.-H. Stefanuto, C. Brasseur, J. Dekeirsschieter, E. Haubruge, E. M. J. Schotsmans, A. S. Wilson, S. Stadler and S. L. Forbes, Chem. Bull. Kazakh Natl. Univ., 2014, 4, 177–186 Search PubMed.
- K. A. Perrault, T. Rai, B. H. Stuart and S. L. Forbes, Anal. Methods, 2014, 7, 690–698 RSC.
- S. Hashimoto, Y. Zushi, A. Fushimi, Y. Takazawa, K. Tanabe and Y. Shibata, J. Chromatogr. A, 2013, 1282, 183–189 CrossRef CAS PubMed.
- T. Ieda, N. Ochiai, T. Miyawaki, T. Ohura and Y. Horii, J. Chromatogr. A, 2011, 1218, 3224–3232 CrossRef CAS PubMed.
- Y. Zushi, S. Hashimoto, A. Fushimi, Y. Takazawa, K. Tanabe and Y. Shibata, Anal. Chim. Acta, 2013, 778, 54–62 CrossRef CAS PubMed.
- S. Hashimoto, Y. Takazawa, A. Fushimi, K. Tanabe, Y. Shibata, T. Ieda, N. Ochiai, H. Kanda, T. Ohura, Q. Tao and S. E. Reichenbach, J. Chromatogr. A, 2011, 1218, 3799–3810 CrossRef CAS PubMed.
- N. Ochiai, T. Ieda, K. Sasamoto, Y. Takazawa, S. Hashimoto, A. Fushimi and K. Tanabe, J. Chromatogr. A, 2011, 1218, 6851–6860 CrossRef CAS PubMed.
- T. Cajka, J. Hajslová, R. Kazda and J. Poustka, J. Sep. Sci., 2005, 28, 601–611 CrossRef CAS PubMed.
- K. A. Perrault, B. H. Stuart and S. L. Forbes, Chromatography, 2014, 1, 120–140 CrossRef PubMed.
- S. L. Forbes and K. A. Perrault, PLoS One, 2014, 9, e95107 Search PubMed.
- M. Brokl, L. Bishop and C. Wright, J. Sep. Sci., 2013, 36, 1037–1044 CrossRef CAS PubMed.
- M. Brokl, L. Bishop, C. G. Wright, C. Liu, K. McAdam and J.-F. Focant, J. Chromatogr. A, 2014, 1370, 216–229 CrossRef CAS PubMed.
- N. Ochiai, T. Ieda, K. Sasamoto, A. Fushimi, S. Hasegawa, K. Tanabe and S. Kobayashi, J. Chromatogr. A, 2007, 1150, 13–20 CrossRef CAS PubMed.
- L. E. DeGreeff and K. G. Furton, Anal. Bioanal. Chem., 2011, 401, 1295–1307 CrossRef CAS PubMed.
- J. Dekeirsschieter, F. J. Verheggen, M. Gohy, F. Hubrecht, L. Bourguignon, G. Lognay and E. Haubruge, Forensic Sci. Int., 2009, 189, 46–53 CrossRef CAS PubMed.
- A. A. Vass, R. R. Smith, C. V Thompson, M. N. Burnett, D. A. Wolf, J. A. Synstelien, N. Dulgerian and B. A. Eckenrode, J. Forensic Sci., 2004, 49, 760–769 CrossRef CAS.
- A. A. Vass, R. R. Smith, C. V Thompson, M. N. Burnett, N. Dulgerian and B. A. Eckenrode, J. Forensic Sci., 2008, 53, 384–391 CrossRef CAS PubMed.
- A. A. Vass, Forensic Sci. Int., 2012, 222, 234–241 CrossRef PubMed.
- M. Statheropoulos, A. Agapiou, C. Spiliopoulou, G. C. Pallis and E. Sianos, Sci. Total Environ., 2007, 385, 221–227 CrossRef CAS PubMed.
- M. Statheropoulos, A. Agapiou, E. Zorba, K. Mikedi, S. Karma, G. C. Pallis, C. Eliopoulos and C. Spiliopoulou, Forensic Sci. Int., 2011, 210, 154–163 CrossRef CAS PubMed.
- E. Rosier, E. Cuypers, M. Dekens, R. Verplaetse, W. Develter, W. Van de Voorde, D. Maes and J. Tytgat, Anal. Bioanal. Chem., 2014, 406, 3611–3619 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |