
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In-gel detection of biotin–protein conjugates with a green fluorescent streptavidin probe†
Alanna E.
Sorenson‡
a,
Samuel P.
Askin‡
a and
Patrick M.
Schaeffer
*ab
aCentre for Biodiscovery and Molecular Development of Therapeutics, James Cook University, Molecular Sciences Building, University Drive, Douglas, QLD 4811, Australia. E-mail: patrick.schaeffer@jcu.edu.au; Tel: +61 07 4781 6388
bComparative Genomics Centre, James Cook University, Douglas, QLD 4811, Australia
First published on 28th January 2015
Abstract
Exploitation of the (strept)avidin–biotin interaction is extremely valuable in a variety of biotechnological applications. Biotin is often covalently linked to proteins or nucleic acids. Determination of the degree of biotinylation of such macromolecules is essential for downstream applications. There is currently a gap in simple yet efficient assays for rapidly quantitating protein biotinylation, as staple methods may produce unclear results or rely on immuno- or competitive assays. We present a simple and reliable electrophoretic method to determine the relative extent of biotinylation of macromolecules. The method relies on complex formation between a biotinylated macromolecule and a streptavidin probe resulting in an electrophoretic mobility shift of the complex detectable by SDS-PAGE. Finally, a green fluorescent protein labelled streptavidin probe was developed to eliminate the need for staining and reduce assay time.
1. Introduction
Biotinylation is the process of covalently attaching a biotin molecule to a protein, DNA or other molecule. This process is rapid, specific and unlikely to interfere with natural molecule function due to biotin's small size (244 Da). The interaction between biotin and streptavidin (Stv) is one of the strongest non-covalent interactions known.1 Stv forms a tetramer, with each monomer capable of binding one biotin or biotinylated compound. The high affinity and specificity, fast on-rate and resistance to heat, pH and proteolysis of the Stv–biotin interaction has led to its exploitation in a wide variety of biotechnological applications including: sensitive detection; labelling; immobilization and purification of biological molecules;2 supramolecular protein complex formation;3 and the study of protein–protein interactions in living cells.4,5 Proteins can be biotinylated either chemically6 or enzymatically.7Enzymatic biotinylation is an established method and requires a 15 amino acid target peptide of biotin protein ligase (BirA), termed AviTag, at the N-terminus, C-terminus or in an internal loop of the protein of interest (POI).8 BirA reacts with the POI comprising an AviTag in the presence of biotin and ATP to site-specifically link biotin to a lysine residue in the AviTag sequence. This can be achieved in vitro9 or in vivo10,11 with reaction times of several hours needed for high-level biotinylation of the AviTag.
Consequently, determination of the degree of biotinylation is important for the optimization of applications using biotinylated proteins, DNA and other macromolecules. One of the most common biotin detection methods is using 2-(4-hydroxyazobenzene) benzoic acid (HABA). Here, the HABA dye is initially bound to avidin and is displaced in the presence of biotin, resulting in a change in absorbance directly proportional to the amount of biotin present.12 More sensitive methods use a similar assay with fluorescent avidin, where the increase in fluorescence upon HABA displacement is proportional to the amount of biotin present.13 Electrochemical techniques have also been developed.14 Evaluation of protein biotinylation can be performed by comparing electrophoretic migration of biotinylated and non-biotinylated proteins.9 This method does not require the removal of biotin from the reaction. However, due to the relatively low molecular weight of biotin, retardation of the migration of the biotinylated protein in SDS-PAGE can be difficult to distinguish. Estimation of in vivo protein biotinylation is also possible by monitoring the supershift of a biotinylated POI in the presence of Stv using SDS-PAGE and western blot analysis4 or by a Stv–peroxidase conjugate on nitrocellulose transblots.15
Here we present, an SDS-PAGE method to determine the relative extent of biotinylation of POI using either Stv or a GFP-tagged Stv (Stv-GFP) as electrophoretic mobility shift inducing probes. Using Stv-GFP, the extent of protein biotinylation could readily be determined by direct UV-transillumination of unstained polyacrylamide gels. In addition, using our recombinant BirA and optimised protocol we were able to achieve >90% biotinylation of an AviTag fused POI in 1 h at room temperature. Using our new workflow, high-level biotinylation of both AviTag-fused Tus16,17 and staphylococcal nuclease18 along with quantitation of their biotin content were achieved in less than 3 h.
2. Materials and methods
2.1. Cloning, expression and purification
![[G with combining low line]](https://www.rsc.org/images/entities/i_char_0047_0332.gif)
![[A with combining low line]](https://www.rsc.org/images/entities/i_char_0041_0332.gif)
![[T with combining low line]](https://www.rsc.org/images/entities/i_char_0054_0332.gif)
![[C with combining low line]](https://www.rsc.org/images/entities/i_char_0043_0332.gif) C-3′ and psJCU322: 5′-TCGAGTTTTTCGTGCCATTCGATTTTCTGAGCCTCGAAGATGTCGTTCAGACCGCCACC-3′ encoding the AviTag and EcoRV (italicized and underlined respectively) recognition sequences were mixed to obtain a final concentration of 1 μM, denatured for 2 min at 80 °C and slowly cooled for optimal annealing. This dsDNA presenting BamHI and XhoI overhangs (italicized) was ligated into the vector and transformed in E. coli DH12S to yield p-AviTag-6HIS (pSA236).
C-3′ and psJCU322: 5′-TCGAGTTTTTCGTGCCATTCGATTTTCTGAGCCTCGAAGATGTCGTTCAGACCGCCACC-3′ encoding the AviTag and EcoRV (italicized and underlined respectively) recognition sequences were mixed to obtain a final concentration of 1 μM, denatured for 2 min at 80 °C and slowly cooled for optimal annealing. This dsDNA presenting BamHI and XhoI overhangs (italicized) was ligated into the vector and transformed in E. coli DH12S to yield p-AviTag-6HIS (pSA236).
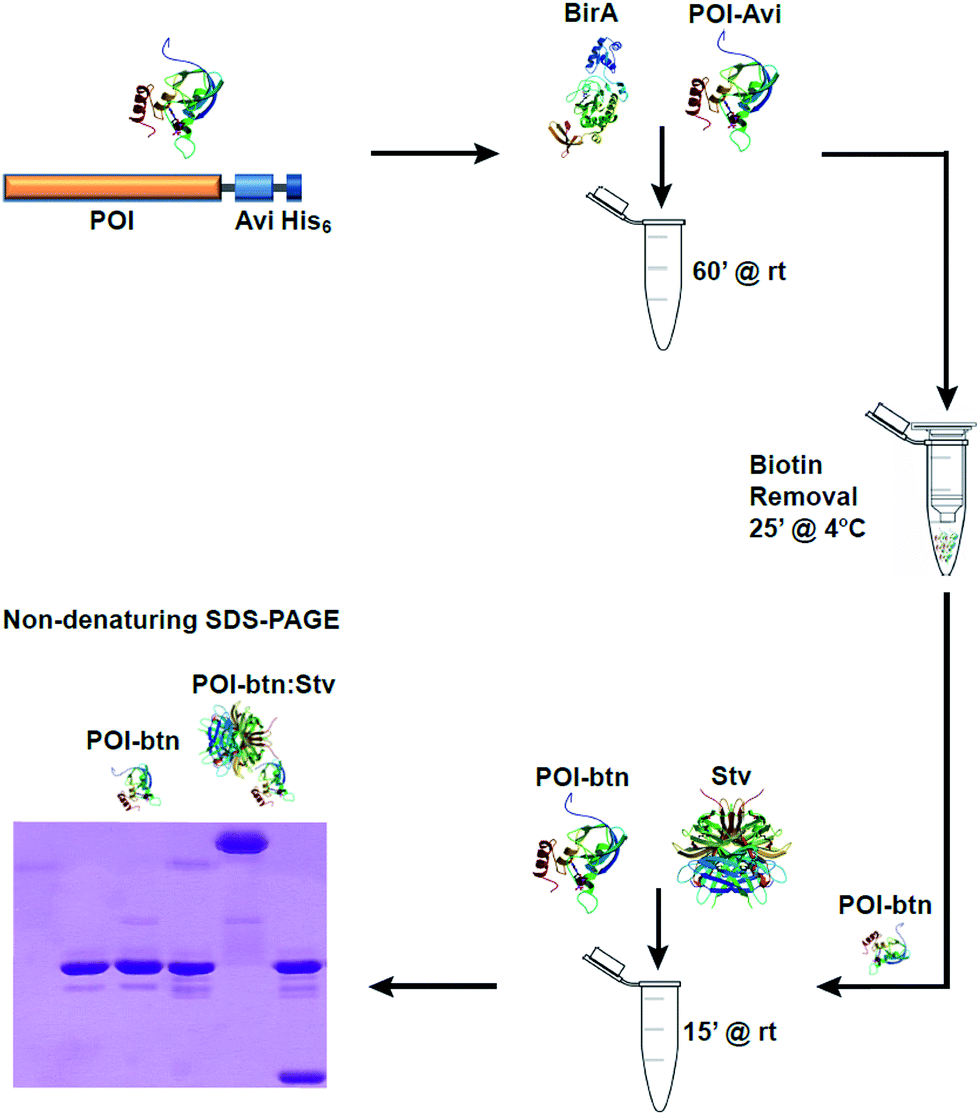 | ||
| Fig. 1 Schematic representation of the biotinylation detection workflow. Recombinant POI-AviTag is incubated at room temperature for 1 h with recombinant BirA in the presence of ATP, biotin and MgCl2 for biotinylation of the AviTag. The biotinylated POI-AviTag is then transferred to a 1.5 ml spin column containing Ni-affinity resin and passed twice through the column. The bound POI-AviTag is eluted in buffer containing 200 mM imidazole by centrifuging at 1000g. Following elution, the biotinylated POI-AviTag (POI-btn) is incubated with excess streptavidin (Stv) for 15 min at room temperature, and the mixture is subjected to SDS-PAGE. | ||
GCTATCAGTAATGTTTCGAA-3′ and psJCU330: 5′-AAAAAATTGACCTGAATCAGCGTTGT-3′. The PCR product was digested with NdeI and BamHI, ligated into pSA236 and transformed in E. coli DH12S to yield pNuc-AviTag-6HIS (pAC240).
ATGGCGTCTATGACCGGTGGTCAGCAGATGGGTAC-3′ and psJCU340: 5′-TACCCATCTGCTGACCACCGGTCATAGACGCCATGAT-3′ coding for the T7 solubility tag20 (MASMTGGQQMG) were mixed to obtain a final concentration of 1 μM, denatured for 2 min at 80 °C and slowly cooled for optimal annealing. The vector pAC229 was digested with EcoRV and NcoI and the annealed dsDNA presenting these overhangs was ligated into the vector to form pBSK-T7tag-Stv (pAC257). The T7tag-Stv sequence was digested with EcoRV and NheI and inserted into the GFP vector pIM013 (pET-uvGFP)21 to form p6HIS-T7tag-Stv-GFP (pAC253).
AAGGATAACACCGTGCCACTG-3′ and psJCU320: 5′-AAAAAATTTTTCTGCACTACGCAGGG-3′. The PCR product was digested with AflII and NheI and ligated into the GFP vector pIM013 (ref. 21) to form p6HIS-BirA-GFP (pSA239). The plasmid pSA239 was digested with NheI and an end-filling reaction performed to introduce a stop codon between the BirA and GFP coding regions to form p6HIS-BirA (pSA267) using a previously described method.21
2.2. Biotinylation and purification of Tus-AviTag and Nuc-AviTag
Biotinylation of AviTag-POI was performed in 50 μl reactions containing 300 nM BirA, 500 μM biotin, 2.5 mM ATP, 1 mM MgCl2 and 30 μM POI-AviTag in 25 mM Tris (pH 8.0). Control reactions without BirA were performed in parallel. Reactions were incubated at room temperature for 1 h. Following incubation, biotin was removed from the reactions by Ni-affinity purification using Profinity IMAC nickel resin (Biorad) and 1.5 ml spin columns. Reactions were bound to 50 μl pre-equilibrated nickel resin slurry for 5 min at 4 °C, then passed through the resin twice by centrifugation at 1000 × g for 1 min at 4 °C. The resin was washed twice with 500 μl of 25 mM Tris (pH 8.0) supplemented with 5% glycerol and 150 mM NaCl by centrifugation at 1000 × g for 1 min at 4 °C. The proteins were eluted from the spin column with 50 μl of the same buffer supplemented with 200 mM imidazole and centrifugation at 1000 × g for 1 min at 4 °C following 5 min incubation at 4 °C. The eluate was then passed a second time through the same spin column as for first elution. All control reactions were processed in identical fashion for comparison to account for protein losses occurred during purification and reaction steps and for normalization.2.3. Detection of biotinylation of POI-AviTag using Stv
Following purification, 7.5 μl of protein elution from spin column (see above) with or without BirA were incubated with 0.75 μl Stv (5 mg ml−1, Invitrogen) at room temperature for 15 min in duplicate. Control reactions were performed similarly by omitting components as indicated in (cf.Fig. 2). One set of reactions were then denatured at 95 °C for 15 min. Reactions (8.25 μl; 225 pmol POI-AviTag) were mixed with non-reducing 2× SDS sample buffer (50 mM Tris–HCl, pH 6.8, 2% SDS, 10% glycerol, 0.01% bromophenol blue), then subjected to 13% SDS-PAGE and stained with Coomassie Blue to determine the extent of biotinylation. Quantitative analysis of biotinylation yields was determined by integration of residual unbound POI-AviTag bands (ImageJ, NIH, USA) and compared to the control reaction (see above).21 Three independent experiments were performed.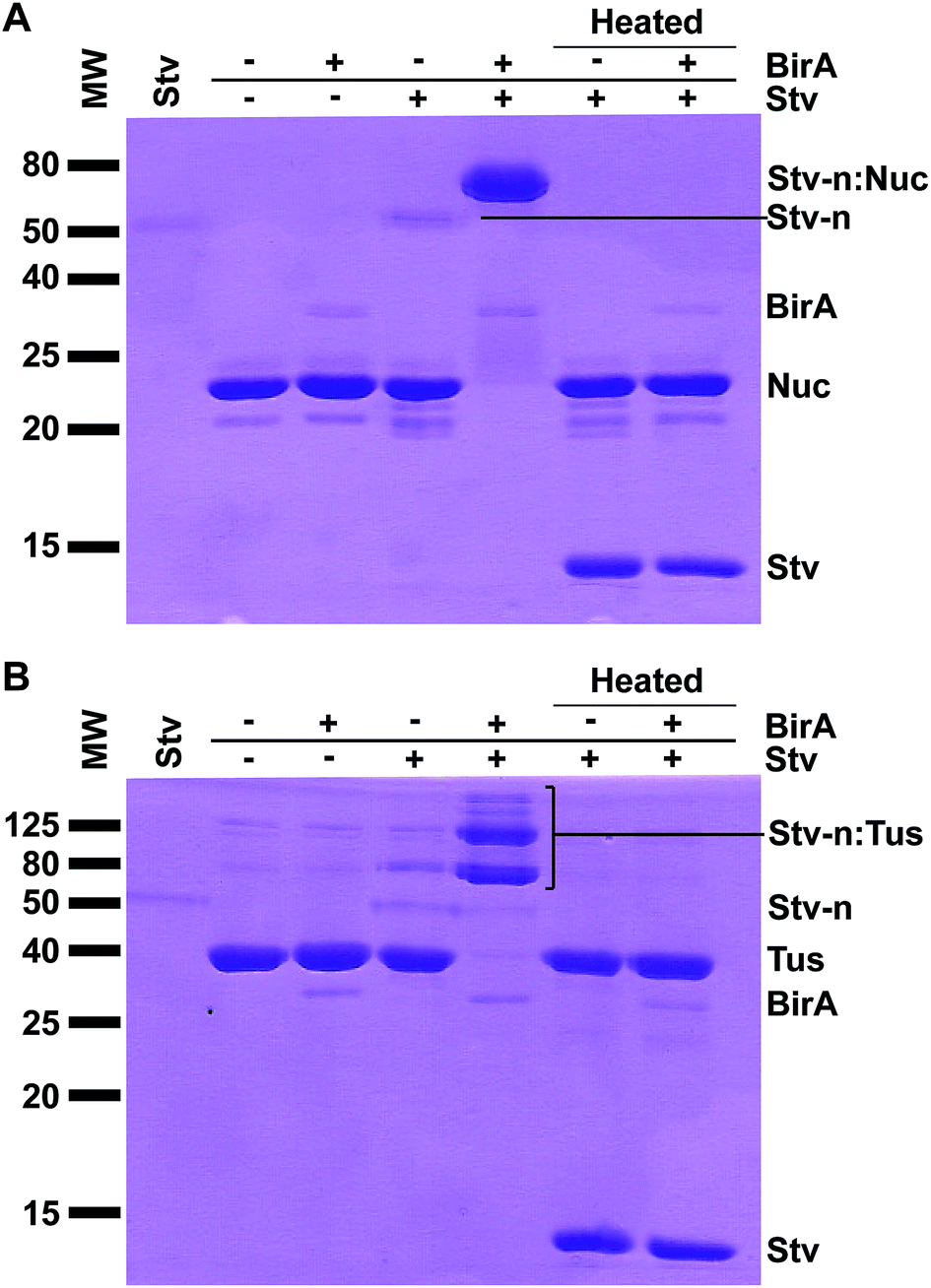 | ||
| Fig. 2 Detection of protein biotinylation by SDS-PAGE and Stv-induced band-shift analysis. Biotinylated Tus- and Nuc-AviTag (225 pmol assuming 100% recovery) were incubated with Stv (750 pmol tetramer) for 15 min at room temperature, then subjected to 13% SDS-PAGE followed by Coomassie staining. (A) Detection of biotinylation of Nuc-AviTag. Samples of Nuc-AviTag (biotinylated or non-biotinylated) with or without addition of Stv were separated by SDS-PAGE. Duplicate reactions as above were also subjected to heat treatment for 15 min at 95 °C prior SDS-PAGE to fully denature Stv. A control of Stv only (native; far left lane) has also been included. (B) Detection of biotinylation of Tus-AviTag. All procedures and presentation are as described for Nuc-AviTag. | ||
2.4. Detection of biotinylation of POI-AviTag using Stv-GFP
Following spin column purification of reactions, a total of 6 reactions (10 μl) were set up for each POI, by mixing 2.5 μl of elutions with and without 7.5 μl Stv-GFP (10 μM in 50 mM Na2HPO4 (pH 7.8), 2 mM β-mercaptoethanol, 300 mM NaCl). Control reactions were performed similarly by omitting components in the biotinylation reactions or addition of competing biotin (2 mM) to Stv-GFP prior to addition to the 2.5 μl biotinylated POI as indicated (cf.Fig. 3). All reactions were carried out at room temperature for 15 min. Reactions (10 μl; 75 pmol POI-AviTag) were mixed with 2× SDS sample buffer and then subjected to 10% SDS-PAGE. The SDS gel was rinsed with milliQ water and visualised over a UV transilluminator. The gel was then stained with Coomassie Blue. Quantitative analysis of biotinylation yields was determined as above.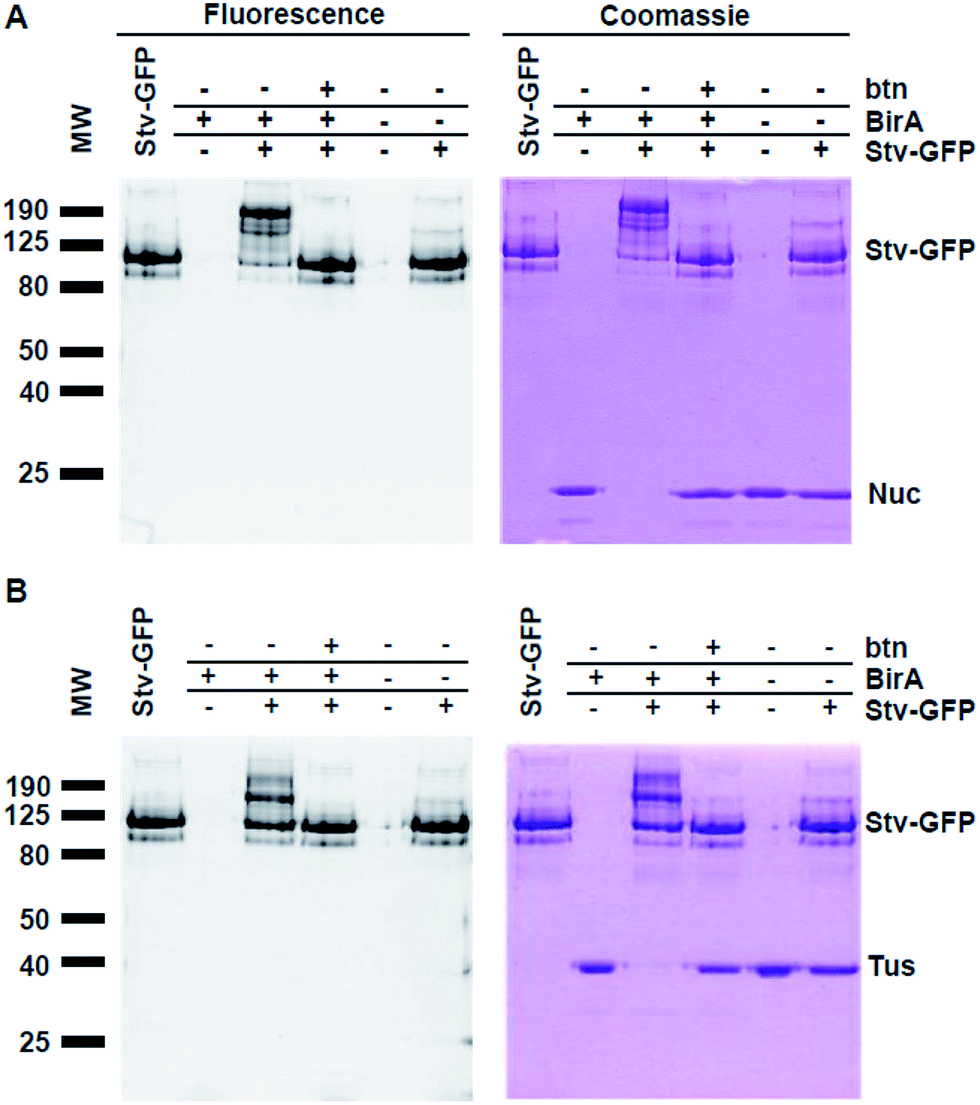 | ||
| Fig. 3 Detection of protein biotinylation by SDS-PAGE and fluorescent Stv-GFP-induced band-shift analysis. Biotinylation reactions of Tus- and Nuc-AviTag (75 pmol assuming 100% recovery) were incubated with Stv-GFP (75 pmol tetramer) for 15 min at room temperature, then subjected to 10% SDS-PAGE followed by fluorescence analysis (left gels displayed in inverted colours) and Coomassie staining (right gels). Background control reactions were performed in the absence of BirA. (A) Detection of biotinylation of Nuc-AviTag. Samples of Nuc-AviTag (biotinylated or non-biotinylated) with or without addition of Stv-GFP were separated by SDS-PAGE. A competition reaction in the presence of Stv-GFP preincubated with excess biotin (500 μM) was performed to confirm that the band shift was due to the formation of the complex through interaction with the biotin. A control lane of Stv-GFP only (native; far left lane) has also been included. (B) Detection of biotinylation of Tus-AviTag. All procedures and presentation are as described for Nuc-AviTag. | ||
2.5. Confirmation of Stv-GFP monomeric form upon heat denaturation
SDS-PAGE was performed to confirm the formation of monomeric Stv-GFP upon heat denaturation of the complex (Fig. S2†).2.6. GFP-electrophoretic mobility shift assay (GFP-EMSA)
GFP-EMSA was performed similarly to a previously described method22 to confirm biotin binding activity by Stv-GFP (Stv-GFP). Following purification, 5 μl of Stv-GFP (5 μM in 50 mM Na2HPO4 (pH 7.8), 2 mM β-mercaptoethanol, 300 mM NaCl) was incubated with or without biotinylated DNA (5 μl of a 5 μM solution in milliQ water; 5′-[biotin]AAAAAAAAAAGGGGATATGTTGTAACTAAAG-3′; Sigma-Aldrich) at room temperature for 15 min. Reactions (10 μl) were mixed with 6× DNA loading dye and separated by agarose (1%) gel electrophoresis in TBE at 80 V for 45 min. The agarose gel was visualized over a UV transilluminator to record GFP-autofluorescence, then stained with GelRed (Biotium, USA) and re-visualized over a UV transilluminator (Fig. S3†).3. Results and discussion
3.1. Protein biotinylation and detection with a Stv probe
Initially, our aim was to enzymatically biotinylate7 two POIs – Tus19,23,24 and staphylococcal nuclease18 – to be used in various biotechnological applications that required accurate quantitation of their extent of biotinylation. To assess the extent of in vivo biotinylation of our POI-AviTag we intended to use the simple SDS-PAGE method described by Li and Sousa.9 Interestingly, we could not detect a shift in electrophoretic mobility for these proteins using this method even after in vitro biotinylation. This was in contrast to previously published analysis of biotinylation, where a small change in shift was observed when comparing biotinylated to non-biotinylated protein by SDS-PAGE.9 One possible explanation for our results was that our recombinant BirA was not functional. Another possible explanation was that the change in electrophoretic mobility caused by the addition of biotin may not have been significant enough to allow detection by SDS-PAGE, and could also be dependent on the size of the protein.Viens et al. had previously reported that upon incubation of cell lysates with Stv, biotinylated proteins were identifiable by western blot analysis through a Stv-induced supershift.4 We decided to eliminate the western blot analysis step as sufficient protein levels were present in our reaction conditions for direct Coomassie Blue detection. Here, excess biotin needed to be removed from the biotinylation reactions. This was achieved via rapid Ni-affinity binding and purification of the biotinylated protein using a quick spin-column protocol in less than 25 min (see Fig. 1 and ESI†). The eluted, biotinylated Tus- (40 kDa) and Nuc-AviTag (26 kDa) were then incubated with Stv for 15 min at room temperature and separated by SDS-PAGE under non denaturing conditions – i.e. samples were not heat denatured prior to loading (Fig. 1).
Previous work on the electrophoretic mobility of Stv has shown that the protein maintains its tetrameric native structure (∼60 kDa) and activity in the presence of SDS-PAGE sample buffers, and requires significant heating to break this interaction.25 A dramatic change in migration was evident for the non heat-denatured reactions where BirA (37 kDa) and Stv were present compared to the same reactions in the absence of BirA (Fig. 2, cf. lanes 4 and 5). When these reactions (Fig. 2, cf. lanes 6 and 7) were heat-denatured prior electrophoresis the protein bands migrated in identical fashion and at their expected subunit sizes as a consequence of denaturation of the complexes. Quantitative determination of biotinylation extent using ImageJ (NIH, USA) indicated 96 ± 2% biotinylation for Nuc-AviTag and 97 ± 2% for Tus-AviTag after 1 h at room temperature. It is interesting to note that endogenous biotinylation that occurred in vivo was not detectable for Nuc-AviTag (Fig. 2A) but trace levels of biotinylation were detected for Tus-AviTag (Fig. 2B).
In the case of the Tus-AviTag biotinylation reaction, two distinct major bands were present at ∼80 kDa and 120 kDa, compared to the Tus-AviTag at ∼40 kDa and Stv at ∼50 kDa (Fig. 2B). These likely corresponded to a single Tus-AviTag and two Tus-AviTag bound to a Stv tetramer, respectively. Other minor higher molecular weight bands were also present, representative of different combinations of Tus-AviTag bound to Stv oligomers (Fig. 2B). It is important to note that the native Stv (Stv-n) and the Stv-n:Tus complexes do not migrate at their expected sizes due to their native configuration. Biotinylated Nuc-AviTag in complex with Stv also exhibited a stark change in migration (Fig. 2A) but only one single band was visible just below 80 kDa, without the higher order bands seen for Tus-AviTag. This was representative of a single Nuc-AviTag bound to a Stv tetramer. This method allows the direct observation of biotin-dependent complex formation and its stoichiometry (Fig. 2, far right lanes). When the Stv:POI complexes were heated at 95 °C for 15 min in SDS-PAGE sample buffer the Stv-induced supershift was abolished, and a distinct Stv band appeared just below 15 kDa, corresponding to the monomeric Stv. It is interesting to note that the native tetrameric Stv band was very faint after Coomassie Blue staining, although after denaturation it was strongly stained. These results confirmed the presence of a Stv-induced shift upon binding to biotinylated proteins, enabling quantitation of the biotinylation level of a POI.
3.2. Detection of biotinylation using a Stv-GFP probe
Although our method is faster and easier than the previously published western blot procedure, we hypothesized that by using a GFP-fused Stv we could eliminate the lengthy protein staining procedure through direct visualization of mobility shifts by UV-transillumination. We have previously shown that GFP tagged proteins retain their fluorescence in unheated SDS-PAGE sample buffer.21,26 The Stv-GFP fusion protein (see ESI†) required an N-terminal T7-tag for soluble expression in E. coli.20 The soluble Stv-GFP was purified by Ni-affinity chromatography and gel filtration (Fig. S2†). The activity of Stv-GFP was assessed through its binding to biotinylated DNA by EMSA (Fig. S3†).For SDS-PAGE-based detection of biotinylation of Tus-AviTag and Nuc-AviTag using the Stv-GFP probe, all steps were repeated as described for the Stv binding analysis (Fig. 1 and 2). However, only 75 pmol of each POI-AviTag were needed for SDS-PAGE. Gels were photographed under UV exposure prior to staining with Coomassie Blue. In contrast to the Stv probe method (Fig. 2), here retardation in the mobility of the fluorescent band corresponding to Stv-GFP was expected upon complex formation with the biotinylated POI (Fig. 3). As described for Fig. 2, binding to Stv-GFP was unique to those reactions where the POI-AviTag was biotinylated in vitro. No endogenous biotinylation that occurred in vivo was detectable by addition of Stv-GFP.
The analysis of GFP fluorescence allowed for specific detection of changes in the mobility of Stv-GFP due to binding to biotinylated proteins. This was supported by the abolition of the POI-AviTag:Stv-GFP interaction upon addition of excess biotin to Stv-GFP prior to the reaction (Fig. 3). While techniques such as western blotting have been previously used to detect this specific interaction,4 here there is no need for any downstream processing or even staining to determine whether a protein has been biotinylated. Furthermore, detection of protein biotinylation using Stv-GFP was easily detectable with 75 pmol POI-AviTag under non-optimal UV excitation conditions, comparing favourably with the linear range of a commercial Fluorescence Biotin Quantitation Kit (Pierce), which has a linear detection range of 10–60 pmol.
In addition to the direct visual detection of protein biotinylation by fluorescence, subsequent staining with Coomassie Blue enables quantitative determination of the extent of biotinylation. Coomassie staining accurately reflected the pattern of fluorescence observed for the Stv-GFP interacting with the Tus- and Nuc-AviTag as expected. Quantitative determination of biotinylation extent using ImageJ (NIH, USA) indicated 88 ± 9% biotinylation for Nuc-AviTag (Fig. 3A) and 97 ± 2% for Tus-AviTag (Fig. 3B) after 1 h at room temperature. These values corresponded well to those obtained for the biotinylation of Tus- and Nuc-AviTag with subsequent binding to Stv, demonstrating the high batch-to-batch reproducibility and time-efficiency of the assay.
4. Conclusion
Biotinylation is an increasingly important tool in many emerging areas of research, including improving the sensitivity of detection of molecules, the formation of supramolecular complexes and the investigation of protein–protein interactions in living cells. Using our recombinant BirA and improved protocol, rapid biotinylation and clean-up of POI-AviTag fusions was achieved, enabling >90% biotinylation in 1 h at room temperature. The short incubation time and low reaction temperature are particularly advantageous for unstable proteins. Most importantly, a simple method was developed to determine the relative extent of biotinylation of POI using fluorescence or standard protein counter-staining. One of the main advantages of the method is that it allows the direct and unambiguous observation of the Stv–POI complex. This is important because it demonstrates that the biotin linked to the protein is available for downstream applications. The assay should also be amenable to the detection and quantitation of biotinylation of other chemically or enzymatically biotinylated macromolecules whether from an in vivo or in vitro source. It could be used for evaluating the biotinylation state of antibodies after random biotinylation using chemical probes with streptavidin in reducing conditions – i.e. after separation of the heavy and light chains. The method could provide a way to identify which chain is biotinylated preferentially and their average degree of biotinylation. A further application of this method could be to determine the topography of biotinylation sites on a protein using partial proteolysis. The Stv-GFP could also be replaced with a commercial fluorescent Stv conjugate but these probes are costly and not homogeneously labeled. The detection methods described in this study enable rapid and unambiguous quantitative determination of a protein's biotinylation level, allowing for optimization of protein biotinylation procedures that are essential for a wide range of applications. As a result, the simplicity and effectiveness of the assay, not being bound to any specialized equipment of reagents show potential for widespread adoption in any laboratory in this field.Conflict of interest
The authors declare that they have no conflict of interest.Acknowledgements
The authors would like to thank the Smart Futures Fund: National and International Research Alliances Program (NIRAP), the National Health and Medical Research Council (NHMRC), James Cook University and the Queensland Tropical Health Alliance for financial support.References
- N. M. Green, Methods Enzymol., 1990, 184, 51–67 CAS.
- N. J. Alves, N. Mustafaoglu and B. Bilgicer, Biosens. Bioelectron., 2013, 49, 387–393 CrossRef CAS PubMed.
- Y. Mori, R. Wakabayashi, M. Goto and N. Kamiya, Org. Biomol. Chem., 2013, 11, 914–922 CAS.
- A. Viens, U. Mechold, H. Lehrmann, A. Harel-Bellan and V. Ogryzko, Anal. Biochem., 2004, 325, 68–76 CrossRef CAS PubMed.
- P. C. Trippier, ChemMedChem, 2013, 8, 190–203 CrossRef CAS PubMed.
- G. Elia, Proteomics, 2008, 8, 4012–4024 CrossRef CAS PubMed.
- M. G. Cull and P. J. Schatz, Methods Enzymol., 2000, 326, 430–440 CAS.
- M. Howarth and A. Y. Ting, Nat. Protoc., 2008, 3, 534–545 CrossRef CAS PubMed.
- Y. Li and R. Sousa, Protein Expression Purif., 2012, 82, 162–167 CrossRef CAS PubMed.
- Y. Li and R. Sousa, Biotechnol. Lett., 2012, 34, 1629–1635 CrossRef CAS PubMed.
- S. S. Ashraf, R. E. Benson, E. S. Payne, C. M. Halbleib and H. Gron, Protein Expression Purif., 2004, 33, 238–245 CrossRef CAS PubMed.
- N. M. Green, Biochem. J., 1965, 94, 23C–24C CAS.
- R. H. Batchelor, A. Sarkez, W. G. Cox and I. Johnson, BioTechniques, 2007, 43, 503–507 CrossRef CAS PubMed.
- Z. Wang, L. Liu, Y. Xu, L. Sun and G. Li, Biosens. Bioelectron., 2011, 26, 4610–4613 CrossRef CAS PubMed.
- J. Zempleni and D. M. Mock, Arch. Biochem. Biophys., 1999, 371, 83–88 CrossRef CAS PubMed.
- M. J. Moreau and P. M. Schaeffer, Mol. BioSyst., 2012, 8, 2783–2791 RSC.
- M. J. Moreau and P. M. Schaeffer, Mol. BioSyst., 2013, 9, 3146–3154 RSC.
- C. B. Anfinsen, Pure Appl. Chem., 1968, 17, 461–517 CrossRef CAS.
- I. Morin, S. P. Askin and P. M. Schaeffer, Analyst, 2011, 136, 4815–4821 RSC.
- A. Gallizia, C. de Lalla, E. Nardone, P. Santambrogio, A. Brandazza, A. Sidoli and P. Arosio, Protein Expression Purif., 1998, 14, 192–196 CrossRef CAS PubMed.
- M. J. Moreau, I. Morin and P. M. Schaeffer, Mol. BioSyst., 2010, 6, 1285–1292 RSC.
- D. B. Dahdah, I. Morin, M. J. Moreau, N. E. Dixon and P. M. Schaeffer, Chem. Commun., 2009, 3050–3052 RSC.
- I. Morin, N. E. Dixon and P. M. Schaeffer, Mol. BioSyst., 2010, 6, 1173–1175 RSC.
- M. J. J. Moreau, I. Morin, S. P. Askin, A. Cooper, N. J. Moreland, S. G. Vasudevan and P. M. Schaeffer, RSC Adv., 2012, 2, 11892–11900 RSC.
- N. Humbert, A. Zocchi and T. R. Ward, Electrophoresis, 2005, 26, 47–52 CrossRef CAS PubMed.
- I. Morin and P. M. Schaeffer, Anal. Biochem., 2012, 420, 121–126 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Figures S1–S4. See DOI: 10.1039/c4ay02666g |
| ‡ Authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |