
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A validated method to measure benzo[a]pyrene concentrations in tobacco by high-performance liquid chromatography-fluorescence detection
M.
Carradus†
*,
K. G.
McAdam
,
J. D. H.
van Heemst
,
C. H. A.
Goss
and
C.
Wright
*
British American Tobacco, Regents Park Road, Millbrook, Southampton SO15 8TL, UK. E-mail: Christopher_Wright@bat.com
First published on 7th January 2015
Abstract
This publication describes a validated method suitable for the quantification of Benzo[a]Pyrene (B[a]P) in tobacco blend and smokeless tobacco products by High-Performance Liquid Chromatography-Fluorescence Detection (HPLC-FLD). Samples were hydrated and extracted with a mixture of hexane and acetone. For the quantification of levels of B[a]P the sample extracts were subjected to adsorption chromatography using base-modified silica to remove co-extracted substances. The concentrated final extract was re-dissolved in acetonitrile and analysed by HPLC with fluorescence detection (FLD). B[a]P and the internal standard, deuterated B[a]P (D12-B[a]P) were resolved chromatographically. The method was validated and determined to be fit for purpose for the quantification of B[a]P in tobacco from 3R4F Kentucky reference cigarettes, a flue-cured Virginia cigarette tobacco blend containing 10% air-cured Burley tobacco, and a Tanzanian dark fire-cured cigarette blend. The method was also validated for smokeless tobacco products including commercially available dry snuff, soft pellet tobacco and pouched snus products over the concentration range of 0.38 ng g−1 to 150 ng g−1, based on extraction of a 0.5–2 g aliquot of sample. The measurement uncertainty at a confidence interval of approximately 95% was estimated from data generated by three analysts using two instruments on three separate occasions using matrix (pouched snus) fortification experiments. The expanded uncertainty of the method was ±21.3% of the mean B[a]P concentration.
Introduction
There are a number of current developments in the area of tobacco product regulation that require validated analytical methods for quantification of toxicants in tobacco and cigarette smoke. For example, the World Health Organisation (WHO) Study Group on Tobacco Product Regulation (TobReg) has recommended limits on the benzo[a]pyrene (B[a]P) and TSNA content of smokeless tobacco products (STPs).1 The same group has also recommended disclosure of the mainstream smoke emissions of 18 toxicants, and mandated lowering of the emissions of 9 of these.2 In the USA the Food and Drug Administration (FDA) has identified over 90 Harmful and Potentially Harmful Constituents (HPHCs) in tobacco products and tobacco smoke,3 and currently requires annual reporting of a subset of these in tobacco products and cigarette smoke emissions.4 B[a]P is one of the priority compounds highlighted by TobReg1 and the FDA,3 and TobReg has recommended a regulatory limit of 5 ng g−1 dry weight of smokeless tobacco,1 based on approximate levels of quantification of currently available analytical methods. Benzo[a]pyrene is also a toxicant identified in smokeless tobacco manufacturer standards, Gothiatek® (a quality established by Swedish Match),5 and the levels established by ESTOC.6 Validated analytical methods are an essential foundation for the effective measurement and potential future regulation of toxicant levels in tobacco products.B[a]P is a Polycyclic Aromatic Hydrocarbon (PAH) and is mainly formed by the incomplete combustion or pyrolysis of organic compounds.7 Approximately 100 PAHs have been detected in tobacco8 and they usually occur as complex mixtures rather than individual compounds. PAH compounds are predominately formed when organic materials are burned at temperatures in the range 500–700 °C, as in the combustion of fossil fuels and cigarettes.7 PAHs may also be introduced into tobacco during leaf growth from environmental sources and the curing process.9 STPs contain variable levels of PAH compounds depending on the type of tobacco used in the product. For instance fire-cured tobaccos, which may contain elevated levels of PAHs, are commonly used in snuffs and certain pellet tobacco products.9
PAHs consist of condensed aromatic ring structures, are lipophilic compounds and are readily absorbed by inhalation, ingestion and dermal exposure.7 The most widely studied PAH is B[a]P, as it is the only PAH identified by IARC as a “known human carcinogen”.10 Data obtained from rat tissue samples have indicated that B[a]P is rapidly distributed throughout the body and is metabolised by the enzyme cytochrome P450 to a reactive epoxide, B[a]P-7,8-diol-9,10-epoxide. The epoxide metabolite is thought to be responsible for the carcinogenic properties of B[a]P.11,12 The structures of the target compound, B[a]P and its deuterated analogue B[a]P (D12-B[a]P), used as internal standard in this study, are shown in Fig. 1.
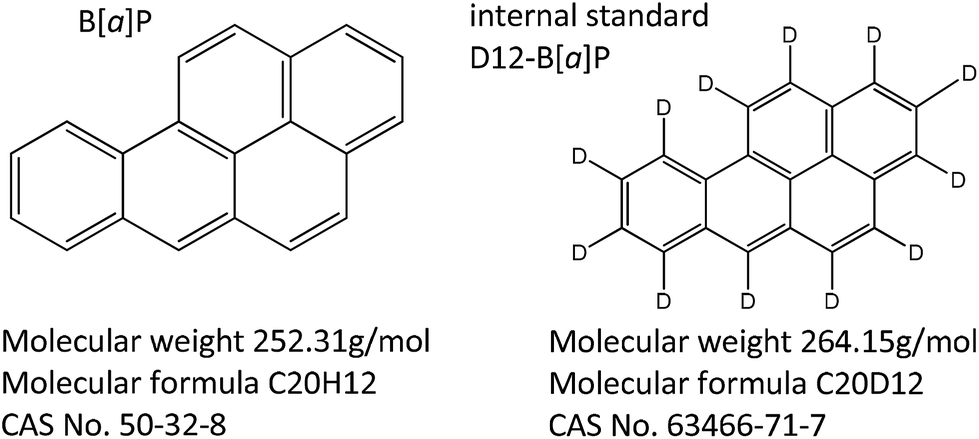 | ||
| Fig. 1 Chemical structures of B[a]P and its deuterated analogue. | ||
There are a number of published methods for the quantification of B[a]P and other PAHs including their measurement in narghile waterpipe tobacco smoke13 and in mainstream cigarette smoke, using Gas Chromatography-Mass Spectrometry (GC-MS);14–16 and of these, CRM 58 (ref. 16) is the only method that has been subjected to extensive inter-laboratory testing and assessment of repeatability and reproducibility. A method for quantification of PAHs (including B[a]P) in mainstream tobacco smoke has been developed using Gas Chromatography-High Resolution Mass Spectrometry (GC-HRMS).17 Alternative techniques in the literature for quantification of PAHs in mainstream cigarette smoke include HPLC-FLD18–21 and HPLC-APPI-MS/MS methods.22
In contrast to tobacco smoke, there are far fewer published methods for B[a]P or PAHs in smokeless tobacco products. Some smokeless tobacco product manufacturers have been operating to Gothiatek® or ESTOC standards for a number of years and therefore unpublished analytical methods are presumably available for this analyte in these matrices. Published methodologies for the determination of 23 PAH compounds in STPs23 and for 21 PAH compounds in STPs9 focus on GC-MS approaches.
HPLC-FLD has been used to determine PAH levels in a similar matrix, tea,24 with levels of sensitivity allowing an LOQ of 0.35 ng g−1 for B[a]P to be achieved. The LOQ of 0.38 ng g−1 for B[a]P in tobacco is considerably below the commonly available LOQ of 5 ng g−1 discussed by TobReg,1 and potentially represents a significant step forward in analytical capability over some of the currently employed methods. The current paper therefore describes the development and validation of a high-performance liquid chromatography-fluorescence detection method for the quantification of B[a]P in both tobacco blends and smokeless tobacco products. The method has been validated in accordance with international standards and guidelines (ICH,25–27 IUPAC,28,29 ISO,30 DG SANCO31 and FDA32). The validation process included a series of experiments designed to test the performance of the method against defined acceptance criteria including precision, expressed as residual standard deviation (RSD) at the LOQ of <20%.31,32 The matrices selected for validation included cigarette tobaccos and STPs with low endogenous B[a]P levels such as commercially available pouched snus, a Swedish-sourced soft pellet STP, and a US-style cigarette tobacco blend containing fire-cured, air-cured and oriental blends. STPs and cigarette tobaccos with high endogenous B[a]P concentration such as commercially available dry snuff product and dark Tanzanian fire-cured tobacco were also examined. Validation of the method for a range of tobacco products ensured method robustness and reproducibility for matrices containing low to high endogenous B[a]P levels.
Experimental
Samples
Smokeless tobacco products (STP)
Reagents and materials
A B[a]P solution with a certified concentration of 1000 μg mL−1 in acetone (QMX Laboratories Ltd, Thaxted, UK) and D12-B[a]P (≥98% isotopic purity) was purchased from Sigma Aldrich (Gillingham, UK). Silica gel 60 Å chromatography grade (70–200 μm) and potassium hydroxide pellets (Fisher Scientific; Loughborough, UK) were used for the preparation of base-modified silica used during the method validation process. Methanol (high-performance liquid chromatography (HPLC) grade) and acetonitrile (HPLC fluorescence grade) with a purity of >99.9%, were purchased from Fisher Scientific (Loughborough, UK) and used in the preparation of matrix samples and for the HPLC mobile phase. Water from an Elga Process Water (High Wycombe, UK) deionised water generator (minimum quality 18.2 MΩ cm−1) was used.Equipment
Cigarette tobacco blends were ground to 1 mm using a Retsch ZM200 mill and the soft pellet tobacco (Oliver Twist) cryomilled prior to extraction using a Retsch Cryomill (Retsch UK Ltd.; Hope, UK). All liquid transfers were made with calibrated pipettes and grade B volumetric glassware. All laboratory consumables were supplied by Fisher Scientific (Loughborough, UK). Sample extraction was carried out using a flat-bed shaker (IKA®-Werke; Staufen, Germany) and a Rotanta 460 centrifuge (Hettich; Tuttlingen, Germany). For production of the base-modified silica a rotary evaporator (Buchi, Oldham, UK) was used. Isolute 70 mL reservoir pack, Isolute 27 mm frits and pre-packed base-modified silica 70 mL 10 g−1 cartridges (Biotage; Uppsala, Sweden).A custom made manifold was used to collect eluent after sample clean up. The manifold was constructed using Lexan® polycarbonate (Gilbert Curry Industrial Plastics; Coventry, UK) (Fig. 2) to enable it to be placed over a Nalgene™ 24 × 30 mm tube rack holder containing 60 mL glass vials (Fisher Scientific; Loughborough, UK). Promega™ One-Way Luer-Lok™ stopcocks (Promega™ UK; Southampton, UK) were inserted into the drilled holes in the manifold to support the 70 mL 10 g−1 potassium silicate cartridge and to control eluent flow.
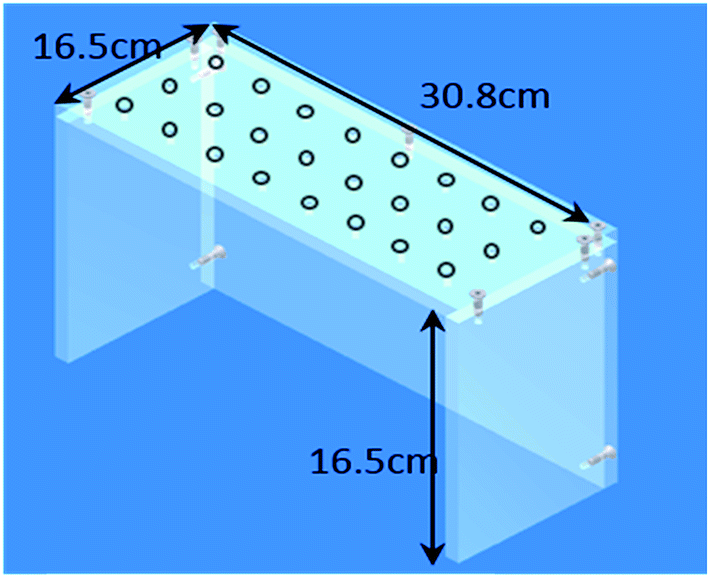 | ||
| Fig. 2 Dimensions of the custom-made manifold. | ||
Turbovap sample concentrators (Biotage; Uppsala, Sweden) with a tube holder for 60 mL vials were used to concentrate the sample extracts. Two Agilent 1200 series HPLC systems (instruments 1 and 2) were coupled to Agilent 1200 Infinity fluorescence detectors and chromatographic separation performed using a Zorbax Eclipse Plus PAH column (250 mm × 2.1 mm with a 5 μm particle size; Agilent Technologies). Data were processed with Agilent Chemstation software (version B.04.03). A Cary 5 double beam spectrophotometer was used for the determination of water in tobacco by near infra-red spectroscopy (Agilent Technologies; Wokingham, UK).
Base-modified silica cartridges
To a 3000 mL round bottomed flask, 500 mL of methanol and 168 g potassium hydroxide were added and the flask attached to a rotary evaporator. The flask was submerged in a water bath and cooled to ambient temperature as the reaction is exothermic. When the potassium hydroxide pellets had dissolved in the methanol, 300 g of silica gel were added with continuous stirring followed by a further addition of 500 mL of methanol. The temperature of the water bath was increased to 40 °C and left to mix for 30 minutes. The methanol was decanted from the flask and further 500 mL methanol added and then left to mix for a further 30 minutes; this was repeated for a further two methanol washes. The excess methanol was decanted off and the slurry poured into a chemical resistant tray and left to dry overnight in a fume hood. Free flowing base-modified silica powder was obtained. A frit was placed in the bottom of a 70 mL reservoir cartridge followed by addition of 10 ± 0.3 g base-modified silica and a frit placed on top of the silica. For routine laboratory use custom made base-modified silica cartridges were produced by Biotage (Uppsala, Sweden).High-performance liquid chromatography-fluorescence detection conditions. HPLC separation was performed at a temperature of 35 °C and run time of 35 min. The injection volume was 10 μL and the eluent flow rate 0.5 mL min−1. Mobile phases used were deionised water (eluent A) and acetonitrile (eluent B). Gradient profiles are shown in Table 1. Excitation wavelength was 290 ± 3 nm and emission wavelength was 440 ± 3 nm.
| Time (min) | Flow (mL min−1) | Eluent A (deionised water) (%) | Eluent B (acetonitrile) (%) |
|---|---|---|---|
| 0 | 0.500 | 20.0 | 80.0 |
| 20.0 | 0.500 | 10.0 | 90.0 |
| 20.5 | 0.500 | 0 | 100.0 |
| 30.0 | 0.500 | 0 | 100.0 |
| 30.1 | 0.500 | 0 | 80.0 |
| 35.0 | 0.500 | 20.0 | 80.0 |
Standard solutions
For the internal standard stock solution, 10 ± 0.1 mg of D12-B[a]P were weighed into a 20 mL amber glass vial. After addition of 5–7 mL acetonitrile, the vial was capped and sonicated for 5 min at 30 ± 5 °C. The solution was cooled to room temperature, quantitatively transferred to an amber glass 10 mL volumetric flask and made up to volume with acetonitrile (final D12-B[a]P stock solution concentration 1000 ng μL−1).The working-standard solutions were prepared by transferring 100 μL D12-B[a]P stock solution to a 100 mL or a 1000 mL volumetric flask and adding acetonitrile to make up to volume (concentrations 1000 ng mL−1 (solution 1) and 100 ng mL−1 (solution 2).
For the B[a]P stock standard solution, 0.25 mL of 1000 μg mL−1 B[a]P in acetone was transferred from an ampoule to a 100 mL volumetric flask and made up to volume with acetonitrile (concentration 2500 ng mL−1; solution 3). A volume of 10 mL B[a]P stock solution was transferred to a 50 mL volumetric flask and made up to volume with acetonitrile (concentration 500 ng mL−1) to make the working standard stock solution (solution 4). Standard solutions were stored at −20 ± 2 °C.
Calibration solutions
Nine calibration standard solutions in the concentration range of 0.25 to 25 ng mL−1 were prepared from D12-B[a]P (solution 1) and B[a]P (solution 4). A volume of 250 μL of D12-B[a]P (solution 1) was transferred to 50 mL volumetric flasks to give a final concentration of 5 ng mL−1 internal standard in each calibration solution. Aliquots of 500 ng mL−1 of B[a]P (solution 4) were added and made up to volume with acetonitrile. The calibration standards were stored at −20 ± 2 °C and were stable for up to 6 months.Linearity and statistical evaluation
First order linear models using ANOVA were performed using Minitab (version 16) statistical software (Minitab Inc., State College, Pennsylvania, USA). The calibration linearity was evaluated for the B[a]P working standard solutions over the entire calibration range. Tests for statistical significance were set at the 95% confidence level. Samples were analysed in triplicate on three separate occasions. A linear correlation fit was applied to the response ratio (the ratio of the B[a]P peak area to the internal-standard peak area), as a function of the analyte concentration ratio (the ratio of B[a]P concentration to the internal-standard concentration). The linear regressions of the scatter plots for the replicates were assessed. The calibration range for the method was determined by the upper and lower concentrations of B[a]P in solvent at which acceptable precision, accuracy and linearity were achieved. The linearity of results for standard fortified matrix extracts were compared with a calibration curve of standards in pure solvent for the assessment of potential matrix effects.In the absence of a blank matrix, the detection limit (LOD) of the method was estimated from calibration curves as LOD = (3.3σ)/slope, where σ = standard deviation of the response.27 The lower limit of quantification of the method was determined by completing six replicate injections of the lowest level of B[a]P standard. The target precision at the LOQ was set at a relative standard deviation of 20% in accordance with DG SANCO and FDA guidelines.31,32
Stability
The stability of B[a]P working-standard solutions was assessed by comparison of the measured concentration of B[a]P in freshly prepared solutions with solutions stored at ambient conditions for 4 days. In addition, the stability of standards in solvent stored at −20 ± 2 °C was assessed periodically over 15 weeks. The stability of modified-Virginia blend sample extracts (n = 5) stored at −20 ± 2 °C was assessed on day 0 and on day 30 by comparison with a freshly made standard solution.Tobacco hydration
A pre-extraction hydration step of a similar dry leaf matrix (tea) was reported to improve the extraction efficiency of pesticide residues from black and green tea samples.35 Experiments were therefore carried out using tobacco from 3R4F reference cigarettes to determine the effect of hydration on B[a]P extraction efficiencies. A 3R4F cigarette tobacco was extracted (1) without hydration; (2) at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 water:tobacco for 18 hours/overnight at in at 5 ± 2 °C, and (3) at a ratio of 4:1 water:tobacco with an hour hydration at ambient temperature. In addition, the efficiency of extraction of B[a]P from STP Oliver Twist was evaluated without hydration and with hydration at a ratio of 1:1 water:tobacco for 18 hours/overnight at 5 ± 2 °C.
:1 water:tobacco for 18 hours/overnight at in at 5 ± 2 °C, and (3) at a ratio of 4:1 water:tobacco with an hour hydration at ambient temperature. In addition, the efficiency of extraction of B[a]P from STP Oliver Twist was evaluated without hydration and with hydration at a ratio of 1:1 water:tobacco for 18 hours/overnight at 5 ± 2 °C.
Sample extraction
Because the method was applied to a wide range of tobacco products with B[a]P content ranging from 0.38 ng g−1 to 150 ng g−1 and in order to avoid potential non-linearity of response for samples with very high B[a]P content, the mass of sample extracted was matched to sample type. For snus samples a mass of 2 g was extracted. For samples containing high proportions of fire-cured tobacco, 0.5 g was extracted and the extracts were diluted prior to analysis. For other samples a mass of 1 g was extracted. Extraction was conducted in three stages in order to ensure exhaustive recovery of the incurred B[a]P and thus quantitative determination.The mass of sample was varied according to the matrix (Table 2). For STPs with low expected endogenous B[a]P concentration (for example Granit White, Lucky Strike Original snus) two pouches equivalent to a mass of 2.0 g were extracted. For tobacco blend and STPs in which high levels of B[a]P were expected (for example products containing a high proportion of fire-cured tobacco, such as Square dry snuff) a lower sample mass was extracted (0.5 g) to ensure that B[a]P responses were within the method calibration limits. For all other samples 1 g of sample was extracted. Samples were hydrated at a ratio of 1:1 water:tobacco and then refrigerated overnight (18 hours) to equilibrate. Granit White and Lucky Strike Original snus were stored in the freezer and defrosted prior to extraction. Optimal extraction efficiency of B[a]P was obtained for samples with a final moisture content of 42–57%. Hydration was required for matrices with low moisture content, but additional hydration of Granit White and Lucky Strike Original snus was determined to be unnecessary due to high intrinsic moisture levels in the product (>40%).
| Sample | Moisture content (%) | Mean (SD) mass of matrix required (g) | Volume of water added (mL) | Final moisture content (%) |
|---|---|---|---|---|
| Kentucky reference cigarette (3R4F) | 10.8 | 1.0 ± 0.1 | 1.0 | 55.4 |
| Modified Virginia blend | 11.7 | 0.5 ± 0.1 | 0.5 | 55.9 |
| Tanzanian fire-cured tobacco | 10.8 | 0.5 ± 0.1 | 0.5 | 55.4 |
| Square dry snuff | 8.1 | 0.5 ± 0.1 | 0.5 | 54.1 |
| Oliver Twist pellet tobacco | 13.1 | 1.0 ± 0.1 | 1.0 | 56.5 |
| Lucky Strike Original snus (two portions) | 49.3 | 2.0 ± 0.3 | 0 | 49.3 |
| Granit White snus (two portions) | 42.2 | 2.0 ± 0.3 | 0 | 42.3 |
In order to remain within the calibration range, samples containing very high B[a]P levels, including the Tanzanian dark fire-cured tobacco and Square dry snuff samples, were fortified with 150 μL of solution 1, equivalent to 150 ng D12-B[a]P. A ten-fold dilution was applied at the end of the extraction step. For all other matrices (including unknown samples) with expected low to medium B[a]P concentrations, 15 ng of D12-B[a]P was added (150 μL of solution 2) and samples were analysed without dilution. The mass of unknown samples was adjusted where necessary to ensure that the B[a]P concentration in the final extract was within the calibration limits.
Two extraction approaches were adopted. For mixtures with low intrinsic moisture content the following approach was adopted. After the addition of D12-B[a]P, samples were equilibrated for 30 minutes at room temperature prior to extraction. The samples were extracted with 25 mL 90:10 (v/v) hexane:acetone for 30 min on a reciprocating shaker at 180 rpm and centrifuged at 4600 rpm for 5 min. The supernatant was decanted into a 60 mL amber vial. The process was repeated two more times to collect the cumulative sample extract. The extract volume was reduced between extraction steps using a Turbovap sample concentrator to obtain a final extract volume of 1–5 mL.
Two snus pouches were fortified with D12-B[a]P and left to equilibrate at ambient temperature for 30 min. Due to the high intrinsic moisture content (>40%) a higher proportion of polar aprotic solvent was required for the initial extraction of pouched snus to ensure optimal extraction efficiency. The extraction efficiencies for B[a]P in pouched snus were 10% higher when a 50:50 (v/v) hexane:acetone solvent system was used for the first extraction step. For blend and the other STPs, extraction efficiencies when using a higher proportion of polar aprotic solvent for initial extraction were reduced with a 14% reduction in extraction efficiency observed for modified Virginia blend). Therefore, two whole snus pouches were first extracted with 50:50 (v/v) hexane:acetone for 30 min on a reciprocating shaker at 180 rpm before centrifugation and concentration, this was then followed by two further extractions with 25 mL 90:10 (v/v) hexane:acetone as described for the other tobacco samples.
Sample clean-up
Each supernatant was transferred to a hexane pre-conditioned base-modified 70 mL 10 g−1 potassium silicate cartridge. The eluent was collected in a 60 mL vial. B[a]P was further eluted from the cartridge with an additional two volumes of 25 mL hexane. The total eluent volume (50–55 mL) was reduced by careful evaporation of the solvent under a flow of nitrogen to just before the point of dryness. The sample was reconstituted in 3 mL acetonitrile and sonicated for 40 min at 25 °C. Dry snuff and fire-cured tobacco were diluted ten-fold prior to analysis because screening measurements indicated that these samples contained B[a]P levels above the upper calibration range.Standard fortified matrix calibration
Fortification standard solutions in acetonitrile were prepared at three levels: low, medium and high (Table 3). Calibration solutions in matrix were prepared as follows: for the low and medium-level fortification solutions, different volumes of 500 ng mL−1 B[a]P working standard stock (solution 4) and for the high fortification standard solution a B[a]P stock of 2500 ng mL−1 (solution 3) was used (Table 3).| Fortification standard | B[a]P standard concentration (ng mL−1) | B[a]P standard volumes (mL) | Volume (mL) | Fortification standard concentration (ng mL−1) |
|---|---|---|---|---|
| Low | 500 | 1 | 50 | 10 |
| Medium | 500 | 10 | 50 | 100 |
| High | 2500 | 10 | 25 | 1000 |
Standard fortified matrix samples were prepared (Table 4) for all tobacco blends and STPs. The final volume of fortified B[a]P matrix samples was 500 μL after the addition of 400 μL of matrix extract.
| Calibration level | Fortification standard concentration (ng mL−1) | Volume of fortification standard (μL) | Acetonitrile (μL) | Target fortification (ng mL−1) |
|---|---|---|---|---|
| 0 | — | 0 | 100 | 0 |
| 1 | 10 | 12.5 | 87.5 | 0.250 |
| 2 | 10 | 25.0 | 75.0 | 0.499 |
| 3 | 10 | 50.0 | 50.0 | 0.999 |
| 4 | 100 | 12.5 | 87.5 | 2.50 |
| 5 | 100 | 25.0 | 75.0 | 4.99 |
| 6 | 100 | 50.0 | 50.0 | 9.99 |
| 7 | 1000 | 10.0 | 90.0 | 20.0 |
The matrix extract for each sample was produced after drying ten extracts each with a volume of 50–55 mL after clean-up to just the point of dryness and reconstitution in 3 mL acetonitrile. The ten samples therefore gave a total extract volume of 30 mL (10 × 3 mL) which was used to produce the standard fortified matrix at each calibration level (n = 4) with two calibration curves produced on each HPLC system (instrument 1 and 2). Unfortified matrix samples were assessed to determine endogenous concentrations of B[a]P as a control.
Accuracy
Accuracy (i.e. closeness of agreement between the result of a measurement and the true value) of the method was assessed by calculation of the ratio between the determined and nominal concentrations. The accuracy was determined for standards in solvent and for each of the fortified matrix extracts after subtraction of the endogenous level. Acceptable accuracies were 50–120% for an analyte concentration of <1 ng g−1, 70–110%, for the range of 1–10 ng g−1 and 80–110% for B[a]P concentration >10 ng g−1.37Repeat extractions of unfortified Granit White snus matrix were analysed (n = 16) to assess the accuracy of the method for samples close to the LOQ (<1 ng g−1). The mean measured endogenous concentration of 2.20 ng g−1 DWB, (precision of 12.2% (n = 16)) was subtracted from B[a]P fortified Granit White snus samples used to determine the accuracy at the three levels of matrix fortification (low, medium and high) with six replicates per level.
Precision
The precision of the method (i.e. the closeness of replicate results) was calculated as the ratio of the standard deviation to the mean corrected concentration of six samples at each fortification level (n = 18 samples) i.e. the relative standard deviation.Intermediate precision
The intermediate precision (i.e. within-laboratory variations arising from different analysts and instruments) was calculated from the measurements made by three different operators using two instruments (instruments 1 and 2) on three separate occasions. The medium fortification standard solution was prepared in solvent (acetonitrile) (Table 4) and used to fortify Granit White extracts. Twenty four Granit White pouches were fortified with 15 ng of internal standard D12-B[a]P (150 μL of solution 2) prior to extraction. To six replicates of each fortification level (unfortified, low, medium and high) the following volumes of B[a]P medium fortification standard 100 ng mL−1 (Table 3) were added: 0 μL, 30 μL, 210 μL and 450 μL equivalent to 0 ng, 3 ng, 21 ng and 45 ng respectively. Unfortified samples were assessed to determine endogenous concentrations of B[a]P as a control.Uncertainty of measurement
Standard uncertainty of measurement (u) (i.e. the parameter characterising the dispersion of the values, was estimated from the mean (![[x with combining macron]](https://www.rsc.org/images/entities/i_char_0078_0304.gif) ) and standard deviation (s) of repeated measurements (n)). The mean of the repeated readings, was calculated using the formula (1) and estimated standard deviation (2)
) and standard deviation (s) of repeated measurements (n)). The mean of the repeated readings, was calculated using the formula (1) and estimated standard deviation (2)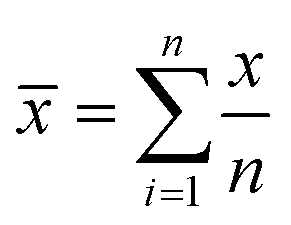 | (1) |
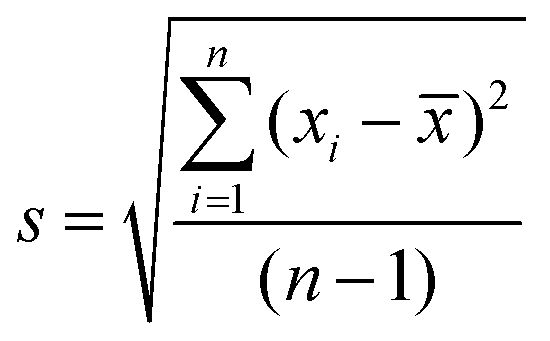 | (2) |
From these values the estimated standard uncertainty of the mean, u was calculated (3) and then the combined standard uncertainty (uc) calculated by squaring the measurement standard uncertainties, adding them all together, and then taking the square root of the total (4).38
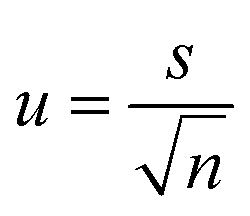 | (3) |
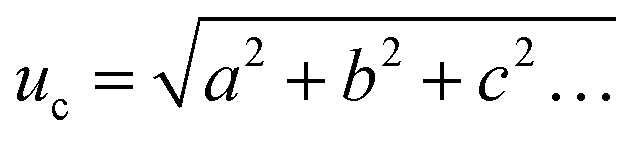 | (4) |
The expanded uncertainty (U) (i.e. the quantity defining an interval around a measured value encompassing a large fraction of the distributed values was calculated using the formula
| U = kuc | (5) |
Results and discussion
Tobacco hydration
Extraction efficiency for B[a]P was 11% higher for 3R4F cigarette blend and 9.4% higher for STP Oliver Twist with tobacco matrix hydration at a ratio of 1:1 water:tobacco (18 hours/overnight in a refrigerator) compared to non-hydrated matrix (Fig. 3). Findings are consistent with published data for pesticide residue extraction in tea.35 At a hydration ratio of 4:1 water:tobacco and equilibration time of one hour, the extraction efficiency of B[a]P was 9.6% lower than the B[a]P concentration determined in non-hydrated samples and 20% lower than in extracts obtained from 3R4F matrix hydrated at a 1:1 water:tobacco ratio. Preliminary data indicated that over hydration at a ratio of 4:1 water:tobacco significantly reduced the extraction efficiency for 3R4F compared to a 1:1 water:tobacco ratio. It is however worth noting the differences in the equilibration time will also influence the extraction efficiency.
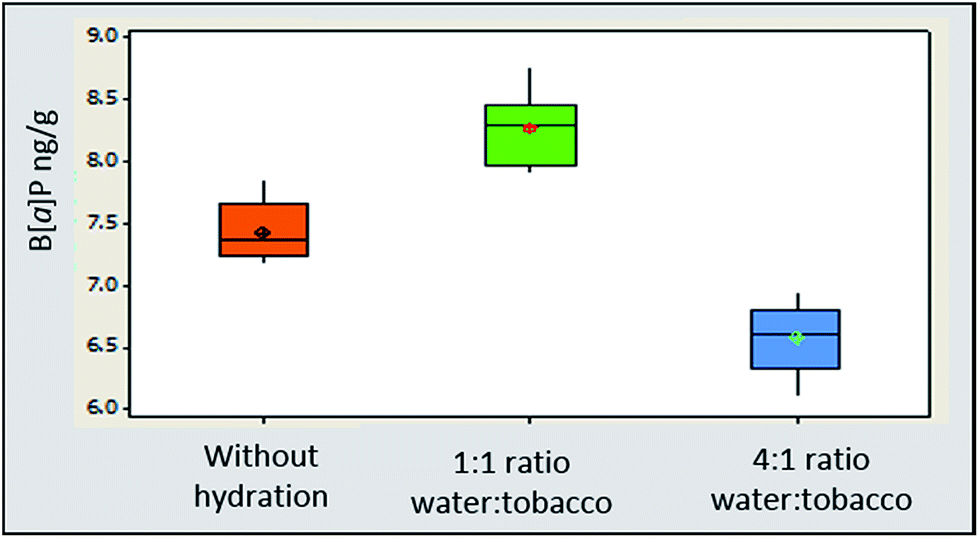 | ||
| Fig. 3 Boxplot of B[a]P concentrations in 3R4F reference tobacco extract expressed on a Wet Weight Basis (WWB) in ng g−1 (1) without hydration (2) 1:1 ratio of water:tobacco hydration and equilibration at 5 ± 2 °C overnight (3) 4:1 water:tobacco hydration with equilibration for one hour at 22 ± 2 °C. | ||
Sample selectivity
B[a]P and D12-B[a]P were chromatographically separated (Fig. 4) and baseline resolved. Assessment of sample matrices with and without fortification with D12-B[a]P enabled the optimization of chromatographic conditions and analytical selectivity.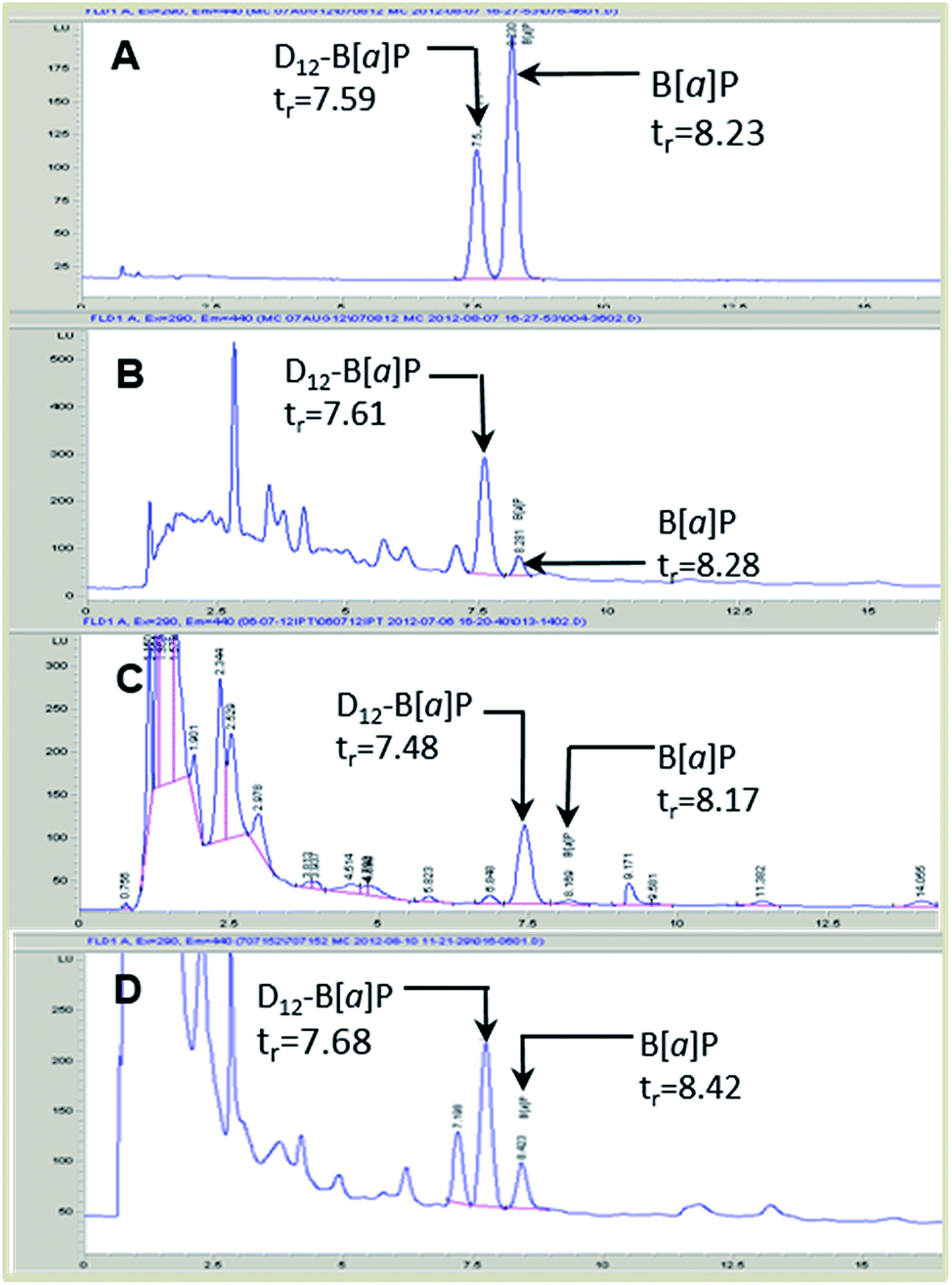 | ||
| Fig. 4 HPLC fluorescence chromatograms of B[a]P and D12-B[a]P in standard stock solutions (A) and in Tanzanian fire-cured tobacco (B), Granit White snus (C) and 3R4F reference tobacco after fortification with 5 ng mL−1 D12-B[a]P. tr, retention time (min). | ||
Stability
Standard solutions were stable at ambient temperature for a week at all calibration levels, with B[a]P concentrations measured as 100–103% of those in fresh solutions. After storage of the standards at −20 °C for 15 weeks, the measured B[a]P concentrations for solutions at calibration levels 3–9 were 95.2–105%. The accuracy of calibration standard levels 1 and 2 were in the range 101–109% at all time-points. For matrix samples, the measured mean concentration of B[a]P in modified Virginia blend extracts (n = 5) reduced by 14.6% after 1 month of storage at −20 °C. The precision of measurement (% RSD) was consistent for freshly prepared extracts and after storage at −20 °C for 15 weeks with values of 8.2% and 7.6% respectively.Linearity and statistical evaluation
The correlation coefficient for calibration standards 1–9 in pure solvent (acetonitrile) was high (R2 > 0.995). The p-value of >0.05 indicated no statistically significant difference at the 95% confidence level between calibration curves for standards in solvent, n = 3. For matrix-matched standards, regression analysis of all calibration curves showed a strong correlation (R2 > 0.996).When the averaged calibration curve for Tanzanian dark fire-cured tobacco was compared with that for solvent, the slopes were not significantly different at the 95% confidence level. However, a statistically significant difference was seen between the intercepts (p < 0.05) at the 95% confidence level. This difference corresponds to the high endogenous level of B[a]P observed in unfortified Tanzanian dark fire-cured tobacco control samples. Significant differences in intercepts between matrix matched standards and standards in solvent were also seen for Square dry snuff, Kentucky reference cigarette, 3R4F (Table 5) which is consistent with the endogenous levels of B[a]P in these matrices. The endogenous levels of B[a]P quantified in unfortified matrix samples are presented in Table 6.
| Matrix | Slope | Intercept p-value |
|---|---|---|
| a Data from instrument 1. b Data from instrument 2. | ||
| Tanzanian fire-cured tobacco | 0.121 | <0.001 |
| Square dry snuffa | 0.703 | <0.001 |
| Kentucky reference cigarette (3R4F) | 0.567 | <0.001 |
| Oliver Twist pellet tobacco | <0.001 | 0.163 |
| Granit White snus | 0.231 | 0.393 |
| Lucky Strike Original snus | 0.440 | 0.332 |
| Modified Virginia blendb | 0.695 | 0.221 |
| Matrix | Water content (%) | B[a]P concentration WWB (ng g−1) | B[a]P concentration DWB (ng g−1) | Relative SD (%) |
|---|---|---|---|---|
| a WWB, wet weight basis; DWB, dry weight basis. | ||||
| Kentucky reference cigarette (3R4F) | 10.8 | 10.7 | 12.0 | 1.22 |
| Modified Virginia blend | 11.7 | 51.1 | 57.9 | 3.84 |
| Tanzanian fire-cured tobacco | 10.8 | 118 | 135 | 6.43 |
| Square dry snuff | 8.1 | 79.7 | 86.7 | 1.39 |
| Oliver Twist pellet tobacco | 13.1 | 0.42 | 0.48 | 11.1 |
| Lucky Strike Original snus | 49.3 | 1.75 | 3.45 | 7.33 |
| Granit White snus | 42.2 | 1.27 | 2.20 | 15.9 |
The p-values for the slopes in all matrices except Oliver Twist pellet tobacco were >0.05 (Table 5), indicating no significant differences between slope gradients for matrix matched standards and standards in solvent at the 95% confidence level. There was a small difference in the magnitude of the slope for B[a]P levels in fortified Oliver Twist matrix compared to standards in solvent. Differences are attributed to the Oliver Twist extract containing endogenous levels of B[a]P close to the LOQ (Table 6) as slightly higher variability was observed at concentrations near the LOQ. Measurements were however within the recommended acceptance limits for the precision of measurement, RSD <20%.31,32
The Oliver Twist unfortified blank matrix (0.48 ng g−1 B[a]P DWB, RSD = 11.1% n = 3) was subtracted from the other fortification levels for statistical analysis which led to a slight difference between the slopes. Although there was a slight difference between slope gradients for matrix and solvent for Oliver Twist pellet tobacco, the other validation parameters were unaffected. Averaged data generated for modified Virginia blend indicated a significant statistical difference between slopes at the 95% confidence level with a p-value = 0 for the slope of averaged matrix matched curves. The observed statistical differences in the slopes for modified Virginia blend may be attributed to instrumental variation as data used to generate the first order linear models using ANOVA were averaged from both instruments 1 and 2. Comparison of calibration curves from a single instrument for modified Virginia blend indicated no significant differences between slopes, with a p-value >0.05 (Table 5). For samples with low endogenous B[a]P concentrations in the unfortified matrix (Oliver Twist chewing tobacco, Granit White snus and LS Original snus) there was no statistically significant difference between intercepts of calibration curves for standards in solvent and fortified matrix at the 95% confidence level. In the absence of blank matrix, the method LOD for B[a]P was estimated as 59 pg from the calibration curves of standards in solvent.27 The LOQ for the analysis based on extraction of Granit White pouched snus matrix was 0.38 ng g−1 B[a]P (WWB) and was within the SANCO31 and FDA32 defined acceptance criteria.
Accuracy and precision
Calibration standards 1–9 in solvent (acetonitrile) exhibited accuracy values of 96.7–114% and precision values of 0.142–2.06% expressed as residual standard deviation (RSD) (Table 7). The accuracy of measurement of the calibration standards 1–9 in solvent (n = 6) ranged from 96.7% to 114% across all calibration levels with a maximum precision (RSD) of 2.06% which met the specified acceptance criteria.15,16,21 The accuracy for calibration standard 1 (114%) and the precision (RSD) was 1.62% which was within the acceptance criteria for concentrations of analyte <1 ng g−1 with accuracy limits of 50–120% and precision of <20%.| B[a]P concentration (ng mL−1) | Mean concentration (n = 6) (ng mL−1) | Mean accuracy (%) | Precision RSD (%) |
|---|---|---|---|
| a RSD, relative standard deviation. | |||
| 0.250 | 0.284 | 114 | 1.51 |
| 0.499 | 0.536 | 107 | 1.17 |
| 1.00 | 0.999 | 100 | 2.06 |
| 2.50 | 2.54 | 102 | 0.713 |
| 4.99 | 5.18 | 104 | 0.142 |
| 9.99 | 9.69 | 96.7 | 0.802 |
| 15.0 | 14.8 | 98.7 | 0.286 |
| 20.0 | 20.1 | 101 | 0.651 |
| 25.0 | 25.2 | 101 | 0.347 |
In all fortified matrices (corrected for endogenous B[a]P concentrations), the accuracy of measurements close to the LOQ ranged from 70% to 120%, with a relative standard deviation <20%. For unfortified Granit White matrix (endogenous B[a]P concentrations close to the LOQ) the precision of measurement (RSD) was 12.2% (n = 16). The average recovery values for D12-B[a]P in Granit White snus were 72.0–83.7% and the accuracy of B[a]P measurement after correction for endogenous B[a]P ranged between 101% and 107%. Therefore, even at calibration level 1, B[a]P could be measured with acceptable accuracy.31,32
The intermediate precision for mass normalised samples at the low level of B[a]P fortification was 1.19 ng g−1 after subtraction of the unfortified B[a]P concentration for Granit White (n = 16). The pooled precision for the low fortification level was 23%. Variability was observed as concentrations were near the LOQ. However the precision (RSD) was ≤20% when data for individual analysts were assessed (range 7.80–14.7%). For medium-level and high-level fortification, the pooled relative standard deviation performance standard of ≤15% was met by all analysts when data were pooled, therefore meeting SANCO and FDA acceptance criteria.31,32
Uncertainty of measurement
The expanded uncertainty for the determination of approximately 95% confidence interval limits was calculated from data generated by the three analysts using instrument 1 and 2 on separate days (Table 8). The percentage-expanded uncertainty was ±22.2% of the mean for low level B[a]P fortification in Granit White samples, ±18.0% for the medium level and ±23.8% for the high level, with an average of ±21.3%. The main factors influencing the uncertainty of measurement was some slight differences in fluorescent detector sensitivity observed from the two different instrumental systems used for method validation. However the main influencing factor on the uncertainty of measurement was the analyst skill (for example possible differences in B[a]P and D12-B[a]P fortification, variability in the pipettes used for fortification and peak integration differences). The uncertainty from each source therefore contributed to the overall expanded uncertainty of measurement.| Fortification level | Mass of B[a]P fortified into matrix (ng) | Mean (n = 18) mass of B[a]P quantified (ng) | Expanded uncertainty (U) (ng) | Expanded uncertainty (%) |
|---|---|---|---|---|
| Low | 2.99 | 2.79 | 0.62 | 22.2 |
| Medium | 21.0 | 19.7 | 3.54 | 18.0 |
| High | 45.0 | 46.7 | 11.1 | 23.8 |
Conclusion
A method has been developed and validated for the determination of B[a]P in Kentucky reference 3R4F cigarette tobacco, a lightly modified Virginia blend cigarette tobacco, Tanzanian dark fire-cured tobacco blend, for STPs including commercially available Square dry snuff, Oliver Twist chewing tobacco and Granit White and Lucky Strike Original snus. The method applies to a concentration range of 0.38 ng g−1 to 150 ng g−1 based on extraction of a 0.5–2 g aliquot of sample. The expanded uncertainty of measurement was ±21.3% of the mean. In the absence of blank matrix, the method LOD for B[a]P was estimated as 59 pg from the calibration curves of standards in solvent27 and the LOQ for the analysis based on extraction of Granit White pouched snus was 0.38 ng g−1 B[a]P (WWB), satisfying the DG SANCO31 and FDA32 defined acceptance criteria.Notes and references
- Report on the scientific basis of tobacco product regulation, report of a World Health Organisation (WHO) Study Group, WHO Technical Report Series No. 955, Geneva World Health Organisation, 2008 Search PubMed.
- D. M. Burns, E. Dybing, N. Gray, S. Hecht, C. Anderson, T. Sanner, R. O'Connor, M. Djordjevic, C. Dresler, P. Hainaut, M. Jarvis, A. Opperhuizen and K. Straif, Tobac. Contr., 2008, 17, 132–141 CrossRef CAS PubMed.
- FDA, Fed. Regist., 2012, 77, 20034–20037 Search PubMed.
- Draft guidance for industry: reporting harmful and potentially harmful constituents in tobacco products and tobacco smoke under federal food, drug and cosmetic act, URL http://www.fda.gov/TobaccoProducts/GuidanceComplianceRegulatoryInformation/ucm297752.htm.
- L. E. Rutqvist, M. Curvall, T. Hassler, T. Ringberger and I. Wahlberg, Harm Reduction Journal, 2011, 8, 11 CrossRef PubMed.
- European Smokeless Tobacco Council, URL http://www.estoc.org/about-smokeless-tobacco.
- L. Toth and W. Blaas, Fleischwirtschaft, 1972, 52, 1419 CAS.
- A. Rodgman and T. A. Perfetti, The Chemical Components of Tobacco and Tobacco Smoke, CRC Press, Boca Raton, F.L., 2nd edn, 2013 Search PubMed.
- K. McAdam, A. Faizi, H. Kimpton, A. Porter and B. Rodu, Chem. Cent. J., 2013, 7, 151 CrossRef PubMed.
- IRAC, IRAC monographs on the evaluation of carcinogenic risk to humans, IRAC Press, Lyon, France, 2007, pp. 55–60 Search PubMed.
- International Programme on Chemical Safety (IPCS), Selected non-heterocyclic polycyclic aromatic hydrocarbons. Environmental Health Criteria, World Health Organization, Geneva, 1998 Search PubMed.
- Chemical Hazard Evaluation and Communication Group, Biomedical and Environmental Information Analysis Section, Health and Safety Research Division, Toxicity summary for benzo[a]pyrene, Risk Assessment Information System, Washington, D.C., 1994 Search PubMed.
- E. Sepetdjian, A. Shihadeh and N. A. Saliba, Food Chem. Toxicol., 2008, 46, 1582–1590 CrossRef CAS PubMed.
- Y. S. Ding, J. S. Trommel, X. J. Yan, D. Ashley and C. H. Watson, Environ. Sci. Technol., 2009, 39, 471–478 CrossRef.
- J. E. Tarrant, K. Mills and C. Williard, J. Chromatogr. A, 2009, 1216, 2227–2234 CrossRef CAS PubMed.
- CORESTA, CORESTA Recommended Method No. 58: Determination of benz[a]pyrene in cigarette mainstream smoke – gas chromatography-mass spectrometry method, Cooperation Centre for Scientific Research Relative to Tobacco, Paris, France, 2004 Search PubMed.
- Q. Zha, N. X. Qian and S. C. Moldoveanu, J. Chromatogr. Sci., 2002, 40, 403–408 CAS.
- N. M. Sinclair and B. E. Frost, Analyst, 1978, 103, 1199–1203 RSC.
- M. Lodovici, V. Akpan, C. Evangelisti and P. Dolara, J. Appl. Toxicol., 2004, 24, 277–281 CrossRef CAS PubMed.
- K. R. R. Mahanama, L. A. Gundel and J. M. Daisey, Int. J. Environ. Anal. Chem., 1994, 56, 289–309 CrossRef CAS.
- B. A. Tomkins, R. A. Jenkins, W. H. Griest and R. R. Reagan, J.–Assoc. Off. Anal. Chem., 1985, 68, 935–940 CAS.
- Y. S. Ding, D. Ashley and C. H. Watson, J. Agric. Food Chem., 2009, 55, 5966–5973 CrossRef PubMed.
- I. Stepanov, P. Villalta, A. Knezevich, J. Jensen, D. Hatsukami and S. S. Hecht, Chem. Res. Toxicol., 2010, 23, 66–73 CrossRef CAS PubMed.
- A. Ishizaki, K. Saito, N. Hanioka, S. Narimatsu and H. Karatoa, J. Chromatogr. A, 2010, 1217, 5555–5563 CrossRef CAS PubMed.
- ICH, ICH harmonized tripartite guideline: validation of analytical procedures: text and methodology Q2(R1), URLhttp://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q2_R1/Step4/Q2_R1__Guideline.pdf.
- ICH guideline for industry, text on validation of analytical procedures: ICH-Q2A, URL http://www.fda.gov/downloads/Drugs/Guidances/ucm073381.pdf.
- ICH Guideline for industry Q2B, text on validation of analytical procedures: methodology, URL http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm073384.pdf.
- IUPAC, Pure Appl. Chem., 2002, 74, 835–855 Search PubMed.
- IUPAC, Pure Appl. Chem., 1999, 71, 337–348 Search PubMed.
- ISO. ISO/IEC 17025: 2005 General requirements for the competence of calibration and testing laboratories, URL http://www.iso.org.
- SANCO, Method validation and quality control procedures for pesticide residue analysis in food and feed: document No. SANCO/10684/2009, URL http://ec.europa.eu/food/plant/protection/resources/qualcontrol_en.pdf.
- FDA, Guidance for industry: bioanalytical method validation, URL http://www.fda.gov/downloads/Drugs/Guidances/ucm070107.pdf.
- E. Roemer, H. Schramke, H. Weiler, A. Buettner, S. Kausche, S. Weber, A. Berges, M. Stueber, M. Muench, E. Trelles-Sticken, J. Pype, K. Kohlgrueber, H. Voelkel and S. Wittke, Beitr. Tabakforsch. Int., 2012, 25, 1 Search PubMed.
- ITGA, Tobacco types, URLhttp://www.tobaccoleaf.org/conteudos/default.asp?ID=18%26IDP=3%26P=5.
- T. Cajka, C. Sandy, V. Bachanova, L. Drabova, K. Kalachova, J. Pulkraova and J. Hajslova, Anal. Chim. Acta, 2012, 743, 51–60 CrossRef CAS PubMed.
- I. Laidlaw, Determination of water in tobacco by near infra-red spectroscopy, URL https://www.chem.agilent.com/Library/applications/uv66.pdf.
- European Commission, Off. J. Eur. Communities: Inf. Not., 2002, L221, 8–32 Search PubMed.
- S. Bell, Measurement Good Practice Guide No. 11, 2001, Issue 2, https://www.wmo.int/pages/prog/gcos/documents/gruanmanuals/UK_NPL/mgpg11.pdf Search PubMed.
Footnote |
| † Present address: Marchwood Scientific Services, 371 Millbrook Road West, Southampton SO15 0HW, UK, E-mail E-mail: maria.carradus@marchwood-scientific.co.uk |
| This journal is © The Royal Society of Chemistry 2015 |