
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chicken, beams, and Campylobacter: rapid differentiation of foodborne bacteria via vibrational spectroscopy and MALDI-mass spectrometry†
Howbeer
Muhamadali
a,
Danielle
Weaver
b,
Abdu
Subaihi
a,
Najla
AlMasoud
a,
Drupad K.
Trivedi
a,
David I.
Ellis
a,
Dennis
Linton
b and
Royston
Goodacre
*a
aSchool of Chemistry, Manchester Institute of Biotechnology, University of Manchester, Manchester, UK. E-mail: roy.goodacre@manchester.ac.uk; Tel: +44 (0)161 306-4480
bFaculty of Life Sciences, University of Manchester, Manchester, UK
First published on 26th October 2015
Abstract
Campylobacter species are one of the main causes of food poisoning worldwide. Despite the availability of established culturing and molecular techniques, due to the fastidious nature of these microorganisms, simultaneous detection and species differentiation still remains challenging. This study focused on the differentiation of eleven Campylobacter strains from six species, using Fourier transform infrared (FT-IR) and Raman spectroscopies, together with matrix-assisted laser desorption ionisation-time of flight-mass spectrometry (MALDI-TOF-MS), as physicochemical approaches for generating biochemical fingerprints. Cluster analysis of data from each of the three analytical approaches provided clear differentiation of each Campylobacter species, which was generally in agreement with a phylogenetic tree based on 16S rRNA gene sequences. Notably, although C. fetus subspecies fetus and venerealis are phylogenetically very closely related, using FT-IR and MALDI-TOF-MS data these subspecies were readily differentiated based on differences in the lipid (2920 and 2851 cm−1) and fingerprint regions (1500–500 cm−1) of the FT-IR spectra, and the 500–2000 m/z region of the MALDI-TOF-MS data. A finding that was further investigated with targeted lipidomics using liquid chromatography-mass spectrometry (LC-MS). Our results demonstrate that such metabolomics approaches combined with molecular biology techniques may provide critical information and knowledge related to the risk factors, virulence, and understanding of the distribution and transmission routes associated with different strains of foodborne Campylobacter spp.
Introduction
Campylobacter species are considered to be the most common causative agents of foodborne diseases globally.1 According to a recent UK-wide survey by the Food Standards Agency,2 almost 73% of chicken sold by major retailers tested positive for the presence of Campylobacter. Overall in the UK, Campylobacter is conservatively estimated to be responsible for over 280![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cases of food poisoning with an estimated 100 fatalities per annum, costing the UK economy £900 million (>US$1.4 billion).3 Although Campylobacter jejuni and Campylobacter coli are the main causes of campylobacteriosis, other species such as Campylobacter fetus,4Campylobacter lari and Campylobacter concisus,5–7 are also human pathogens. It is also perhaps worth noting that the frequency of C. concisus detected in human diarrhoeal cases is equal to that of C. coli and C. jejuni, in many studies,7–9 which may explain the remaining undiagnosed cases of gastroenteritis.10 Therefore, development and application of new analytical technologies capable of rapid detection and differentiation of these organisms may provide critical information towards the understanding of distribution and risk factors associated with different strains and their transmission routes.
000 cases of food poisoning with an estimated 100 fatalities per annum, costing the UK economy £900 million (>US$1.4 billion).3 Although Campylobacter jejuni and Campylobacter coli are the main causes of campylobacteriosis, other species such as Campylobacter fetus,4Campylobacter lari and Campylobacter concisus,5–7 are also human pathogens. It is also perhaps worth noting that the frequency of C. concisus detected in human diarrhoeal cases is equal to that of C. coli and C. jejuni, in many studies,7–9 which may explain the remaining undiagnosed cases of gastroenteritis.10 Therefore, development and application of new analytical technologies capable of rapid detection and differentiation of these organisms may provide critical information towards the understanding of distribution and risk factors associated with different strains and their transmission routes.
Although the main source of Campylobacter infection is contaminated chicken,11 probably due to its ubiquitous consumption as an inexpensive protein source and the high body temperature of avian species (41–45 °C),12 there are other sources of infection, including raw milk,13 contaminated water,14–16 and direct contact with animals (e.g. pets).17–20 The application of appropriate detection and identification techniques may also provide valuable information on the attribution of sporadic infections to sources, assisting in the design of rigorous prevention and control strategies, which have also been successfully demonstrated by several recent studies.21–23 Due to the relatively fastidious and biochemically unreactive nature of Campylobacter spp., traditional methods of culture isolation and biochemical testing are laborious, and a number of assays have been developed to allow more rapid detection and identification. These include: enzyme-linked immunosorbent assays (ELISA),24–26 latex agglutination tests,27 as well as molecular techniques such as polymerase chain reaction (PCR),28 multilocus sequence typing (MLST),29 and pulsed-field gel electrophoresis (PFGE).30 Immunoassays, such as ELISA, are arguably the simplest and, currently, most rapid methods, but cannot differentiate between the most common species (C. jejuni, C. coli and C. upsaliensis).25 Whilst DNA-based technologies are considered as reliable tools for bacterial identification and detection, they are time consuming;31 require highly skilled personnel and most importantly, do not provide any real information regarding the phenotypic characteristics of the sample under investigation, and cannot readily differentiate between live and dead cells. In addition, most PCR assays are tailored for the detection of common species, with very few allowing for the simultaneous identification of the more ‘emerging’ foodborne Campylobacter species.7,32
During the past two decades, the application of metabolomics-based approaches and technologies into various fields of microbiology has opened up exciting new opportunities. For an overview on metabolomics and its applications in different areas of microbiology, the reader is directed to the following excellent reviews.33–39 Metabolic fingerprinting is generally described as a rapid, untargeted, semi-quantitative approach for the detection of intracellular metabolites.40,41 Various studies have demonstrated the applications of Raman and Fourier transform infrared (FT-IR) spectroscopies as metabolic fingerprinting techniques requiring minimal sample preparation. Furthermore, these are both vibrational spectroscopy methods which can be considered as holistic techniques, providing biochemical (metabolic) fingerprints of bacterial cells which can be used for detection,42 identification,43,44 classification,45,46 or as a diagnostic tool for industrial and clinical applications.47
Recently, the use of matrix-assisted laser desorption ionisation time-of-flight (MALDI-TOF) mass spectrometry for Campylobacter speciation,48–51 amongst other important bacteria,52–56 has also attracted a lot of attention. In comparison to molecular techniques, MALDI-TOF–MS requires minimal sample preparation, while offering a rapid and uniform approach for identification and classification of a wide range of bacteria.52
This study is focused on employing MALDI-TOF-MS together with Raman and FT-IR spectroscopies, combined with multivariate statistical analysis for differentiation of Campylobacter down to subspecies level. The classifications achieved via these techniques were compared with 16S rRNA sequence-based phylogenetic analysis, for confirmation and comparison purposes.
Materials and methods
Bacterial strains and growth conditions
Bacterial strains tested in this study are listed in Table 1. All Campylobacter strains were grown on Columbia agar (Oxoid Ltd) supplemented with 5% horse blood (TCS Biosciences). Cultures of C. jejuni, C. coli and C. lari were grown in a microaerobic VA500 workstation (85% N2, 10% CO2, and 5% O2) (Don Whitley Scientific Ltd) at 42 °C except for C. fetus which was grown at 37 °C. C. concisus and C. hyointestinalis were grown at 37 °C in a microaerobic atmosphere containing 5% hydrogen (generated using CampyGen™ gas generation system and sodium borohydride). Harvested cells were suspended in Mueller Hinton broth to an optical density (OD600 nm) of 11–12. Cells were then plated onto blood agar plates as replicates (n = 5) and incubated for 48 h as described above. All the strains were grown using the same batch of culturing plates, in order to reduce any potential unwanted phenotypic variation.| Bacterial strain | Original source/Reference | Abbreviation |
|---|---|---|
| C. jejuni NCTC 11168H | Human faeces97 | C. j-11168 |
| C. jejuni 81–176 | Human faeces98 | C. j-81176 |
| C. jejuni 81116 | Human faeces99 | C. j-81116 |
| C. coli RM2228 | Chicken carcass100 | C. c-RM2228 |
| C. coli DW1 | Retail chicken meat | C. c-DW1 |
| C. coli DW6 | Retail chicken meat | C. c-DW6 |
| C. lari RM2100 | Human faeces101 | C. l |
| C. hyointestinalis subsp. hyointestinalis NCTC 11608 | Porcine intestine102 | C. h |
| C. fetus subsp. fetus NCTC 10842 | Foetal sheep brain103 | C. f-fetus |
| C. fetus subsp. venerealis NCTC 10354 | Heifer vaginal mucus103 | C. f-ven |
| C. concisus NCTC 11485 | Human gingival sulcus104 | C. con |
Sample preparation
Bacterial slurries were prepared by harvesting the biomass from the surface of each plate using sterile inoculating loops and resuspended in 1 mL of sterile normal saline solution (0.9% wt/vol NaCl). From this stage onwards all samples were kept on ice. The prepared bacterial slurries were washed by centrifugation at 5000g for 5 min at 4 °C using a Thermo MicroCL 17R centrifuge (Thermo Scientific, UK). The supernatants were removed and cell pellets were resuspended in 1 mL of normal saline solution. OD600 nm of all samples were recorded using an Eppendorf BioSpectrometer (Eppendorf, Cambridge, UK) and used for sample normalisation. All samples were stored at −80 °C until further analysis.FT-IR analysis
20 μL aliquots from each of the samples were spotted onto a pre-washed Bruker 96-well silicon plate (Bruker Ltd, Coventry, UK), as previously described.57,58 The samples were heated to dryness (30 min) at 55 °C using a standing oven. FT-IR analysis of the samples was carried out using a Bruker Equinox 55 infrared spectrometer. Spectral data were collected in the mid-IR range (4000–600 cm−1) with 64 spectral co-adds and 4 cm−1 resolution.59 The extended multiplicative signal correction (EMSC)60 method was employed to scale the FT-IR spectral data, followed by removal of CO2 vibrations (2400–2275 cm−1). A total of 45 FT-IR spectra were collected for each strain, which included five biological replicates, three analytical replicates and three machine replicates.Raman analysis
Calcium fluoride (CaF2) disks were washed three times using 70% ethanol and rinsed twice using deionised water. 5 μL aliquots from each of the samples were spotted onto prewashed CaF2 discs and air-dried in a desiccator at room temperature following a previously published protocol.61 Raman analysis was carried out using a 785 nm laser on a Renishaw inVia Raman microscope (Renishaw Plc., Gloucestershire, UK). All spectra were acquired using 20 s exposure time, in the 466–1878 cm−1 range, with three accumulations and 600 l mm−1 grating and laser power adjusted on the sample to ∼30 mW. Spectral data were collected using the GRAMS WiRE 3.4 software (Galactic Industries Corp. Salem, NH). A total of five Raman spectra were collected for each of the bacterial strains. All Raman spectra were baseline corrected, and then scaled using the EMSC method.60MALDI analysis
Bacterial cell pellets were resuspended in 700 μL of water containing 0.1% TFA. The matrix was prepared by dissolving 10 mg of sinapinic acid (SA) in 500 μL of 2% trifluoroacetic acid (TFA) and 500 μL of acetonitrile (ACN), followed by mixing an equal volume (10 μL) of matrix and bacterial slurry. The mixture was then vortexed for 3 s and 2 μL from this mixture was spotted onto a MALDI-TOF-MS stainless steel plate, and air dried for 60 min at ambient temperature (21 ± 1 °C). An AXIMA-Confidence MALDI-TOF-MS (Shimadzu Biotech, Manchester, UK) equipped with a nitrogen pulsed UV laser with a wavelength of 337 nm was employed for the analysis.55 The laser power was set at 120 mV, and 70 profiles were collected with 20 shots from each profile using what is known as a square raster pattern, taking approximately 3 min for each sample. The MALDI device was operated using a linear TOF and positive ionization mode, and the mass-to-charge (m/z) of the samples ranged from 500–15000. A protein mixture of insulin (5735 Da), cytochrome c (12362 Da), and apomyoglobin (16952 Da) (Sigma-Aldrich) was used to calibrate the MALDI-TOF-MS device. Five biological replicates and three analytical replicates were collected from each strain, resulting in a total number of 165 MALDI-TOF-MS spectra (11 isolates from Campylobacter × 5 biological replicates × 3 analytical replicates). All biological replicates were analysed on five consecutive days, with each replicate on a separate day.
LC-MS analysis
Bacterial cell pellets were resuspended in 1 mL of cold (−20 °C) methanol:chloroform (1:2) solution and vortexed for 15 min at ambient temperature, followed by addition of 0.5 mL of water to aid the extraction of lipids by phase separation. All samples were centrifuged at 5000g for 3 min at −9 °C. Equal volume (500 μL) from the bottom layer of each sample was transferred to a new 2 mL Eppendorf microcentrifuge tube and dried at 40 °C. Dried pellets were normalised according to OD600 nm of each sample by reconstituting in different volumes of LC-MS grade water:methanol (1:4 v/v). Quality control (QC) samples were prepared by combining 20 μL from each of the normalised extracts. Samples were transferred to LC-MS clear vials with a fixed 200 μL insert.
Analysis was carried out on an Accela UHPLC auto sampler system using a Hypersil Gold C18 reverse phase column (L = 100 mm, ID = 2.1 mm, particle size 1.9 μm) coupled to an electrospray LTQ-Orbitrap XL hybrid mass spectrometry system (Thermo Fisher, Bremen, Germany). Excalibur and TunePlus software were used for instrument operation and tuning and calibration was carried out following manufacturer’s instruction. 10 μL of each sample was injected on to the column and a methanol/water (with 0.1% formic acid) solvent gradient (Table S1†) was used for separation of the metabolites over the column. Samples were analysed in positive ESI mode using the following settings: 1 micro scan per 400 ms, 100–1000 m/z range, ESI ion source trasfer tube set at 275 °C, tube lens voltage = 110 V, capillary V = 35 V, sheath gas = 40, aux gas = 5, sweep gas = 1, resolution = 30000 in centroid mode.
Initially, RAW data files were converted into netCDF format within the software conversion option of Excalibur and deconvolution via the XCMS (http://masspec.scripps.edu/xcms/xcms.php) based algorithm in R. A data matrix formatted in Excel (.csv) that indicates retention time vs. mass and peak areas linked to each sample injection was created as an output. QC data from this file was then used to check for analytical reproducibility. Out of the deconvolved feature set those that had percentage coefficient of variance (%CV) of more than 20% in sample groups were removed for robustness in data. Statistical analysis was performed on the resulting matrix containing retention time and m/z pairs for features.
Multivariate statistical analysis
All statistical analysis was carried out in MATLAB version 2013a (The Mathworks Inc., Natwick, US). Principal component analysis (PCA), an unsupervised exploratory statistical approach,62 was applied to all collected data to reduce the dimensionality of the data. The supervised method of principal component-discriminant function analysis (PC-DFA) was also employed,63,64 which functions by minimising within class variance, while maximising between class variance, to discriminate between the groups according to a priori knowledge (strain information). To simplify the interpretation of the PC-DFA scores plots, hierarchical cluster analysis (HCA) was applied on the mean of DFA scores from each class of data, to visualise the distance between different clusters. In order to validate the models generated by PC–DFA, the projection approach was applied,65,66 by which 60% of the samples from each class were randomly selected and used to generate the model (training set), while the remaining 40% were projected into the PC-DFA space to test the model.All collected data in this study are available upon request from the authors.
Molecular identification
Genomic DNA was extracted using ArchivePure DNA Cell/Tissue Kit (5 PRIME, Inc.). 16S ribosomal RNA (rRNA) gene regions were amplified using Phusion® High-Fidelity PCR master mix (New England Biolabs Inc.) with primers 8F (5′-AGA GTT TGA TCC TGG CTC AG-3′)67 and C1288R (5′- CAT TGT AGC ACG TGT GTC-3′).68 Amplicons were sequenced on an Applied Biosystems 3730 DNA analyser (Life Technologies) using primers C412F (5′- GGA TGA CAC TTT TCG GAG C-3′),68 and 805R (5′-GAC TAC CAG GGT ATC TAA T-3′).69 Approximately 1040 bases of sequence data from each strain were aligned using ClustalW,70 and a neighbour-joining tree constructed using MEGA 6 software.71Results
All spectra collected during the analysis are presented (Fig. 1) and illustrate visually the results obtained from the three analytical methods applied. In relation to results from both of the vibrational spectroscopies, complementary biochemical information from the FT-IR (Fig. 1a) and Raman (Fig. 1b) spectra are readily apparent. By contrast the data from MALDI-TOF-MS has a very different structure in that it is measuring discrete signals, largely from proteins (including ribosomal subunits)72 and peptides. The next stage was therefore to investigate whether these different physicochemical approaches could be used to separate out these bacteria into groups that corresponded with the expected species classifications.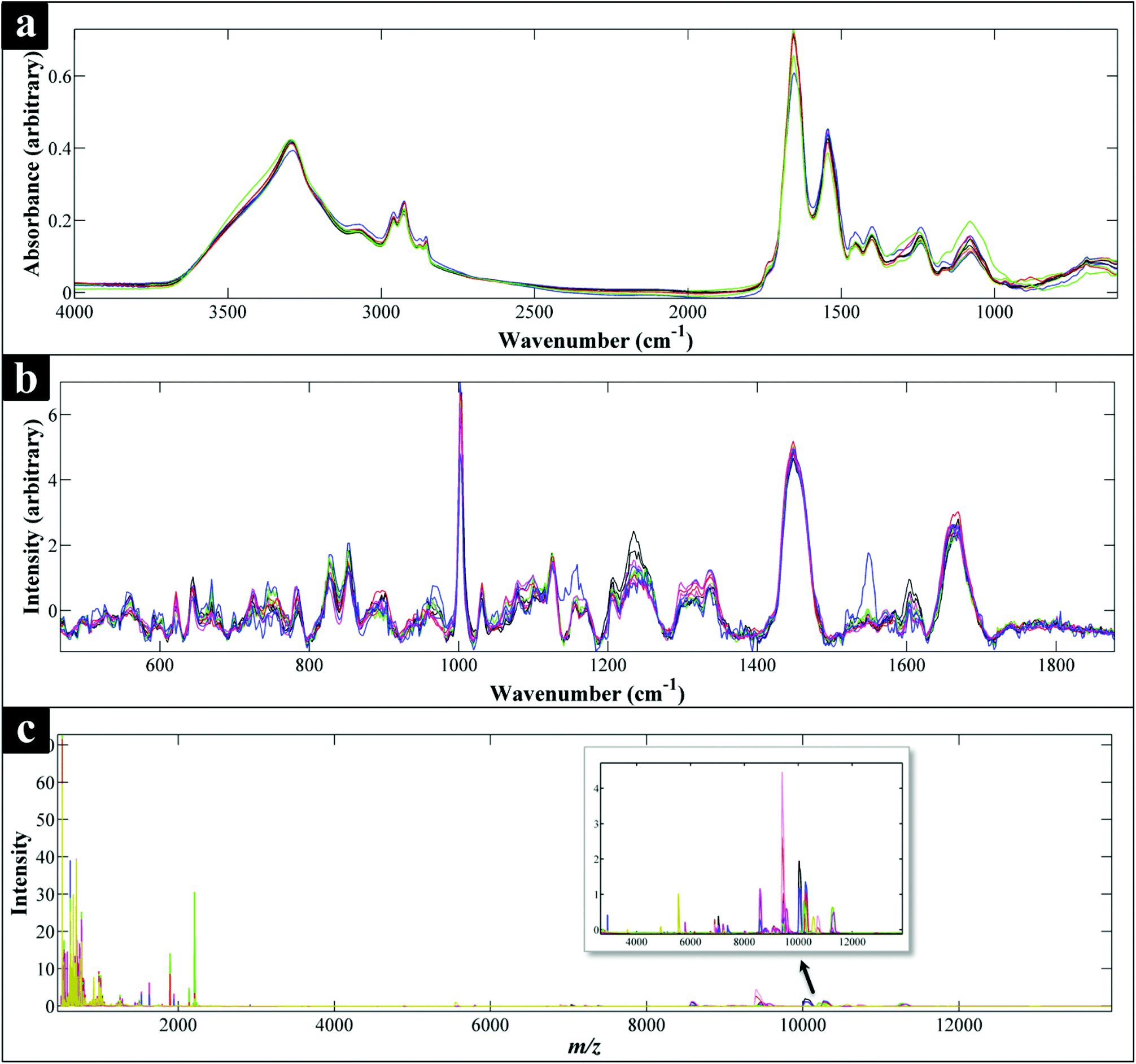 | ||
| Fig. 1 Comparison of FT-IR (a), Raman (b) and MALDI-TOF-MS (c) spectra of all the Campylobacter strains examined in this study. Each spectrum represents the average spectra of all the replicates of each Campylobacter strain. All spectral data have been scaled,60 while the Raman data (b) have also been baseline corrected. (c) Also shows an expansion from ca. 3000–1300 m/z to allow additional mass spec. features to be visualised. | ||
FT-IR analysis
PC–DFA scores plot of the FT-IR data (Fig. 2b), displayed clear separation of C. concisus from all other strains according to DF1, while DF2 separated the rest of the samples. The C. jejuni and C. coli strains formed a tight cluster, while C. lari, C. fetus subspecies fetus and C. hyointestinalis also clustered closely (see Table 1 for abbreviations of species used in Fig. 2 and 3). As DF1 was dominated by C. concisus, and to discriminate between the remaining strains further, a 3D PC–DFA scores plot of the data was constructed using scores from the first three discriminant functions, which clearly separated all the species based on DF2 and DF3 (Fig. 2a). The generated PC-DFA model was validated using a test set projection validation approach (Fig. S1a†), explained in the methods section and employed in other studies,65,66,73 which displayed tight clustering of the training and test sets. The congruent clustering of the test spectra with the spectra used to construct the model does indeed suggest that the clustering pattern seen was wholly representative of the bacterial phenotypes, and that such clustering into the respective bacterial groups was reproducible.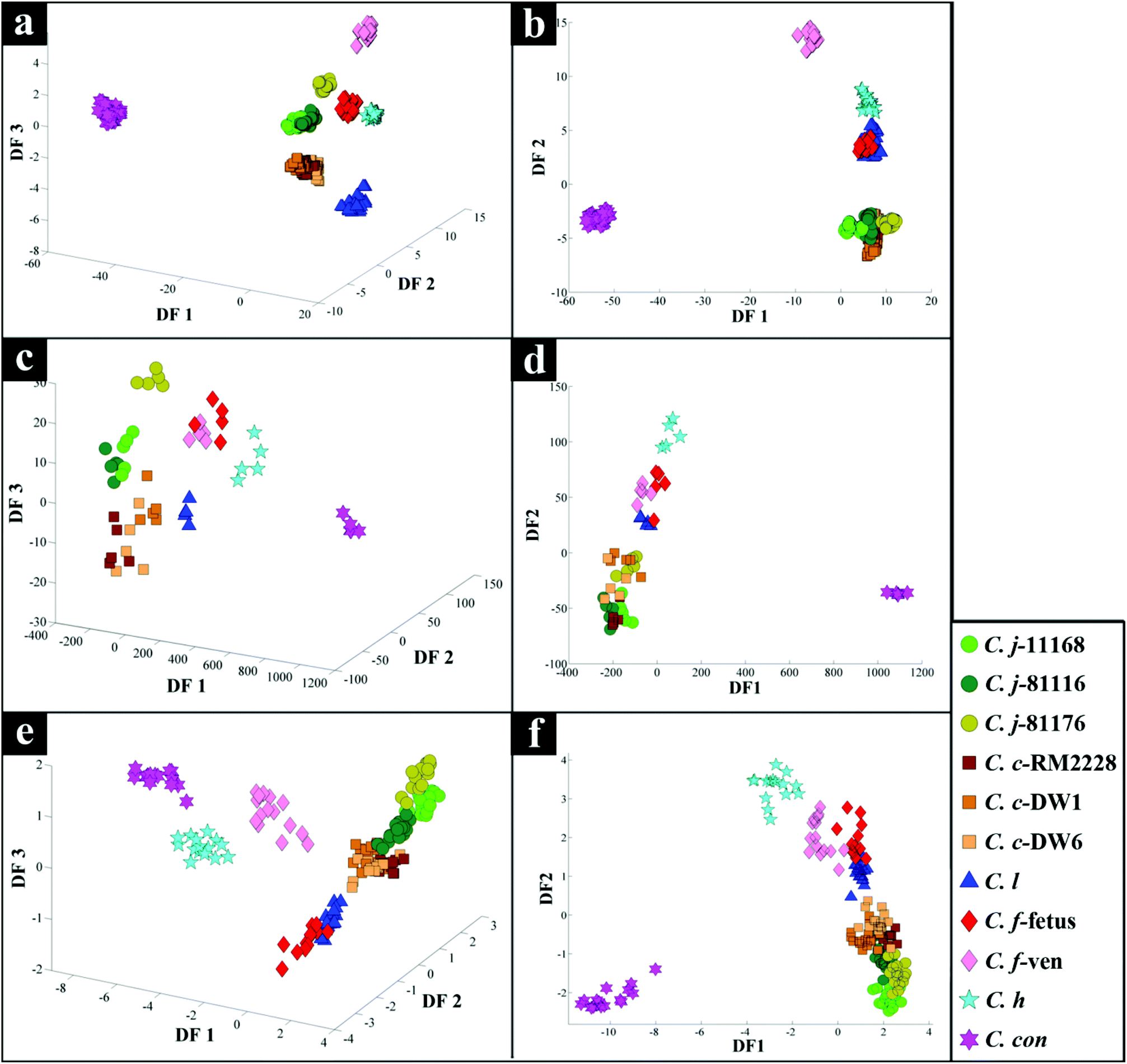 | ||
| Fig. 2 PC-DFA scores plots of spectral data generated via FT-IR (a, b) and Raman (c, d) spectroscopic analysis, and mass spectral data collected from MALDI-TOF-MS analysis (e, f) of different Campylobacter species and sub-species examined in this study. Coloured symbols represent different Campylobacter strains. | ||
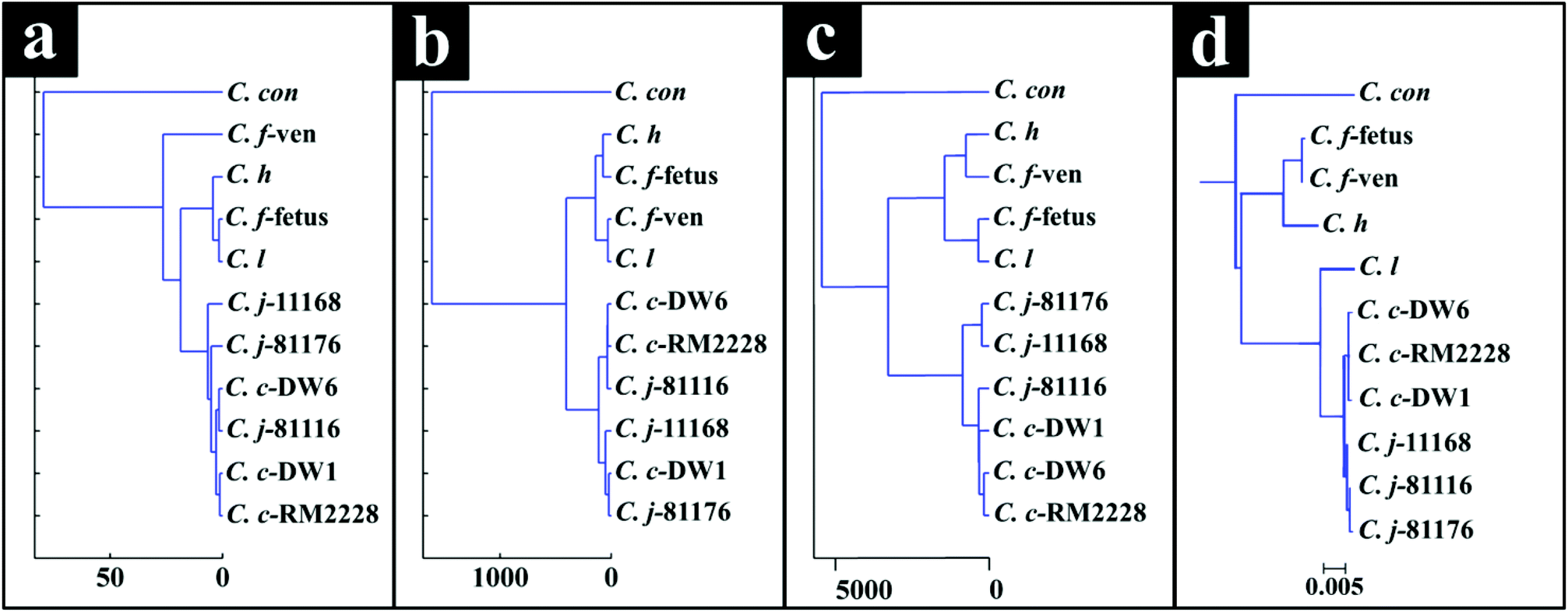 | ||
| Fig. 3 Dendograms generated by HCA using means of the PC-DFA scores of each class (strain) of sample for the FT-IR (a), Raman (b) and MALDI-TOF-MS (c) spectral data. A phylogenetic tree of all the strains was also generated from 16S rRNA gene sequence of all strains using neighbour-joining from the full sequence data (d). | ||
According to DF1 loadings plot (Fig. S2†), the main vibrational regions that contribute toward the separation of C. concisus from other strains in this study include 2920 and 2851 cm−1 (CH2 from lipids: 2920 cm−1 for asymmetric C–H stretching and 2851 cm−1 for symmetric C–H stretching), 1736 cm−1 (esters, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching), amide I (proteins and peptides, 1638 cm−1, CO stretching), and 1200–900 cm−1 (polysaccharides). Yet, on DF2 and DF3 loadings plots (Fig. S2†), the most important areas seem to be amides and the fingerprint region (1500–500 cm−1).
O stretching), amide I (proteins and peptides, 1638 cm−1, CO stretching), and 1200–900 cm−1 (polysaccharides). Yet, on DF2 and DF3 loadings plots (Fig. S2†), the most important areas seem to be amides and the fingerprint region (1500–500 cm−1).
Interestingly, although C. fetus subspecies fetus and C. fetus subspecies venerealis are subspecies from within the same species and are phylogenetically closely related, according to DF2 axis they cluster some considerable distance away from each other (Fig. 2a). This is also evident from the HCA dendogram (Fig. 3a), where the subspecies cluster as two distinct groups. To compare and to explore the differences further between the biochemical fingerprints of just these strains, their FT-IR spectral data were examined by PCA. The PCA scores plot (Fig. 4a) displayed clear separation of C. fetus subspecies fetus and C. fetus subspecies venerealis based on PC1, accounting for 69.1% of the total explained variance (TEV). According to PC1 loadings plot (Fig. 4b), the main variants are in the lipids (2920 and 2851 cm−1) and the fingerprint region (1500–500 cm−1). FT-IR spectra of C. fetus subspecies fetus and C. fetus subspecies venerealis (Fig. 4c), confirmed the PCA findings, as C. fetus subspecies venerealis displayed higher spectral intensities in the lipid region, while C. fetus subspecies fetus exhibited more spectral features in the fingerprint region.
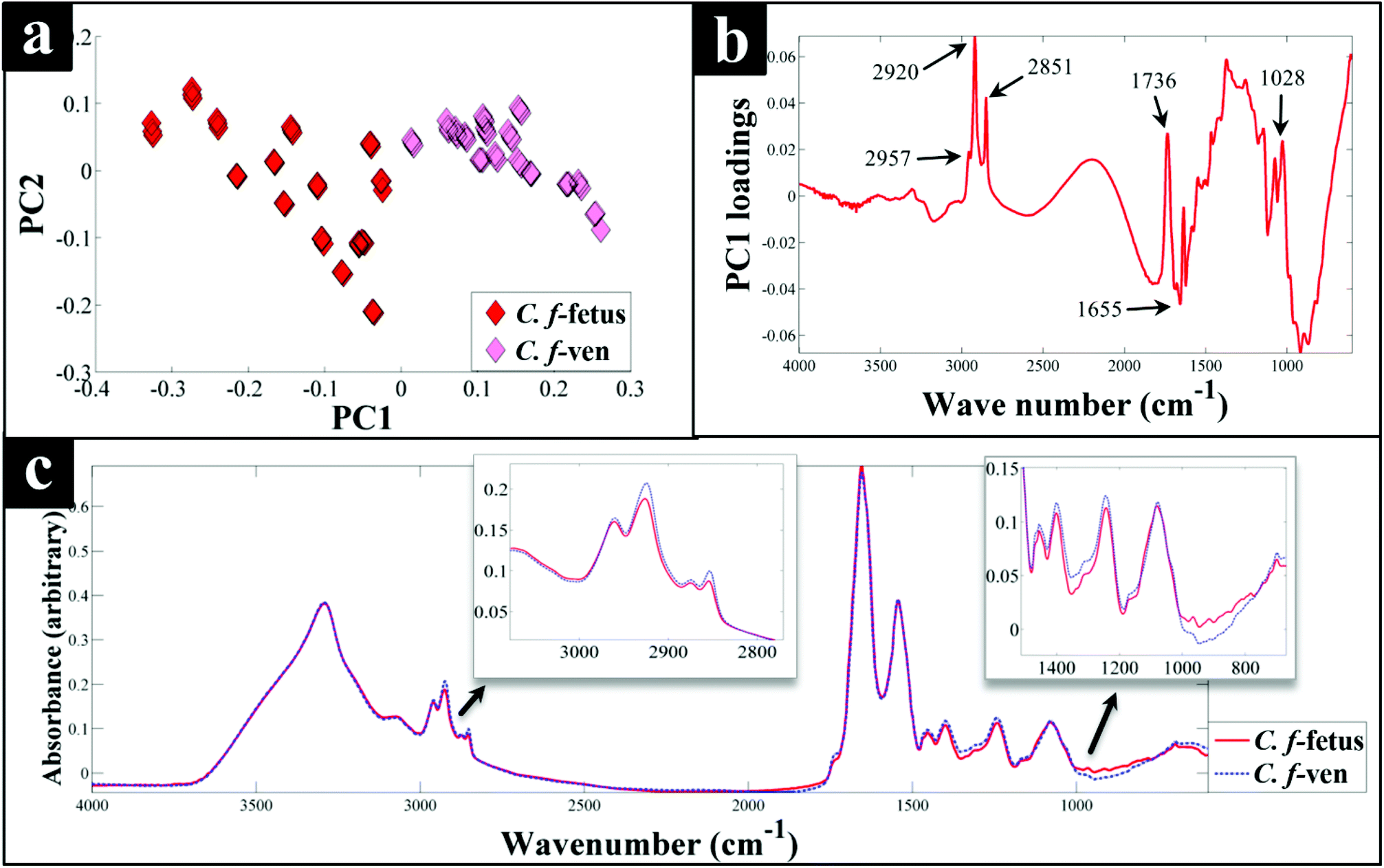 | ||
| Fig. 4 PCA scores plot of C. fetus subsp. fetus and venerealis FT-IR spectral data (a), the correspnding PC1 loadings plot (b), and their corresponding FT-IR spectra (c) with zooms of important spectral areas. The FT-IR spectra plotted for each of the strains are from an average of 45 replicates. | ||
Previous studies have demonstrated the temperature dependence changes in the FT-IR CH2 symmetric vibrational frequency at 2851 cm−1 to be linked with membrane fluidity resulting from changes (so called homeoviscous adaptation) in the composition of saturated and unsaturated fatty acids.74–76 With this in mind, the ratio of 2851/2920 cm−1 for all the samples were compared using box whisker plots (Fig. S3†), which revealed a clear difference between samples incubated at 42 °C (C. coli, C. jejuni and C. lari) and those incubated at 37 °C (C. fetus, C. hyointestinalis and C. concisus). However, no significant differences between C. fetus subsp. fetus and C. fetus subsp. venerealis were observed, emphasising the fact that the PC-DFA (Fig. 2) and PCA (Fig. 4) clustering patterns are not temperature dependent but due to other phenotypic differences.
Raman analysis
PC-DFA scores plot of the Raman spectral data (Fig. 2d) displayed similar clustering patterns to that of the FT-IR data (Fig. 2b), where C. concisus was discriminated from all other strains based on DF1. The 3D PC-DFA scores plot of the Raman spectral data (Fig. 2c), also exhibited a very similar clustering pattern to the FT-IR results, where all the remaining species were discriminated based on DF2 and DF3. This is perhaps not surprising, as FT-IR and Raman are considered as complementary vibrational spectroscopy techniques. The generated PC-DFA model was validated by adopting a similar approach to the FT-IR data, which again revealed acceptable reproducibility (Fig. S1b†).However, unlike the FT-IR findings (Fig. 2a and b), C. fetus subspecies fetus and C. fetus subspecies venerealis clustered closely on the Raman PC–DFA plot (Fig. 2c and d), which is also evident from the HCA dendogram of the Raman cluster analysis (Fig. 3b). Initial justification of these results pointed towards the restricted spectral range (466–1878 cm−1) used for the Raman analysis, which mainly focused on the amide and fingerprint regions (leaving out the fatty acid region) (Fig. 1b).77 To test this hypothesis, PC-DFA of the FT-IR spectral data was also restricted to 600–1878 cm−1. However, the two C. fetus subsp. were again clearly separated (Fig. S6†), suggesting that the inability to separate these two subspecies using the Raman spectral data is not entirely due to the restriction of the acquired spectral range, but is perhaps due to other underlying factors (e.g. the complementarity of Raman to IR being based on molecular bond polarisability during the vibration compared with net change in dipole moment in the molecule, respectively).
The DF1 loadings plot (Fig. S3†), revealed the peaks at 1551 cm−1 (amide II, combination of C–N stretching and N–H bending), 1141 cm−1 (ester, C–O–C symmetric stretching, often associated with lipids) and 1004 cm−1 (phenylalanine, ring breathing) as the significant variance. Whilst, DF2 and DF3 loadings plots (Fig. S4†) displayed major differences in the fingerprint region, which is consistent with the FT-IR findings (Fig. S2†).
MALDI-TOF-MS analysis
The scores plot obtained from the PC-DFA of the MALDI-TOF-MS data (Fig. 2f), was also in agreement with the Raman and FT-IR clustering patterns, and displayed complete separation of C. concisus from the remaining strains according to DF1. Similar to that of the FT-IR, the MALDI-TOF-MS 3D PC–DFA scores plot (Fig. 2e), revealed the distinct separation of C. fetus subspecies fetus from C. fetus subspecies venerealis on the DF3 axis. The PC-DFA loadings plots (Fig. S5†) indicated that despite the detection of proteins in the higher m/z region (2000–14000) (Fig. 1c), which could be linked to various ribosomal subunits,72 the most significant region contributing towards the discrimination of the strains in this study was between m/z of 500–2000 which is the area that contains information from peptides and potentially lipids. However, comparison of the m/z features detected in this region did not match any of the significant lipids identified in the LC-MS lipid profiles (Table S2†).
The HCA dendogram (Fig. 3c), also produced three main clusters, where C. concisus exhibited a significant distance from all other strains while C. jejuni and C. coli strains were closely linked.
LC-MS analysis
To investigate the FT-IR findings further, where the lipid region was most discriminatory, LC–MS of lipophilic extracts was employed to examine and compare lipid profiles of C. fetus subsp. fetus and venerealis. PCA scores plot (Fig. 5a) of the lipid profiles confirmed the FT-IR findings (Fig. 4a), and revealed clear separation of the two subspecies. PC1 loadings plot (Fig. 5b) was employed to identify the most significant variables contributing to this separation. These initial results revealed major differences in the intensities (Fig. 5c) of several classes of lipids including: phosphatidylcholine, phosphatidylethanolamine and phosphatidic acids, which are involved in glycerophospholipid metabolism and cellular membrane biosynthesis.78 Perhaps this is not surprising as Lambert and colleagues79 also reported major differences in the fatty acid content of Campylobacter strains, which allowed for the differentiation of 365 strains of Campylobacter down to subspecies level.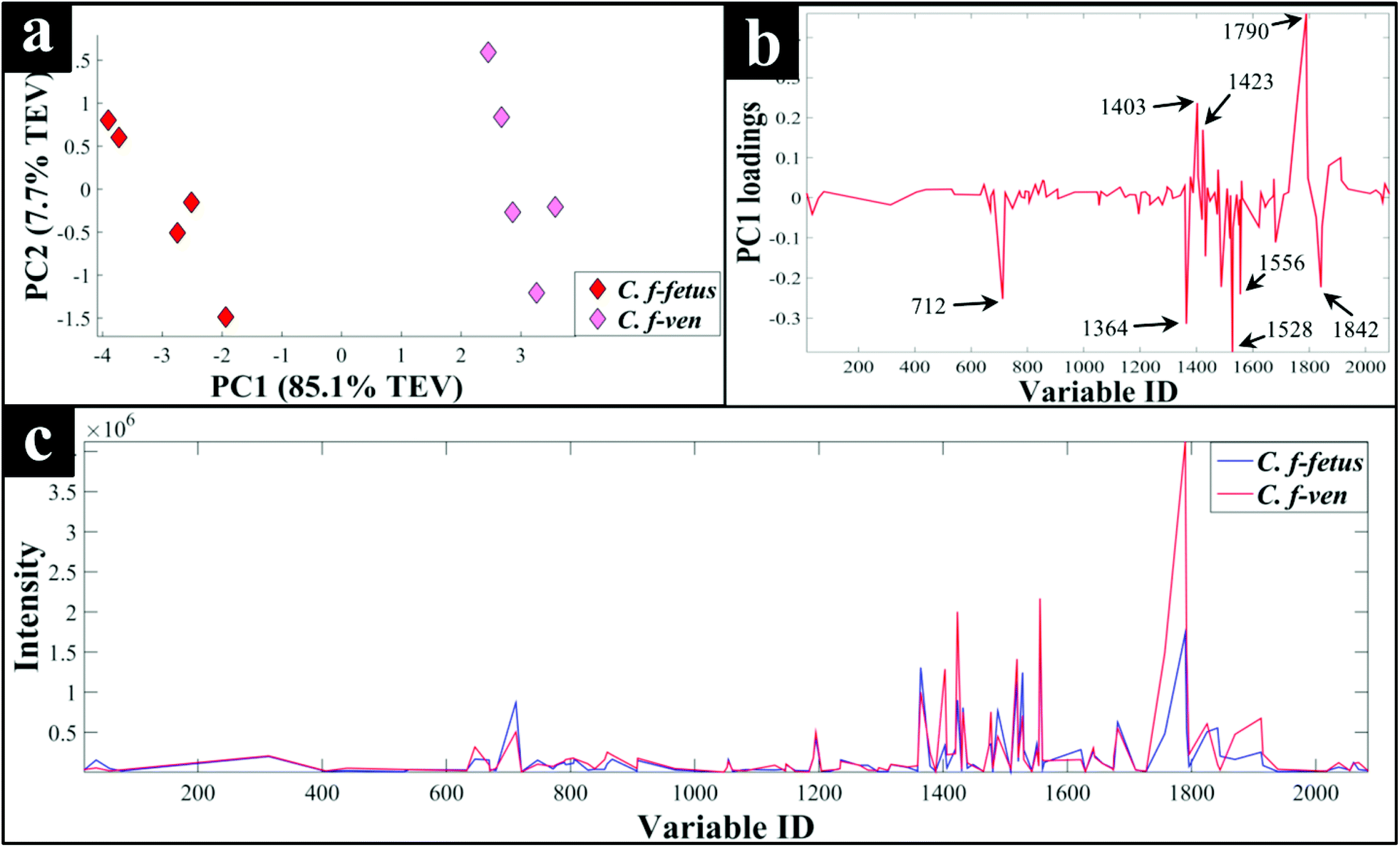 | ||
| Fig. 5 PCA scores plot of C. fetus subsp. fetus and venerealis lipid profiles obtained via LC-MS analysis (a), the corresponding PC1 loadings plot (b), and the lipid profile of each strain (c). The lipid profiles are presented as average lipid intensities of five biological replicates for all detected lipids with %CV of 20% and below. A full list of the significant lipids identified by PCA, and their corresponding identifications can be found in Table S2.† | ||
Discussion
Campylobacteriosis is one of the most prevalent foodborne diseases in the UK,80 costing the UK economy around £900 million.3 Furthermore, it is estimated that preceding infection with Campylobacter spp. may also cause nearly 15% of all cases of Guillain-Barré syndrome,81 an autoimmune-mediated disorder of the peripheral nervous system, which is the most common cause of neuromuscular paralysis.82 Therefore, unequivocal detection and identification of Campylobacter spp. is crucial for both appropriate treatment of Campylobacter infections, routine epidemiological surveillance, and food safety.15,83 Despite the availability of established culturing and molecular techniques for classification and identification of Campylobacter spp., these techniques are generally considered either time consuming or else specific for the most commonly isolated species (C. jejuni and C. coli). Thus, in this study FT-IR and Raman spectroscopies along with MALDI-TOF-MS were employed as metabolic fingerprinting and proteomic fingerprinting approaches (which could also be described as spectral phenotyping) to characterise a set of closely related Campylobacter species representing the key human and veterinary pathogens. The ability of these rapid analytical techniques to discriminate among these taxonomic groups was compared to the standard molecular technique (phylogenetic analysis of 16S rRNA sequencing) for differentiation of Campylobacter spp. down to subspecies level.The PC-DFA results obtained from all three analytical techniques displayed clear separation between all Campylobacter spp. examined in this study (Fig. 2a–f). In addition, the HCA dendograms generated using the PC-DFA scores (Fig. 3a–c), were generally in agreement with the 16S rRNA phylogenetic tree (Fig. 3d). The HCA dendograms of the Raman spectroscopy (Fig. 3b) and MALDI-TOF-MS (Fig. 3c) data exhibited three main clusters: (i) all C. jejuni and C. coli strains, (ii) C. hyointestinalis, C. fetus subspecies fetus, C. fetus subspecies venerealis and C. lari, and (iii) a single member cluster comprising C. concisus. Although the dendogram of the FT-IR (Fig. 3a) spectral data revealed similar grouping pattern, both C. fetus subspecies were differentiated into a completely separate group (away from cluster (ii) detailed above), thereby resulting in four groups in the FT-IR analyses. These FT-IR findings agreed with the PC-DFA results of MALDI-TOF-MS data (Fig. 2e), where C. fetus subspecies fetus and C. fetus subspecies venerealis were differentiated according to DF3 axis. By contrast, the phylogenetic tree generated using the 16S rRNA sequence, did not display any significant differences between these two strains. Further investigation of the FT-IR spectral data however, did reveal distinct differences in the lipid (2920 and 2851 cm−1) and the fingerprint region (1500–500 cm−1) of the two C. fetus subspecies (Fig. 4). Comparison of the lipid profiles obtained via our initial LC–MS analysis (Fig. 5), were in complete agreement with the FT-IR findings (Fig. 4), revealing significant differences in the lipid content of C. fetus subsp. fetus and venerealis.
Although C. jejuni and C. coli account for more than 90% of campylobacteriosis cases,84 the contribution of C. fetus is not uncommon, accounting for around 2.4% of cases.85C. fetus is also the most common cause of Campylobacter bacteremia (19–53%),86,87 with a reported fatality rate of around 14%,88,89 as well as being associated with thrombophlebitis. C. fetus is also an important veterinary pathogen causing significant economic losses. C. fetus subsp. fetus is commensal and an opportunistic pathogen of livestock and can cause abortion in these animals and be transmitted to humans via consumption of contaminated food, while C. fetus subsp. venerealis causes an economically significant disease known as bovine venereal campylobacteriosis resulting in reduced fertility and abortion.90 Therefore, the ability to rapidly identify the correct C. fetus subspecies is vital.91
However, rapid and accurate identification of C. fetus subspecies is a significant issue within the field of Campylobacter research. Identifying C. fetus subsp. can be problematic as some strains, such as C. fetus subsp. venerealis Biovar intermedius, behave as C. fetus subsp. fetus upon traditional phenotypic tests.92,93 There have also been several attempts to develop molecular assays (predominantly PCR-based) for routine diagnostics, but most have been deemed unreliable upon further scrutiny.94,95
It is perhaps worth noting that although the classification results achieved from all three analytical techniques were in general agreement, the results obtained from PC-DFA of the MALDI-TOF-MS and FT-IR spectral data, not only allowed for the differentiation of the Campylobacter spp., but they also provided the clear separation of C. fetus subsp. fetus from C. fetus subsp. venerealis. In addition, whilst the FT-IR spectral data provided higher reproducibility, the application of MALDI-TOF-MS provides much higher sensitivity and detailed information for identification of the significant peptides or proteins (predominantly ribosomes, and hence the congruence with 16S rRNA) specific to different species. These findings highlight the potential of these techniques as rapid, high throughput and universal approaches for simultaneous detection and differentiation of different Campylobacter spp. down to subspecies levels. In addition, techniques such as FT-IR and Raman spectroscopies also have potential as handheld detection methods,96 to be deployed within food supply chains to detect particular species of this important foodborne pathogen, and identify points prone to contamination within the complex food processing supply and distribution systems.
Acknowledgements
DW was supported by a Society for Applied Microbiology Studentship. RG is indebted to UK BBSRC (BB/L014823/1) for funding our Raman microspectroscopy.References
- G. M. Ruiz-Palacios, Clin. Infect. Dis., 2007, 44, 701–703 CrossRef PubMed.
- A microbiological survey of Campylobacter contamination in fresh, whole, UK-produced chilled chicken at retail sale, https://www.food.gov.uk/sites/default/files/campylobacter-retail-survey-q3-results.pdf, accessed 29.07.2015.
- Acting on Campylobacter together, https://www.food.gov.uk/science/microbiology/campylobacterevidenceprogramme, accessed 10.08.2015.
- J. A. Wagenaar, M. A. P. van Bergen, M. J. Blaser, R. V. Tauxe, D. G. Newell and J. P. M. van Putten, Clin. Infect. Dis., 2014, 58, 1579–1586 CrossRef PubMed.
- L. Zhang, H. Lee, M. C. Grimm, S. M. Riordan, A. S. Day and D. A. Lemberg, World J. Gastroenterol., 2014, 20, 1259–1267 CrossRef PubMed.
- N. O. Kaakoush and H. M. Mitchell, Front. Cell. Infect. Microbiol., 2012, 2, 4 Search PubMed.
- S. M. Man, Nat. Rev.: Gastroenterol. Hepatol., 2011, 8, 669–685 CrossRef CAS PubMed.
- J. Engberg, S. L. W. On, C. S. Harrington and P. Gerner-Smidt, J. Clin. Microbiol., 2000, 38, 286–291 CAS.
- S. L. W. On, J. Microbiol. Methods, 2013, 95, 3–7 CrossRef CAS PubMed.
- N. O. Kaakoush, N. P. Deshpande, M. R. Wilkins, C. G. Tan, J. A. Burgos-Portugal, M. J. Raftery, A. S. Day, D. A. Lemberg and H. Mitchell, PLoS One, 2011, 6, e29045 CAS.
- C. R. Friedman, R. M. Hoekstra, M. Samuel, R. Marcus, J. Bender, B. Shiferaw, S. Reddy, S. D. Ahuja, D. L. Helfrick, F. Hardnett, M. Carter, B. Anderson and R. V. Tauxe, Clin. Infect. Dis., 2004, 38(Suppl. 3), S285–S296 CrossRef PubMed.
- T. Humphrey, S. O'Brien and M. Madsen, Int. J. Food Microbiol., 2007, 117, 237–257 CrossRef PubMed.
- A. E. Heuvelink, C. van Heerwaarden, A. Zwartkruis-Nahuis, J. J. Tilburg, M. H. Bos, F. G. Heilmann, A. Hofhuis, T. Hoekstra and E. de Boer, Int. J. Food Microbiol., 2009, 134, 70–74 CrossRef CAS PubMed.
- J. A. Painter, R. M. Hoekstra, T. Ayers, R. V. Tauxe, C. R. Braden, F. J. Angulo and P. M. Griffin, Emerg. Infect. Dis., 2013, 19, 407–415 CrossRef PubMed.
- E. V. Taylor, K. M. Herman, E. C. Ailes, C. Fitzgerald, J. S. Yoder, B. E. Mahon and R. V. Tauxe, Epidemiol. Infect., 2013, 141, 987–996 CrossRef CAS PubMed.
- T. Pitkänen, J. Microbiol. Methods, 2013, 95, 39–47 CrossRef PubMed.
- G. Kapperud, E. Skjerve, N. H. Bean, S. M. Ostroff and J. Lassen, J. Clin. Microbiol., 1992, 30, 3117–3121 CAS.
- J. Neimann, J. Engberg, K. Molbak and H. C. Wegener, Epidemiol. Infect., 2003, 130, 353–366 CrossRef CAS PubMed.
- B. Chaban, M. Ngeleka and J. E. Hill, BMC Microbiol., 2010, 10, 73 CrossRef PubMed.
- B. N. Parsons, C. J. Porter, R. Ryvar, J. Stavisky, N. J. Williams, G. L. Pinchbeck, R. J. Birtles, R. M. Christley, A. J. German, A. D. Radford, C. A. Hart, R. M. Gaskell and S. Dawson, Vet. J., 2010, 184, 66–70 CrossRef CAS PubMed.
- O. Sahin, C. Fitzgerald, S. Stroika, S. Zhao, R. J. Sippy, P. Kwan, P. J. Plummer, J. Han, M. J. Yaeger and Q. Zhang, J. Clin. Microbiol., 2012, 50, 680–687 CrossRef CAS PubMed.
- L. Mughini Gras, J. H. Smid, J. A. Wagenaar, A. G. de Boer, A. H. Havelaar, I. H. M. Friesema, N. P. French, L. Busani and W. van Pelt, PLoS One, 2012, 7, e42599 Search PubMed.
- S. Ann, G. B. Michael, W. Nick, C. M. Jonathan, M. Petra, M. C. Donald, J. L. Robin and P. F. Nigel, Emerging Infectious Dis. J., 2011, 17, 1007 CrossRef PubMed.
- P. A. Granato, L. Chen, I. Holiday, R. A. Rawling, S. M. Novak-Weekley, T. Quinlan and K. A. Musser, J. Clin. Microbiol., 2010, 48, 4022–4027 CrossRef PubMed.
- O. A. Oyarzabal and C. Battie, Immunological methods for the detection of Campylobacter Spp. - current applications and potential use in biosensors, INTECH Open Access Publisher, 2012 Search PubMed.
- L. Liu, S. K. Hussain, R. S. Miller and O. A. Oyarzabal, J. Food Prot., 2009, 72, 2428–2432 Search PubMed.
- R. S. Miller, L. Speegle, O. A. Oyarzabal and A. J. Lastovica, J. Clin. Microbiol., 2008, 46, 3546–3547 CrossRef PubMed.
- B. A. Oyofo, S. A. Thornton, D. H. Burr, T. J. Trust, O. R. Pavlovskis and P. Guerry, J. Clin. Microbiol., 1992, 30, 2613–2619 CAS.
- K. E. Dingle, F. M. Colles, D. R. A. Wareing, R. Ure, A. J. Fox, F. E. Bolton, H. J. Bootsma, R. J. L. Willems, R. Urwin and M. C. J. Maiden, J. Clin. Microbiol., 2001, 39, 14–23 CrossRef CAS PubMed.
- E. M. Ribot, C. Fitzgerald, K. Kubota, B. Swaminathan and T. J. Barrett, J. Clin. Microbiol., 2001, 39, 1889–1894 CrossRef CAS PubMed.
- M. U. Ahmed, L. Dunn and E. P. Ivanova, Foodborne Pathog. Dis., 2012, 9, 375–385 CrossRef PubMed.
- W. Yamazaki-Matsune, M. Taguchi, K. Seto, R. Kawahara, K. Kawatsu, Y. Kumeda, M. Kitazato, M. Nukina, N. Misawa and T. Tsukamoto, J. Med. Microbiol., 2007, 56, 1467–1473 CrossRef CAS PubMed.
- M. L. Reaves and J. D. Rabinowitz, Curr. Opin. Biotechnol., 2011, 22, 17–25 CrossRef CAS PubMed.
- J. Han, L. C. Antunes, B. B. Finlay and C. H. Borchers, Future Microbiol., 2010, 5, 153–161 CrossRef CAS PubMed.
- M. J. Werf Van Der, R. H. Jellema and T. Hankemeier, J. Ind. Microbiol. Biotechnol., 2005, 32, 234–252 CrossRef PubMed.
- V. Mapelli, L. Olsson and J. Nielsen, Trends Biotechnol., 2008, 26, 490–497 CrossRef CAS PubMed.
- C. L. Winder, W. B. Dunn and R. Goodacre, Trends Microbiol., 2011, 19, 315–322 CrossRef CAS PubMed.
- D. I. Ellis, V. L. Brewster, W. B. Dunn, J. W. Allwood, A. P. Golovanov and R. Goodacre, Chem. Soc. Rev., 2012, 41, 5706–5727 RSC.
- R. Goodacre, S. Vaidyanathan, W. B. Dunn, G. G. Harrigan and D. B. Kell, Trends Biotechnol., 2004, 22, 245–252 CrossRef CAS PubMed.
- O. Fiehn, Comp. Funct. Genomics, 2001, 2, 155–168 CrossRef CAS PubMed.
- D. I. Ellis and R. Goodacre, Analyst, 2006, 131, 875–885 RSC.
- N. Nicolaou, Y. Xu and R. Goodacre, Anal. Chem., 2011, 83, 5681–5687 CrossRef CAS PubMed.
- K. Maquelin, C. Kirschner, L. P. Choo-Smith, N. van den Braak, H. P. Endtz, D. Naumann and G. J. Puppels, J. Microbiol. Methods, 2002, 51, 255–271 CrossRef CAS PubMed.
- D. Naumann, D. Helm and H. Labischinski, Nature, 1991, 351, 81–82 CrossRef CAS PubMed.
- X. Lu, Q. Huang, W. G. Miller, D. E. Aston, J. Xu, F. Xue, H. Zhang, B. A. Rasco, S. Wang and M. E. Konkel, J. Clin. Microbiol., 2012, 50, 2932–2946 CrossRef CAS PubMed.
- C. Kirschner, K. Maquelin, P. Pina, N. A. N. Thi, L. P. Choo-Smith, G. D. Sockalingum, C. Sandt, D. Ami, F. Orsini, S. M. Doglia, P. Allouch, M. Mainfait, G. J. Puppels and D. Naumann, J. Clin. Microbiol., 2001, 39, 1763–1770 CrossRef CAS PubMed.
- D. I. Ellis, D. P. Cowcher, L. Ashton, S. O'Hagan and R. Goodacre, Analyst, 2013, 138, 3871–3884 RSC.
- M. A. Winkler, J. Uher and S. Cepa, Anal. Chem., 1999, 71, 3416–3419 CrossRef CAS PubMed.
- E. Bessede, O. Solecki, E. Sifre, L. Labadi and F. Megraud, Clin. Microbiol. Infect., 2011, 17, 1735–1739 CrossRef CAS PubMed.
- R. E. Mandrell, L. A. Harden, A. Bates, W. G. Miller, W. F. Haddon and C. K. Fagerquist, Appl. Environ. Microbiol., 2005, 71, 6292–6307 CrossRef CAS PubMed.
- R. Kolinska, M. Drevinek, V. Jakubu and H. Zemlickova, Folia Microbiol., 2008, 53, 403–409 CrossRef CAS PubMed.
- P. R. Murray, Clin. Microbiol. Infect., 2010, 16, 1626–1630 CrossRef CAS PubMed.
- P. Seng, M. Drancourt, F. Gouriet, B. La Scola, P. E. Fournier, J. M. Rolain and D. Raoult, Clin. Infect. Dis., 2009, 49, 543–551 CrossRef CAS PubMed.
- H. Moura, A. R. Woolfitt, M. G. Carvalho, A. Pavlopoulos, L. M. Teixeira, G. A. Satten and J. R. Barr, FEMS Immunol. Med. Microbiol., 2008, 53, 333–342 CrossRef CAS PubMed.
- N. AlMasoud, Y. Xu, N. Nicolaou and R. Goodacre, Anal. Chim. Acta, 2014, 840, 49–57 CrossRef CAS PubMed.
- A. E. Clark, E. J. Kaleta, A. Arora and D. M. Wolk, Clin. Microbiol. Rev., 2013, 26, 547–603 CrossRef CAS PubMed.
- H. Muhamadali, Y. Xu, D. I. Ellis, J. W. Allwood, N. J. W. Rattray, E. Correa, H. Alrabiah, J. R. Lloyd and R. Goodacre, Appl. Environ. Microbiol., 2015, 81, 3288–3298 CrossRef CAS PubMed.
- H. Muhamadali, Y. Xu, D. I. Ellis, D. K. Trivedi, N. J. W. Rattray, K. Bernaerts and R. Goodacre, Microb. Cell Fact., 2015, 14, 157–157 CrossRef PubMed.
- C. L. Winder, S. V. Gordon, J. Dale, R. G. Hewinson and R. Goodacre, Microbiology, 2006, 152, 2757–2765 CrossRef CAS PubMed.
- H. Martens, J. P. Nielsen and S. B. Engelsen, Anal. Chem., 2003, 75, 394–404 CrossRef CAS PubMed.
- H. Muhamadali, M. Chisanga, A. Subaihi and R. Goodacre, Anal. Chem., 2015, 87, 4578–4586 CrossRef CAS PubMed.
- S. Wold, K. Esbensen and P. Geladi, Chemom. Intell. Lab. Syst., 1987, 2, 37–52 CrossRef CAS.
- H. J. H. Macfie, C. S. Gutteridge and J. R. Norris, J. Gen. Microbiol., 1978, 104, 67–74 CrossRef CAS PubMed.
- P. S. Gromski, H. Muhamadali, D. I. Ellis, Y. Xu, E. Correa, M. L. Turner and R. Goodacre, Anal. Chim. Acta, 2015, 879, 10–23 CrossRef CAS PubMed.
- R. M. Jarvis and R. Goodacre, Anal. Chem., 2004, 76, 40–47 CrossRef CAS PubMed.
- B. S. Radovic, R. Goodacre and E. Anklam, J. Anal. Appl. Pyrolysis, 2001, 60, 79–87 CrossRef CAS.
- P. A. Eden, T. M. Schmidt, R. P. Blakemore and N. R. Pace, Int. J. Syst. Bacteriol., 1991, 41, 324–325 CrossRef CAS PubMed.
- D. Linton, R. J. Owen and J. Stanley, Res. Microbiol., 1996, 147, 707–718 CrossRef CAS PubMed.
- D. P. Herlemann, M. Labrenz, K. Jurgens, S. Bertilsson, J. J. Waniek and A. F. Andersson, ISME J., 2011, 5, 1571–1579 CrossRef CAS PubMed.
- M. A. Larkin, G. Blackshields, N. P. Brown, R. Chenna, P. A. McGettigan, H. McWilliam, F. Valentin, I. M. Wallace, A. Wilm, R. Lopez, J. D. Thompson, T. J. Gibson and D. G. Higgins, Bioinformatics, 2007, 23, 2947–2948 CrossRef CAS PubMed.
- S. Kumar, M. Nei, J. Dudley and K. Tamura, Brief. Bioinform., 2008, 9, 299–306 CrossRef CAS PubMed.
- D. Ziegler, J. F. Pothier, J. Ardley, R. K. Fossou, V. Pfluger, S. de Meyer, G. Vogel, M. Tonolla, J. Howieson, W. Reeve and X. Perret, Appl. Microbiol. Biotechnol., 2015, 99, 5547–5562 CrossRef CAS PubMed.
- N. N. Kaderbhai, D. I. Broadhurst, D. I. Ellis, R. Goodacre and D. B. Kell, Comp. Funct. Genomics, 2003, 4, 376–391 CrossRef CAS PubMed.
- P. Dekeyser, J. P. Butzler, J. Sternon and M. Gossuind, J. Infect. Dis., 1972, 125, 390–392 CrossRef CAS PubMed.
- J. P. Butzler, P. Dekeyser, M. Detrain and F. Dehaen, J. Pediatr., 1973, 82, 493–495 CrossRef CAS PubMed.
- M. B. Skirrow, Br. Med. J., 1977, 2, 9–11 CrossRef CAS PubMed.
- J. M. Hollas, Modern spectroscopy, John Wiley & Sons, 2005, 4th edn Search PubMed.
- T. W. Cullen, J. A. Madsen, P. L. Ivanov, J. S. Brodbelt and M. S. Trent, J. Biol. Chem., 2012, 287, 3326–3336 CrossRef CAS PubMed.
- M. A. Lambert, C. M. Patton, T. J. Barrett and C. W. Moss, J. Clin. Microbiol., 1987, 25, 706–713 CAS.
- N. J. C. Strachan and K. J. Forbes, Lancet, 2010, 376, 665–667 CrossRef.
- C. C. Tam, L. C. Rodrigues, I. Petersen, A. Islam, A. Hayward and S. J. O'Brien, J. Infect. Dis., 2006, 194, 95–97 CrossRef PubMed.
- J. Kaldor and B. R. Speed, Br. Med. J., 1984, 288, 1867–1870 CrossRef CAS PubMed.
- V. Fussing, E. Moller Nielsen, J. Neimann and J. Engberg, Clin. Microbiol. Infect., 2007, 13, 635–642 CrossRef CAS PubMed.
- I. A. Gillespie, S. J. O'Brien, J. A. Frost, G. K. Adak, P. Horby, A. V. Swan, M. J. Painter and K. R. Neal, Emerg. Infect. Dis., 2002, 8, 937–942 CrossRef PubMed.
- S. Bullman, D. Corcoran, J. O'Leary, D. O'Hare, B. Lucey and R. D. Sleator, FEMS Immunol. Med. Microbiol., 2011, 63, 248–253 CrossRef CAS PubMed.
- A. Fernandez-Cruz, P. Munoz, R. Mohedano, M. Valerio, M. Marin, L. Alcala, M. Rodriguez-Creixems, E. Cercenado and E. Bouza, Medicine, 2010, 89, 319–330 CrossRef PubMed.
- J. Pacanowski, V. Lalande, K. Lacombe, C. Boudraa, P. Lesprit, P. Legrand, D. Trystram, N. Kassis, G. Arlet, J. L. Mainardi, F. Doucet-Populaire, P. M. Girard and J. L. Meynard, Clin. Infect. Dis., 2008, 47, 790–796 CrossRef PubMed.
- L. Gazaigne, P. Legrand, B. Renaud, B. Bourra, E. Taillandier, C. Brun-Buisson and P. Lesprit, Eur. J. Clin. Microbiol. Infect. Dis., 2008, 27, 185–189 CrossRef CAS PubMed.
- R. L. Guerrant, R. G. Lahita, W. C. Winn, Jr. and R. B. Roberts, Am. J. Med., 1978, 65, 584–592 CrossRef CAS PubMed.
- H. Sprenger, E. L. Zechner and G. Gorkiewicz, Eur. J. Microbiol. Immunol., 2012, 2, 66–75 CrossRef CAS PubMed.
- W. M. Kalka-Moll, M. A. P. Van Bergen, G. Plum, M. Krönke and J. A. Wagenaar, Clin. Microbiol. Infect., 2005, 11, 341–342 CrossRef CAS PubMed.
- S. M. Salama, M. M. Garcia and D. E. Taylor, Int. J. Syst. Bacteriol., 1992, 42, 446–450 CrossRef CAS PubMed.
- M. A. van Bergen, K. E. Dingle, M. C. Maiden, D. G. Newell, L. van der Graaf-Van Bloois, J. P. van Putten and J. A. Wagenaar, J. Clin. Microbiol., 2005, 43, 5888–5898 CrossRef CAS PubMed.
- A. McGoldrick, J. Chanter, S. Gale, J. Parr, M. Toszeghy and K. Line, J. Microbiol. Methods, 2013, 94, 199–204 CrossRef CAS PubMed.
- L. van der Graaf-van Bloois, W. G. Miller, E. Yee, M. Rijnsburger, J. A. Wagenaar and B. Duim, J. Clin. Microbiol., 2014, 52, 4183–4188 CrossRef PubMed.
- D. I. Ellis, H. Muhamadali, S. A. Haughey, C. Elliott and R. Goodacre, Analytical Methods, 2015 10.1039/C5AY02048D.
- A. V. Karlyshev, D. Linton, N. A. Gregson and B. W. Wren, Microbiology, 2002, 148, 473–480 CrossRef CAS PubMed.
- R. V. Tauxe, C. M. Patton, P. Edmonds, T. J. Barrett, D. J. Brenner and P. A. Blake, J. Clin. Microbiol., 1985, 21, 222–225 CAS.
- S. R. Palmer, P. R. Gully, J. M. White, A. D. Pearson, W. G. Suckling, D. M. Jones, J. C. L. Rawes and J. L. Penner, Lancet, 1983, 321, 287–290 CrossRef.
- D. E. Fouts, E. F. Mongodin, R. E. Mandrell, W. G. Miller, D. A. Rasko, J. Ravel, L. M. Brinkac, R. T. DeBoy, C. T. Parker, S. C. Daugherty, R. J. Dodson, A. S. Durkin, R. Madupu, S. A. Sullivan, J. U. Shetty, M. A. Ayodeji, A. Shvartsbeyn, M. C. Schatz, J. H. Badger, C. M. Fraser and K. E. Nelson, PLoS Biol., 2005, 3, e15 Search PubMed.
- J. Benjamin, S. Leaper, R. J. Owen and M. B. Skirrow, Curr. Microbiol., 1983, 8, 231–238 CrossRef.
- C. J. Gebhart, G. E. Ward, K. Chang and H. J. Kurtz, Am. J. Vet. Res., 1983, 44, 361–367 CAS.
- M. Véron and R. Chatelain, Int. J. Syst. Evol. Microbiol., 1973, 23, 122–134 Search PubMed.
- A. C. R. Tanner, S. Badger, C.-H. Lai, M. A. Listgarten, R. A. Visconti and S. S. Socransky, Int. J. Syst. Evol. Microbiol., 1981, 31, 432–445 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5an01945a |
| This journal is © The Royal Society of Chemistry 2016 |