
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ion mobility coupled to native mass spectrometry as a relevant tool to investigate extremely small ligand-induced conformational changes†
Johann
Stojko
ab,
Sonia
Fieulaine
c,
Stéphanie
Petiot-Bécard
ab,
Alain
Van Dorsselaer
ab,
Thierry
Meinnel
c,
Carmela
Giglione
*c and
Sarah
Cianférani
*ab
aBioOrganic Mass Spectrometry Laboratory (LSMBO), IPHC, Université de Strasbourg, 25 rue Becquerel, 67087 Strasbourg, France. E-mail: sarah.cianferani@unistra.fr
bIPHC, CNRS, UMR7178, 67087 Strasbourg, France
cInstitute for Integrative Biology of the Cell (I2BC), CEA, CNRS, Univ. Paris-Sud, Université Paris-Saclay, 91198, Gif-sur-Yvette cedex, France. E-mail: carmela.giglione@i2bc.paris-saclay.fr
First published on 17th September 2015
Abstract
We evaluate the potential of native mass spectrometry (MS) and ion mobility (IM-MS) for the screening of protein![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ligand complexes when very subtle conformational changes are involved. As a proof of concept, we investigate the interactions between a peptide deformylase (PDF1B), a promising target for the development of new antibiotics, and three of its specific inhibitors that bind in different modes. First, real-time native MS reveals two types of ligands, both interacting in a 1:1 stoichiometry with PDF1B but with different affinities and gas phase stabilities. Conformational IM-MS screening then highlights two very close but significantly distinct ligand-induced conformations with collision cross sections that differ by less than 1%. Real-time IM-MS is used to monitor not only the dynamics of ligand binding to apoPDF1B but also the switching between holo conformations. This study provides additional evidence that the most potent ligands inhibit peptide deformylases through a slow-tight binding mechanism, in agreement with previous structural and enzymology studies. Furthermore, this approach, wherein the characteristics obtained by native MS are combined with IM-MS conformational screening, prove valuable in characterizing extremely subtle dynamic conformational changes induced when ligands bind to protein assemblies. We discuss the promise and limitations of IM-MS in the context of detection of very small conformational changes induced upon ligand binding.
:ligand complexes when very subtle conformational changes are involved. As a proof of concept, we investigate the interactions between a peptide deformylase (PDF1B), a promising target for the development of new antibiotics, and three of its specific inhibitors that bind in different modes. First, real-time native MS reveals two types of ligands, both interacting in a 1:1 stoichiometry with PDF1B but with different affinities and gas phase stabilities. Conformational IM-MS screening then highlights two very close but significantly distinct ligand-induced conformations with collision cross sections that differ by less than 1%. Real-time IM-MS is used to monitor not only the dynamics of ligand binding to apoPDF1B but also the switching between holo conformations. This study provides additional evidence that the most potent ligands inhibit peptide deformylases through a slow-tight binding mechanism, in agreement with previous structural and enzymology studies. Furthermore, this approach, wherein the characteristics obtained by native MS are combined with IM-MS conformational screening, prove valuable in characterizing extremely subtle dynamic conformational changes induced when ligands bind to protein assemblies. We discuss the promise and limitations of IM-MS in the context of detection of very small conformational changes induced upon ligand binding.
Introduction
The use of orthogonal biophysical techniques (fluorescence, X-ray, NMR, calorimetry, etc.) or enzymatic assays has turned drug research into a multifaceted approach, involving high-throughput screening (HTS) of large libraries of molecules, structure-based drug discovery, and lead optimization or fragment-based drug discovery.1,2 Mass spectrometry (MS) and its applications in the pharmaceutical industry have evolved continuously over the past 20 years, mainly through the outstanding instrumental breakthroughs that have been achieved. Mass spectrometry is now routinely used to evaluate the purity of recombinant proteins,3 to characterize monoclonal antibodies4,5 and, in proteomics, to identify new candidate biomarkers.6,7 Mass spectrometry is also pivotal throughout the drug-discovery process—for the quality control of compound libraries, the evaluation of compound purity, and for solubility, stability, and ADME-Tox studies.8Subtle changes that occur in response to ligand binding in multiprotein complexes are often difficult to identify without high resolution X-ray analysis. Native MS has nonetheless emerged as a valuable alternative through the insights it provides into the dynamic changes that result from ligand binding.9,10 This technique has indeed been incorporated into the drug discovery workflow, notably during the early stages, in structure- or fragment-based approaches. Automation11 and a coupling to high resolution native MS12 allows the method to be used for the primary screening of focused libraries containing a few thousand molecules. Native MS, which provides information on binding stoichiometries, specificities, affinities13,14 and even gas phase stabilities, is now considered robust and reliable enough to validate hit compounds identified by HTS. In this context, native MS provides strong support for the validation and characterization of leads derived from close analogues and virtual screen hits. Native MS has also been used to characterize a variety of protein:ligand systems,15–17 facilitating the discovery of small modulator molecules for orphan nuclear receptors18–20 for instance, or identifying the calixarenes that bind to Staphylococcus aureus leucotoxines21 and the ligands of transthyretin.22
However, classical MS screens cannot reveal the subtle changes that occur in response to ligand binding in proteins, being sensitive only to overall changes in binding characteristics. Historically, NMR, high resolution X-ray crystallography, computational studies and kinetic measurements have been used to detect ligand-induced conformational changes in proteins. These are now complemented by emerging techniques such as those involving single molecules23 and ion mobility MS (IM-MS), the latter offering a combined binding and conformational characterization of the system. In ion mobility experiments, ions are separated according to their charge and collision cross section (CCS) using an electric field in a controlled gas environment.24,25 The IM component thereby adds a gas phase separation dimension based on the mass, charge and shape of the ions. Many studies have demonstrated the potential of IM-MS as an integrative tool for structural biology programs.26–28 However, this technique is mainly used to assess the global shape of the protein (globular versus helical,29 for instance) and only rarely has IM-MS been used to characterize the conformation of protein/ligand systems. Typically indeed, the conformational changes have to be rather large (ΔCCS > 5%) to be detected;30 in a recent study however,31 the subtle conformational changes (ΔCCS = 2%) induced by inhibitor binding were successfully detected by fine tuning the instrumental settings. More recently, Ruotolo and coworkers have outlined new possibilities for IM-MS kinase/ligand screening,32 while Nyon et al. have successfully used IM-MS to monitor changes in the conformational dynamics of α1-antitrypsin upon ligand binding.33
Here, we report the dynamic characterization of protein:ligand complexes at both binding and conformational levels. Peptide deformylases (PDFs) are promising therapeutic target for the development of antibacterial and anticancer inhibitors.34–36 Four subfamilies are usually defined (viz. PDF1A, PDF1B, PDF2 and PDF3) according to their sequence and structural singularities.37,38 The two very well documented enzymes, Escherichia coli PDF and Arabidopsis thaliana PDF1B39–41 belong to the second of these families, the most common in eubacteria. We have characterized the binding of three bacterial PDF inhibitors (Fig. 1) to A. thaliana PDF1B (simply named PDF1B in the following) by IM-MS. This system is interesting because previous enzymatic, biophysical and crystallographic studies42 have revealed the slow-tight binding (STB) of a number of potent inhibitors.43 This is consistent with the induced-fit binding model, whereby a ligand induces a conformational change that either enhances substrate catalysis or strengthens inhibitor binding.44
 | ||
| Fig. 1 Chemical structures of the PDF1B ligands used in the present study. | ||
According to the binding stoichiometries, specificities and gas phase stabilities obtained by native MS, the PDF inhibitors investigated here are classed as either slow-tight or slow-moderate binders to PDF1B. A comparison of the CCSs derived from crystallography and IM-MS then highlights the existence of two distinct conformational changes induced by ligand binding. Altogether, these data further suggest that the STB inhibition of PDFs occurs through an induced-fit mechanism.
Results
Characterization of individual PDF1B![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :ligand complexes by native MS deciphers binding properties and kinetics
:ligand complexes by native MS deciphers binding properties and kinetics
933 ± 1 Da, Fig. 2a), in agreement with previous biochemical data.41 To assess the binding stoichiometries, titration experiments were performed with the three ligands (viz. actinonin and the chemically designed compounds 21 and 6b, see Fig. 1 and the Experimental section), using native MS. The addition of actinonin in two-fold excess and after 10 min incubation shifts the main mass signal to 22318 ± 1 Da, corresponding to PDF1B + Ni2+ + actinonin (Fig. 2b). Similar 1:1 binding stoichiometries are obtained in the presence of compounds 21 and 6b (Fig. 2c). Note also that non-specific ligand multi-addition occurs when the inhibitors are present in large excess (from 1:2 to 1:10 stoichiometries, Fig. S2 in the ESI†).
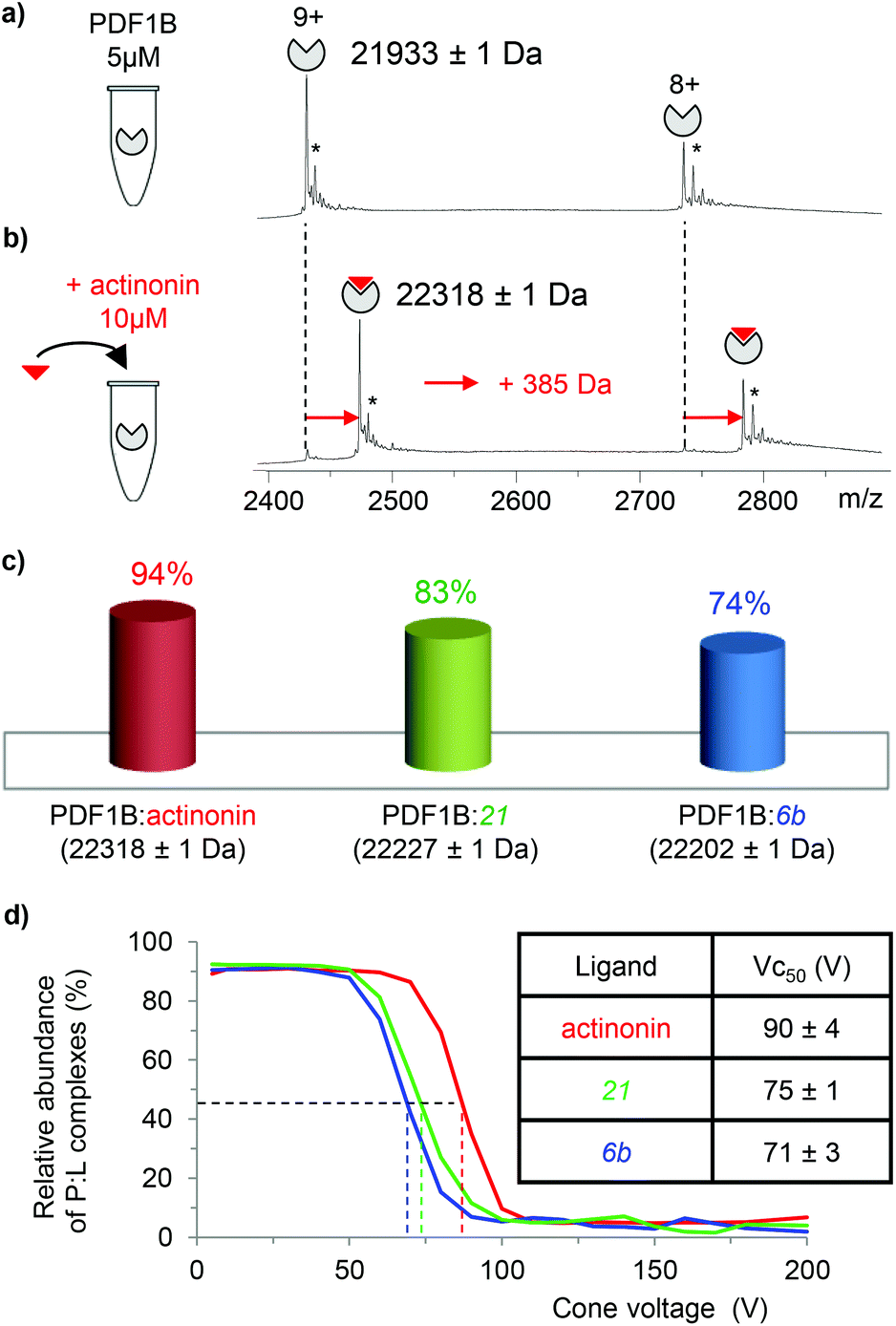 | ||
| Fig. 2 Native mass spectrometry (MS) characterization of PDF1B:ligand interactions. Native mass spectra of PDF1B (5 μM) (a) alone or (b) after 10 min incubation with 10 μM actinonin (the asterisks indicate signals from Ni2+ adducts). (c) Relative proportions of the PDF1B:ligand complexes, as determined by native MS from the intensities of the 8+ and 9+ charge states after incubation of 5 μM PDF1B with 10 μM ligand. (d) Protein:ligand ratios for PDF1B:actinonin (red), PDF1B:21 (green) and PDF1B:6b (blue) complexes as a function of the cone voltage in the ion source. The dashed lines indicate the corresponding Vc50 values. | ||
To further specify the dynamics and affinity of the PDF1B:ligand interactions, the relative proportions of free and bound forms were quantified by native MS at different protein:ligand ratios and incubation times. For all the molecules and protein:ligand ratios, a 1:1 PDF1B:ligand complex forms in a minute (Fig. S2†). The proportion of the bound form at saturation depends on the ligand however, with 94%, 83% and 74% of the signal arising from the 1:1 complex under a two-fold molar excess of actinonin and compounds 21 and 6b, respectively (Fig. 2c; the results are similar with a ten-fold excess, see Fig. S2†). These results show, in agreement with a previous study,43 that the affinity of PDF1B is higher for actinonin than it is for compounds 21 or 6b.
:ligand complexes were investigated by recording the accelerating voltage required to dissociate 50% of the complex (Vc50)45 during in source collision-induced dissociation experiments. A clear correlation was found between Vc50 values and the contribution of electrostatic and H-bond based interactions in the total binding energy of the complexes in several studies.45,46Fig. 2d shows that Vc50 is significantly higher for actinonin (90 ± 4 V) than it is for compounds 21 (75 ± 1 V) and 6b (71 ± 3 V), which indicates that the polar stabilization of PDF1B:actinonin is greater than that of the other two complexes. This is consistent with the interaction networks suggested by the crystal structures. Indeed, while actinonin binds to PDFs through hydrophobic contacts and a number of hydrogen bonds,47,48 these are fewer in the complexes formed with 6b.42
Real-time characterization of PDF1B:ligand mixtures by native MS identifies two ligand affinity classes
The dynamics of ligand binding were investigated further by monitoring pairwise incubations of the ligands over time by native MS. These competition experiments were performed by mixing the ligands either simultaneously (direct competition) or sequentially (indirect competition) with the enzyme.
:2:2 PDF1B:ligand A:ligand B ratio), the major component always corresponds to the 1:1 PDF1B:actinonin complex (Fig. 3b and e). After 30 min moreover, the signals from the 1:1 PDF1B:6b or PDF1B:21 complexes are no longer observed (Fig. 3c and f). When the two compounds are in direct competition with each other however, both 1:1 complexes are present, with initially a significantly higher proportion of PDF1B:6b than PDF1B:21 (56% and 31% respectively, with 13% of the apo protein, Fig. 3h and j). The two complexes are found in equal proportions after 10 min, and at 30 min, an equilibrium is reached between 1:1 PDF1B:6b (37%) and PDF1B:21 (49%) (Fig. 3i and j). The inversion of relative ligand binding affinities on the minute time scale is quite unusual, but can be explained by the inhibition mode of these two compounds. We know that initially during ligand recognition, the affinity of 6b for various PDFs is higher (KI6b ≪ KI21, data not shown). However, compound 21 induces a conformational change in the enzyme upon binding, resulting in a final complex of moderate but nonetheless higher affinity than that of the encounter complex (KI*21 < KI21, manuscript in preparation), characterized by a very low dissociation constant. The PDF1B:21 complex is thereby likely more stable than PDF1B:6b is, displacing the equilibrium towards the former as shown in Fig. 3j.
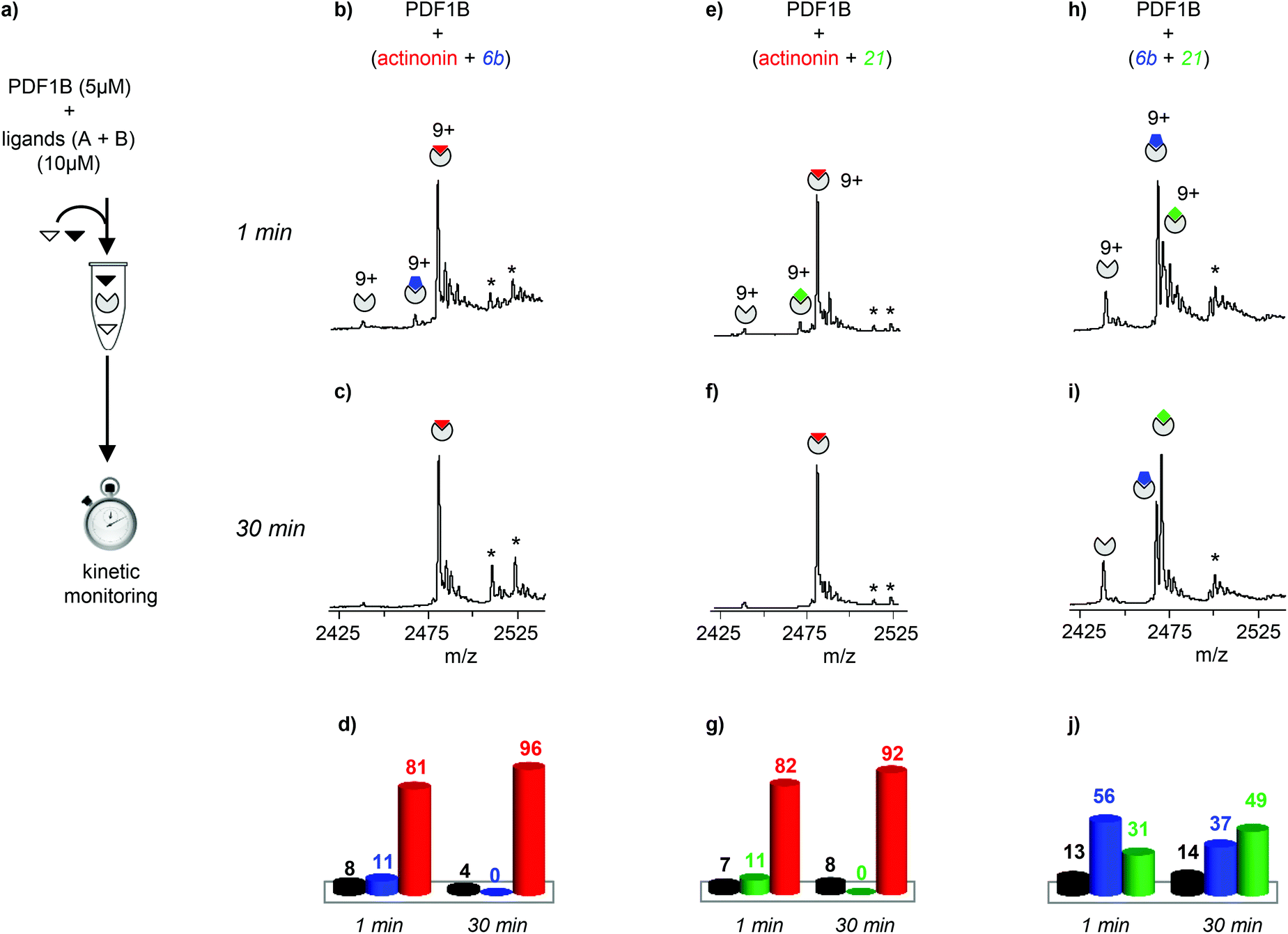 | ||
| Fig. 3 (a) Schematic outline of the direct competition experiments—5 μM PDF1B is added to equimolar mixtures of two of the three ligands tested here (actinonin and compounds 6b and 21, see Fig. 1) at 10 μM—monitored in real time by native mass spectrometry (MS). (b, c, e, f, h, i) Native mass spectra (zoom on the 9+ charge state) obtained after (b, e, h) 1 min and (c, f, i) 30 min pairwise incubation of (b, c) actinonin/6b, (e, f) actinonin/21, and (h, i) 6b/21. (d, g, j) Relative proportions, as determined by native MS, of the different species present after 1 and 30 min pairwise incubation of (d) actinonin/6b, (g) actinonin/21, and (j) 6b/21. Asterisks represent non-specific binding of a second ligand molecules. | ||
Note furthermore that the direct competition experiments show no signs of simultaneous specific binding of two different ligands, strongly suggesting that actinonin, 21 and 6b compete for the same binding site, a situation that was not clear in previous crystallographic studies with 21 found in an unusual pocket but was not confirmed by enzymatic assay.42,43
:1 complexes was then monitored by real-time native MS (Fig. 4a). As expected from the respective affinities of the ligands, even at high excess (up to 1:2:10 PDF1B:actinonin:6b/21, data not shown), the designed compounds do not displace actinonin from PDF1B (Fig. S3 in the ESI†). Reciprocally, actinonin readily displaces 6b and 21 in 1:1 complex with PDF1B. For PDF1B:6b indeed, ∼92% of the detected species are 1:1 PDF1B:actinonin after just 1 min of actinonin incubation (Fig. 4c and e), the initial ligand being almost fully replaced after 30 min (∼97% 1:1 PDF1B:actinonin, Fig. 4d and e). The behavior is similar starting with a PDF1B:21 complex (Fig. 4g–i). Meanwhile, Fig. 4k–m (Fig S3g–i in the ESI†) shows that compound 6b (21) replaces 21 (6b) in complex with PDF1B. Interestingly, the equilibrium after 30 min in both cases involves major (51%, 56%) and minor (34%, 30%) populations of PDF1B:21 and PDF1B:6b, respectively.
 | ||
| Fig. 4 (a) Indirect competition experiments monitored by real-time native mass spectrometry (MS)—PDF1B is first incubated with ligand A for 10 min before ligand B is added. (b, c, d, f, g, h, j, k, l) Native mass spectra (9+ charge state) of PDF1B incubated with (b, c, d) 6b and then actinonin, (f, g, h) 21 and then actinonin, and (j, k, l) 21 and then 6b, in each case (b, f, j) 0 min, (c, g, k) 1 min, and (d, h, l) 30 min after adding the second compound. (e, i, m) Relative proportions, as determined by native MS, of the different species present 1 and 30 min after adding the second ligand in sequential incubations of (e) 6b and then actinonin, (i) 21 and then actinonin, and (m) 21 and then 6b. Asterisks represent non-specific binding of a second ligand molecules. | ||
In summary, this binding screening by real-time native MS shows that actinonin has a much higher affinity for PDF1B than compounds 6b and 21 do. The actinonin complex is also more stable in the gas phase than the other two because of a more extensive network of polar interactions.
IM-MS characterization of individual PDF1B:ligand complexes identifies two subtly different ligand induced conformations
Actinonin and certain other compounds inhibit PDFs through the STB mechanism. Inhibition proceeds through a competitive mechanism which is indicative of overlapping binding sites. Crystal structures of various complexes indicate indeed that the binding sites are identical and fully overlapping.42 The ligand first binds to PDF, inducing next a conformational change that favors further hydrogen bonding between the two partners, thereby locking the complex in a much more stable state.42 We used IM-MS to search for conformational changes resulting from actinonin, 6b or 21 binding. The major difficulty of this analysis consisted in finding instrumental parameters that ensure a high IM resolution and sensitivity without altering or activating the protein:ligand complexes. The IM-MS settings were fine-tuned using a sub-stoichiometric PDF1B:actinonin mixture (1:0.5 protein:ligand ratio) incubated for 10 min, allowing the simultaneous detection of the apo and holo forms (Fig. 5). The IM cell parameters (wave height, wave velocity and He/N2 pressure ratio, see the Experimental section), were optimized to enable the separation of the arrival time distributions (ATDs) of the apo and holo forms of PDF1B (resolution factor, Rs = 0.6, Fig. 5a) in the +9 charge states. The drift time increases upon actinonin binding (ΔtD = +0.44 ms). Ligands 21 and 6b were next analyzed using the same IM-MS parameters and under identical experimental conditions (Fig. 5b and c). The drift time also increases when 21 and 6b bind, with ΔtD = +0.33 ms recorded for both compounds. Although the absolute values of tD vary slightly, ΔtD between the apo and holo forms remains constant from one analysis to the next and from day to day. The drift time shift is thereby a reliable marker of the conformational changes induced by ligand binding.
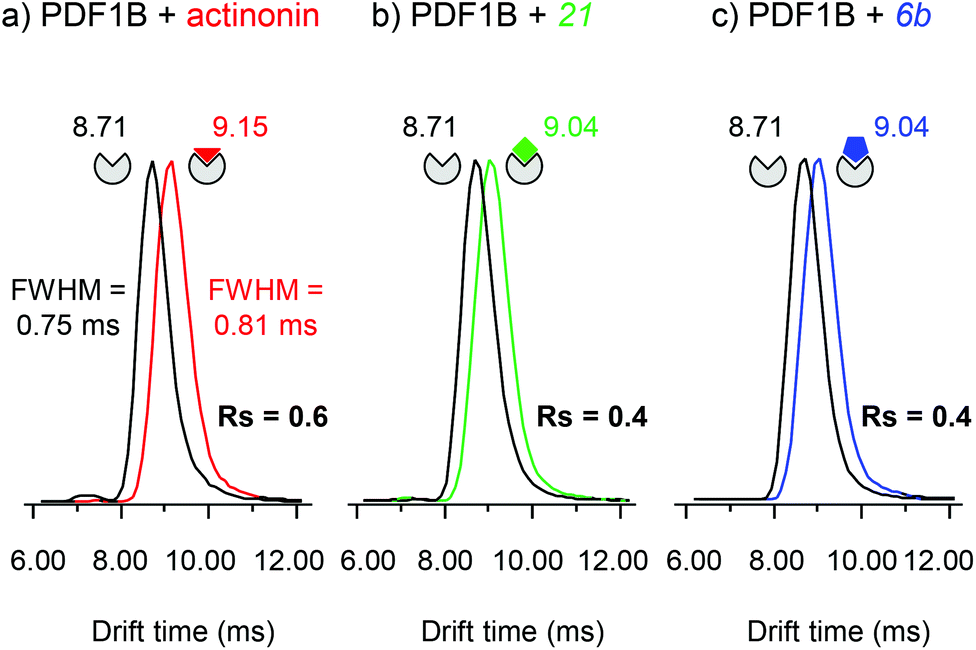 | ||
| Fig. 5 Ion-mobility mass spectrometry characterization of the conformational changes induced upon ligand binding on PDF1B. Extracted arrival drift time distributions (ATDs) of the 9+ charge states of PDF1B in the presence of sub-stoichiometric (1:0.5) (a) actinonin, (b) compound 21, and (c) compound 6b after 10 min incubation. The ATDs corresponding to PDF1B, PDF1B:actinonin, PDF1B:21 and PDF1B:6b are shown in black, red, green and blue, respectively. | ||
The corresponding TWCCSN2s calculated before and after binding for the two main charge states (8+ and 9+) are listed in Table 1. As for the drift time, each ligand induces a slight but constant and reproducible change in the TWCCSN2 for both charge states. The gas phase conformations of apo- and holo-PDF1B differ little in terms of their TWCCSN2 (Table 1 and Fig. 5). The most important change in TWCCSN2 is observed upon actinonin binding, +1.1 ± 0.1% for the 9+ charge state, compared with +0.8 ± 0.1% and +0.7 ± 0.1%, respectively when compounds 21 and 6b bind. These very small differences in TWCCSN2 are nonetheless significant and reveal the presence of three distinct conformations corresponding to apoPDF1B, a more compact PDF1B:actinonin structure, and the intermediate, semi-compact holo forms with 6b and 21.
| apoPDF1B | PDF1B:actinonin |
PDF1B:21 |
PDF1B:6b |
|
|---|---|---|---|---|
| t D 9+ (ms) | 8.67 ± 0.06 | 9.11 ± 0.06 | 9.00 ± 0.06 | 8.97 ± 0.13 |
| ΔtD 9+ (ms) | 0 | +0.44 ± 0.00 | +0.33 ± 0.00 | +0.33 ± 0.00 |
| TWCCSN2 9+ (Å2) | 1948 ± 7 | 1971 ± 8 | 1959 ± 10 | 1957 ± 5 |
| Δ TWCCSN2 9+ (Å2) | 0 | 22 ± 1 (+1.1%) | 17 ± 1 (+0.8%) | 15 ± 3 (+0.7%) |
| t D 8+ (ms) | 11.40 ± 0.17 | 12.06 ± 0.13 | 11.84 ± 0.13 | 11.69 ± 0.19 |
| ΔtD 8+ (ms) | 0 | 0.66 ± 0.11 | +0.40 ± 0.06 | +0.37 ± 0.06 |
| TWCCSN2 8+ (Å2) | 1844 ± 8 | 1868 ± 14 | 1853 ± 14 | 1846 ± 6 |
| Δ TWCCSN2 8+ (Å2) | 0 | 24 ± 5 (+1.3%) | 14 ± 2 (+0.8%) | 13 ± 2 (+0.7%) |
We next attempted to compare our experimentally IM-MS measured TWCCSN2 with those predicted from different X-ray crystallography models (Table 2). Several algorithms have been developed in this context,49–53 which typically exploit either the hard sphere projection approximation (PA), trajectory method (TM) or exact hard sphere scattering (EHSS). As has already been reported, the PA leads to underestimated CCSs, while the EHSS method yields slight overestimates. Thus TM is most reliable for systems with a molecular weight greater than 1.5 kDa.54,55 For the PDF1B system, the CCSs predicted via any of these algorithms suggest that there is no conformational change upon actinonin binding—the ligand which other data show has the strongest structural effect on PDF1B. Conversely, a significant compaction (1.4% to 1.6%) is predicted for 6b binding with both PA and EHSS. Finally, the TM method outputs the most variable predictions and suggests that none of the ligands alter the conformation of PDF1B. In summary, the different algorithms tested here for PDF1B ligand binding yield contradictory results and fail to predict the experimentally verified changes in conformation. Several reasons can account for these observations among which: (i) the fact that extremely small conformational changes (<1% in CCS) are expected in PDF1B case; (ii) each algorithm has been developed for particular applications and molecular size ranges and differ in the assumptions and approximations made in modelling the collisions between the protein and the IM gas; (iv) the fact that our TWCCSN2 measurements were performed in N2 whereas simulation are done in He and maybe also (v) the fact that our simulations does not take into account the presence of a buried Ni2+ atom.
| Method | apoPDF1B (PDB: 3PN2) | PDF1B:actinonin (PDB: 3M6P) |
PDF1B:6b (PDB: 3O3J) |
|
|---|---|---|---|---|
| a Projection approximation. b Trajectory method. c Exact hard sphere scattering. d Not significant. Uncertainty values represent the standard deviation as a result of 6 repeated MobCal runs. | ||||
| CCS (Å2) | PAa | 1562 ± 2 | 1565 ± 1 | 1540 ± 2 |
| EHSSb | 1987 ± 3 | 1988 ± 2 | 1955 ± 3 | |
| TMc | 1972 ± 33 | 1977 ± 26 | 1937 ± 20 | |
| ΔCCS (Å2) | PA | 0 | N.S.d | −22 (−1.4%) |
| EHSS | 0 | N.S. | −32 (−1.6%) | |
| TM | 0 | N.S. | N.S | |
Real-time IM-MS characterization of PDF1B:ligand mixtures reveals conformational switches
Having evidenced two different ligand binding-induced conformational changes in the enzyme, we used IM-MS to study the dynamics of ligand displacement through the same series of direct and indirect competitions as described above.
:actinonin:6b 1:2:2 mixture, reveals the same three conformations as identified in the analysis of individual PDF1B:ligand complexes, namely apoPDF1B, PDF1B:6b and PDF1B:actinonin, with drift time shifts of +0.33 and +0.44 ms between the latter two and apoPDF1B, respectively. The relative populations of the three conformers deduced from the ATDs (Fig. 6c; 9, 17 and 74% for apoPDF1B, PDF1B:6b and PDF1B:actinonin, respectively) match those measured by native MS (Fig. 3d; 8, 11 and 81%, respectively). Moreover, after 30 min incubation, the intensities of the ATDs reflect the displacement of 6b by actinonin (Fig. 6b and c), as already revealed by native MS. Similar results are obtained for actinonin and compound 21 in direct competition (Fig. 6d–f).
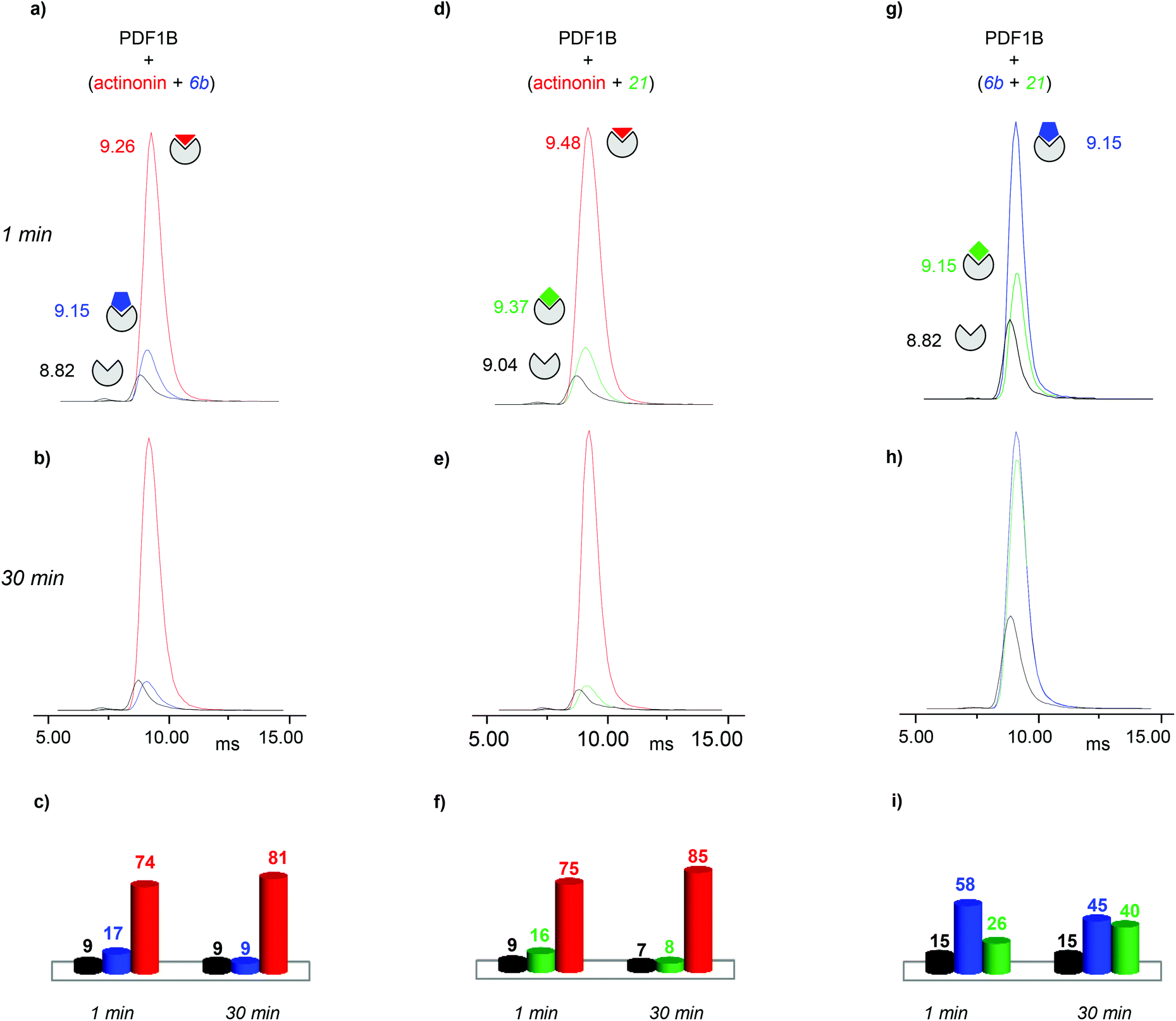 | ||
| Fig. 6 Direct competition experiments monitored by real-time ion-mobility mass spectrometry. (a, b, d, e, g, h) Arrival time distributions (ATDs) extracted from the 9+ charge state of apoPDF1B (black), PDF1B:actinonin (red), PDF1B:21 (green) and PDF1B:6b (blue) measured for mixtures of (a, b) actinonin/6b, (d, e) actinonin/21, and (g, h) 6b/21. Relative proportions, calculated from the ATD intensities of the different species present after 1 (a, d, g) and 30 min (b, e, h) pairwise incubation of (c) actinonin/6b, (f) actinonin/21 and (i) 6b/21. | ||
Since 6b and 21 both induce a ΔtD of +0.33 ms upon binding, the two conformations are identical in terms of their IM-MS arrival times (Fig. 6g and h). The variation in intensity of the corresponding ATDs nonetheless reveals the displacement over time of 6b by 21 (Fig. 6i). These IM-MS results confirm that ligands 21 and 6b have not only similar affinities for PDF1B, but also induce similar global conformational changes in the enzyme.
:21 complex show a similar trend (data not shown). Conversely, pre-formed PDF1B:actinonin remains unaltered by the addition of 6b (Fig. S4 in the ESI†) or 21 (data not shown). These IM-MS results suggest that actinonin blocks PDF1B in a specific conformation that prevents the binding of 21 or 6b (and the associated changes in conformation).
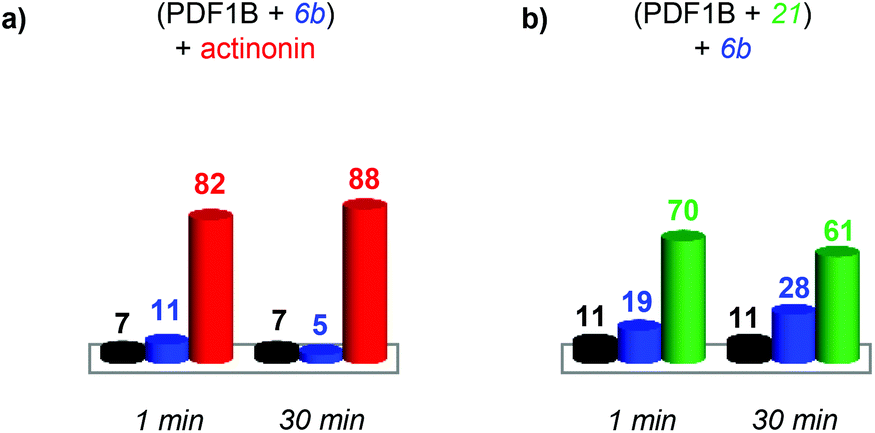 | ||
| Fig. 7 Indirect sequential competition experiments monitored by real-time ion-mobility mass spectrometry. Relative proportions, calculated from the arrival time distribution intensities of the 9+ and 8+ charge states, of apoPDF1B (black), PDF1B:actinonin (red), PDF1B:21 (green) and PDF1B:6b (blue) 1 and 30 min after adding the second ligand in sequential incubations of (a) 6b and then actinonin and (b) 21 and then 6b. | ||
As for the direct competition experiments, when 21 and 6b compete, only two conformations are detected (the one corresponding to apoPDF1B and the one corresponding to PDF1B:6b/21). Pre-incubated PDF1B:21 is gradually replaced by PDF1B:6b until an equilibrium is reached after 30 min, when respectively 61% and 28% of the total ATD intensity arises from the two complexes (Fig. 7b). The reverse experiment shows that 21 also displaces 6b, equilibrium being reached at 46% PDF1B:21 and 40% PDF1B:6b after 30 min (Fig. S4†).
Discussion and conclusions
IM-MS adds evidence for the existence of slow-tight binding to PDF1B
Enzyme-ligand interactions are often associated with conformational changes that precede and/or follow binding. Such transitions between conformational states are however difficult to capture. Based on a combination of convincing crystallographic and enzymatic data, the conformational changes that PDF1B undergoes upon actinonin binding have been explained by an induced-fit mechanism,42 whereby ligands bind to the active site of apoPDF1B in a locally open conformation (state O, Fig. 8). The binding-induced remodeling of the active site yields an intermediate conformation (state I), populated in a concentration/affinity dependent manner (k3, k4, Fig. 8). Binding to the I state eventually leads to a conformation with a completely closed active site (state C, Fig. 8), with low dissociation rates (k5, k6). Structural snapshots of these very subtle and local conformational transitions have been obtained by X-ray crystallography for rational PDF1B mutants, allowing the sequence of events triggered by ligand binding to be reconstructed (see Movies S1 and S2 in ref 42). First, actinonin binds to the enzyme by aligning its long aliphatic chain in the hydrophobic S1′ pocket of PDF1B, triggering a conformation change as key sidechains adjust and reshape the pocket. This local rearrangement is followed by a global closure of the enzyme and the formation of a crucial hydrogen bond. The present paper investigates the putative benefits of native MS and IM-MS in understanding the response of PDFs to ligand binding. Indeed, although enzymatic data suggest that other classes of PDFs bind ligands through induced-fit, the transient nature of the encounter complex means that PDF1B is the only system for which this mechanism has been evidenced at the atomic-scale. | ||
| Fig. 8 Schematic representation of the slow tight binding (STB) mechanism for PDF1B. In apoPDF1B, the active site is mainly in an open conformation (State O). Upon ligand binding, the active site closes either partially or completely. Non-STB ligands induce a partial locking of the active site (State I) as they bind. Conversely, STB ligands adopt a precise orientation in the catalytic site, leading to its complete closure and to the formation of a more stable complex (PDF1B:L*, state C). ΔCCSO→I and ΔCCSO→C are the differences between the collision cross sections of the open (O) and semi-closed (I), and open and closed (C) conformations, respectively. | ||
Native MS demonstrates that 21 and 6b have similar binding stoichiometries, specificities, affinities and gas phase stabilities, and bind PDF1B more weakly than actinonin does. In addition, the native MS data show no evidence of simultaneous 6b and 21 binding, strongly suggesting that the two compounds bind to the same active site, in agreement with enzymatic data. The PDF1B:21 structure in which 21 targets an alternative pocket on the protein surface is thereby most probably a crystallographic artifact.42
The detection here of three distinct conformational states by IM-MS (apoPDF1B, PDF1B:6b/21 and PDF1B:actinonin) proves the ability of this technique to evidence extremely small ligand binding-induced conformational changes, while the nature of the abovementioned states is strong evidence in favor of a binding-induced inhibition mechanism. From our study, PDF1B ligands can be distinguished in two types: (i) actinonin which has higher gas phase stability, affinity and induces a more substantial global conformational change and (ii) 6b/21 with lower stabilities, affinities and smaller conformational changes. So, for PDF1B:ligand systems, the more extensive network of polar interactions that stabilizes actinonin in the gas phase (Fig. 2d) might also confer a higher affinity to PDF1B and further more substantial global conformational change upon binding.
One should note also that IM-MS is sensitive to the change in the global conformation of the protein induced by ligand binding. While X-ray data suggest that the active site contracts upon ligand binding, the slight global extension revealed here by IM-MS (tD and the TWCCSN2 increase) is seemingly in contradiction. However, an overall comparison of the X-ray structures of PDF1B bound to either actinonin (PDB code: 3M6P) or 6b (PDB: 3O3J) highlight other subtle conformational changes away from the active site. The structures of apoPDF1B and PDF1B:actinonin differ mainly in the vicinity of the active site (Fig. S5 in the ESI,† pairwise alpha carbon deviations of up to 1.2 Å), but also along the adjacent secondary structure elements (pairwise deviations ranging from 0.6 to 1 Å). The latter reflects the slight global extension of PDF1B when actinonin binds. This example illustrates the complementarity of high- and low-resolution biophysical techniques—in this case X-ray diffraction and IM-MS—to decipher global and local conformational changes.
In conclusion, this combination of native and IM-MS offers direct evidence that the inhibition of PDF1B occurs through STB mediated by an induced-fit process. The conformational screening of individual PDF1B:ligand complexes reveals two very subtle but distinct conformational changes, respectively induced by actinonin and 21/6b binding.
Advantages and pitfalls of IM-MS for ligand screening when subtle conformational changes are targeted
While native MS is now a routine technique for protein:ligand screening in structural biology and drug discovery programs, IM-MS is still in its infancy. Native MS provides a range of valuable information (binding stoichiometries and specificities, gas phase stabilities, solution affinities) through a series of well-established and easy to perform assays (titration, competition, and collision induced dissociation experiments). Similarly, IM-MS may become established as a primary conformational screening method, the ligands being classified in terms of their respective CCSs. This simple approach may however prove inadequate when extremely subtle conformational changes are expected. The complexity of the system studied here makes it an ideal basis for a discussion of the current limitations of IM-MS.
:ligand conformational screening. Since ligand binding does not alter the charge state of PDF1B, differences in the ionic separation for the 9+ state of the apo and holo forms could be interpreted in terms of changes in conformation. However, because high charge states are susceptible to Coulombic repulsion-induced unfolding,56 the IM-MS parameters had to be carefully optimized to maintain the protein in its non-activated conformational state and avoid misinterpretation.
For the PDF1B:ligand system, in contrast with prediction algorithms, IM-MS data can be reliably and unambiguously interpreted in terms of conformational changes.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :ligand screening.
Since different proteins can have identical CCSs, the interpretation of IM-MS data becomes more problematic for larger molecules. Similarly, the discriminatory power of IM-MS decreases with the amplitude of the conformational change induced by ligand binding, limiting the effectiveness of this approach for small protein:ligand systems in particular. Collision-induced unfolding (CIU) experiments, in which drift time distributions are recorded as the protein unfolds at increasing internal energies,57 offer a potential solution to this problem. Fingerprint distributions have been obtained for the unfolding of reference ligand-binding proteins57 and have been proposed as a means of identifying subtle ligand-induced differences in tertiary structure.58,59 For the PBF1B:ligand system however, the CIU fingerprints for actinonin, 21 and 6b binding are indistinguishable (Fig. S6 in the ESI†).
:21/6b over time into a predominantly PDF1B:actinonin population after actinonin is added to the mixture. As a complement to real-time native MS, real-time IM-MS thereby constitutes an extra source of information on protein:ligand binding dynamics. Importantly, the conformational populations measured here by IM-MS are in good agreement with those obtained by native MS. Real-time IM-MS should be of broad interest for the study of multi-step binding mechanisms with relatively slow kinetics, on the minute time-scale, in the sensitivity range of native MS.
:ligand screening.
Since different proteins can have identical CCSs, the interpretation of IM-MS data becomes more problematic for larger molecules. Similarly, the discriminatory power of IM-MS decreases with the amplitude of the conformational change induced by ligand binding, limiting the effectiveness of this approach for small protein:ligand systems in particular. Collision-induced unfolding (CIU) experiments, in which drift time distributions are recorded as the protein unfolds at increasing internal energies,57 offer a potential solution to this problem. Fingerprint distributions have been obtained for the unfolding of reference ligand-binding proteins57 and have been proposed as a means of identifying subtle ligand-induced differences in tertiary structure.58,59 For the PBF1B:ligand system however, the CIU fingerprints for actinonin, 21 and 6b binding are indistinguishable (Fig. S6 in the ESI†).
:21/6b over time into a predominantly PDF1B:actinonin population after actinonin is added to the mixture. As a complement to real-time native MS, real-time IM-MS thereby constitutes an extra source of information on protein:ligand binding dynamics. Importantly, the conformational populations measured here by IM-MS are in good agreement with those obtained by native MS. Real-time IM-MS should be of broad interest for the study of multi-step binding mechanisms with relatively slow kinetics, on the minute time-scale, in the sensitivity range of native MS.
In conclusion, real-time native MS combined with IM-MS is demonstrated as an alternative and complementary approach to enzymology and crystallography for the structural elucidation of dynamic interaction processes. This approach is amenable to automation and could thereby satisfy the demand for higher throughput screening methods from the biopharmaceutical industry, providing a time-resolved view of binding-induced conformational changes.
Experimental section
Chemicals
Actinonin was purchased from Sigma (St. Louis, MO, USA) while compounds 6b and 21 were chemically synthetized as described in the literature.43 Each component was solubilized to 100 mM in ethanol and then diluted stepwise in a 100 mM ammonium acetate (pH 7.5) buffer. The final ethanol concentration used in the experiments never exceeded 0.25% (v/v).PDF1B expression and purification
Mature Arabidopsis thaliana PDF1B (PDF1B) was expressed and purified as described elsewhere.41,42 Briefly, PDF1B expression was induced in JM101Tr cells after IPTG induction. The cells were lysed and the debris removed by centrifugation. The lysis supernatant was applied on a Q-Sepharose anion exchange column (GE Healthcare) previously equilibrated in buffer A (50 mM Tris-HCl pH 7.5, 5 mM nickel acetate) and elution was performed with buffer B (buffer A + 1 M NaCl). Partially purified protein was then applied on a Superdex 75 column (GE Healthcare Life Sciences) and eluted in buffer C (buffer A + 0.1 M NaCl). The PDF1B-containing fractions were pooled, concentrated and stored at −80 °C in buffer C. All purification procedures were performed at 4 °C.Buffer exchange for native MS experiments
Prior to any native MS experiment, PDF1B was exchanged twice against a 100 mM ammonium acetate (NH4Ac) pH 7.5 buffer using microcentrifuge gel-filtration columns (Zeba 0.5 mL, ThermoScientific, Rockford, IL, USA). The protein concentration was determined by UV absorption at 280 nm (protein extinction coefficient: 8940 M−1 cm−1), using a NanoDrop 2000 spectrophotometer (Thermo Scientific).Titration experiments
Reactions were trigged by mixing 5 μM PDF1B with 5–50 μM solutions of each ligand separately. Each titration point was monitored by native MS and IM-MS after 1, 5, 10, 15, 30 and 60 min incubation at room temperature (RT). The relative proportions of each species and conformer were deduced from either the relative peak intensities measured from the raw mass spectrum for native MS or from IM-MS ATD intensities (taking into account 8+ to 9+ charge states).Vc50 experiments
PDF1B (5 μM) was incubated with 2 molar excesses of each ligand (10 μM) for 10 min at RT. In-source collision induced dissociation was achieved by increasing the cone voltage of the instrument from 10 to 200 V. The 1:1 PDF1B:ligand population, deduced from native MS peak intensities of the main charge states, was plotted as a function of this voltage, with Vc50 being the value at which 50% of the initial 1:1 PDF1B:ligand population is dissociated. Each experiment was performed in triplicate.
Competition experiments
Direct competition experiments were performed by adding 5 μM PDF1B to mixtures of two ligands, each at 10 μM (1:2:2 molar ratios). Competition experiments were monitored by native MS and IM-MS after 1, 5, 10, 15, 30 and 60 min incubation at RT.
For the indirect competition experiments, a first ligand (10 μM) was pre-incubated with 5 μM PDF1B for 10 min (1:2 molar ratio). A second ligand was then added at either 10 or 50 μM (corresponding to 1:2:2 and 1:2:10 molar ratios, respectively), and the evolution of the species was monitored by native MS and IM-MS at the abovementioned incubation times.
All direct and indirect competition experiments were performed in triplicates.
Native MS characterization
Native MS analyses were carried out on a hybrid electrospray quadrupole ion-mobility time-of-flight mass spectrometer (Synapt G2 HDMS, Waters, Manchester, UK) coupled to an automated chip-based nanoESI infusion source (Triversa Nanomate, Advion, Ithaca, NY) operating in positive ion mode. External calibration was performed with the multiply charged ions produced by a 2 μM horse heart myoglobin solution diluted in a 1:1 (v/v) water:acetonitrile mixture acidified with 1% (v/v) formic acid. The MS parameters were carefully adjusted to optimize the sensitivity while preserving the non-covalent complex. In particular, the pressure during the first pumping stage was increased to 6.6 mbar using a throttling valve and the acceleration voltage applied on the sample cone was set to 30 V. The data obtained were processed using MassLynx 4.0 (Waters, Manchester, UK). The relative quantities of each species were deduced from the peak intensities of the 9+ and 8+ charge states.
IM-MS analysis
The IM-MS experiments were performed on the abovementioned spectrometer. The IM separation parameters had to be fine-tuned to retain the native complexes without extensive ion activation before IM separation, while achieving sufficient conformational resolution and ion desolvation.61 Specifically, the trap and transfer collision energies were set to 4 V and the trap bias voltage to 20 V. The IM wave height and velocity were adjusted to 34 V and 1225 m s−1, respectively, while the cooling (QHe) and separation (QN2) gas flow rates were set to 80 and 20 mL min−1, respectively. The IM resolution factor (Rs) was used to estimate the separation between two extracted conformations (A and B) at a given m/z, with arrival drift times (tAd and tBd) and half widths (IA1/2 and IB1/2):
The ion TWCCSN2 were determined from calibration values obtained from triplicate IM-MS analyses of native ions of known DTCCSHe,62 namely equine cytochrome C (1767 and 2061 m/z) and bovine β-lactoglobulin (2042, 2297, 2625, 2827, 3063 m/z), performed under conditions similar to those in this study. The IM-MS data were processed using MassLynx 4.0. The drift times of the charge states of individual species were deduced from the IM extraction of these ions. The populations of the conformers were deduced from the absolute intensities (peak apex) of the respective smoothed ATDs corresponding to the 9+ and 8+ charge states.
Conformational studies of crystallographic structures
The crystallographic structures of apoPDF1B (PDB code: 3PN2), PDF1B:actinonin (3M6P) and PDF1B:6b (3O3J) were visualized using Pymol (DeLano Scientific, San Carlos, CA, USA). The alpha carbons of the apoPDF1B and PDF1B:actinonin structures were aligned using the software Visual Molecular Dynamics.63 The theoretical CCS values reported above were obtained from the same three structures using Mobcal,64,65 ignoring solvent and metal atoms and altering the starting coordinates using random seed numbers. The uncertainties quoted correspond to the standard deviations of six Mobcal replicates.
Acknowledgements
The authors would like to thank Julien Marcoux for fruitful discussions and Matteo Degiacomi for the sequence alignments. This work was supported by the French National Research Agency (ANR-10-BLANC-1510), the Centre National de la Recherche Scientifique, the University of Strasbourg (UdS), the “Fondation pour la Recherche Médicale”, the Alsace Region, and the GIS IBiSA (funding for a Synapt G2 HDMS mass spectrometer). S. P.-B. was supported by a studentship from the CNRS and NovAliX; J. S. acknowledges the Institut de Recherches Servier for supporting his doctoral fellowship.References
- R. A. Carr, M. Congreve, C. W. Murray and D. C. Rees, Drug Discovery Today, 2005, 10, 987–992 CrossRef CAS.
- L. Oster, S. Tapani, Y. Xue and H. Kack, Drug Discovery Today, 2015, 20, 1104–1111 CrossRef PubMed.
- M. Carini, L. Regazzoni and G. Aldini, Curr. Pharm. Biotechnol., 2011, 12, 1548–1557 CAS.
- A. Beck, S. Sanglier-Cianferani and A. Van Dorsselaer, Anal. Chem., 2012, 84, 4637–4646 CrossRef CAS PubMed.
- A. Beck, E. Wagner-Rousset, D. Ayoub, A. Van Dorsselaer and S. Sanglier-Cianferani, Anal. Chem., 2013, 85, 715–736 CrossRef CAS PubMed.
- S. Shao, T. Guo and R. Aebersold, Biochim. Biophys. Acta, 2015, 1854, 519–527 CrossRef CAS PubMed.
- A. P. Drabovich, E. Martinez-Morillo and E. P. Diamandis, Biochim. Biophys. Acta, 2015, 1854, 677–686 CrossRef CAS PubMed.
- D. Lebert, G. Picard, C. Beau-Larvor, L. Troncy, C. Lacheny, B. Maynadier, W. Low, N. Mouz, V. Brun, C. Klinguer-Hamour, M. Jaquinod and A. Beck, Bioanalysis, 2015, 1–15, DOI:10.4155/bio.15.56.
- V. Katta and B. T. Chait, J. Am. Chem. Soc., 1991, 113, 8534–8535 CrossRef CAS.
- B. Ganem, Y. T. Li and J. D. Henion, J. Am. Chem. Soc., 1991, 113, 6294–6296 CrossRef CAS.
- K. Benkestock, C. K. Van Pelt, T. Akerud, A. Sterling, P. O. Edlund and J. Roeraade, J. Biomol. Screening, 2003, 8, 247–256 CrossRef CAS PubMed.
- H. J. Maple, O. Scheibner, M. Baumert, M. Allen, R. J. Taylor, R. A. Garlish, M. Bromirski and R. J. Burnley, Rapid Commun. Mass Spectrom., 2014, 28, 1561–1568 CrossRef CAS PubMed.
- E. N. Kitova, A. El-Hawiet, P. D. Schnier and J. S. Klassen, J. Am. Soc. Mass Spectrom., 2012, 23, 431–441 CrossRef CAS PubMed.
- J. M. Daniel, G. McCombie, S. Wendt and R. Zenobi, J. Am. Soc. Mass Spectrom., 2003, 14, 442–448 CrossRef CAS.
- V. Vivat Hannah, C. Atmanene, D. Zeyer, A. Van Dorsselaer and S. Sanglier-Cianferani, Future Med. Chem., 2010, 2, 35–50 CrossRef PubMed.
- K. J. Pacholarz, R. A. Garlish, R. J. Taylor and P. E. Barran, Chem. Soc. Rev., 2012, 41, 4335–4355 RSC.
- C. Schmidt and C. V. Robinson, FEBS J., 2014, 281, 1950–1964 CrossRef CAS PubMed.
- C. Stehlin-Gaon, D. Willmann, D. Zeyer, S. Sanglier, A. Van Dorsselaer, J. P. Renaud, D. Moras and R. Schule, Nat. Struct. Biol., 2003, 10, 820–825 CrossRef CAS PubMed.
- C. Bovet, A. Wortmann, S. Eiler, F. Granger, M. Ruff, B. Gerrits, D. Moras and R. Zenobi, Protein Sci., 2007, 16, 938–946 CrossRef CAS PubMed.
- F. Bitsch, R. Aichholz, J. Kallen, S. Geisse, B. Fournier and J. M. Schlaeppi, Anal. Biochem., 2003, 323, 139–149 CrossRef CAS PubMed.
- B. J. Laventie, C. Potrich, C. Atmanene, M. Saleh, O. Joubert, G. Viero, C. Bachmeyer, V. Antonini, I. Mancini, S. Cianferani-Sanglier, D. Keller, D. A. Colin, T. Bourcier, G. Anderluh, A. van Dorsselaer, M. Dalla Serra and G. Prevost, Biochem. J., 2013, 450, 559–571 CrossRef CAS PubMed.
- S. E. Kolstoe, P. P. Mangione, V. Bellotti, G. W. Taylor, G. A. Tennent, S. Deroo, A. J. Morrison, A. J. Cobb, A. Coyne, M. G. McCammon, T. D. Warner, J. Mitchell, R. Gill, M. D. Smith, S. V. Ley, C. V. Robinson, S. P. Wood and M. B. Pepys, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20483–20488 CrossRef CAS PubMed.
- N. S. Hatzakis, Biophys. Chem., 2014, 186, 46–54 CrossRef CAS PubMed.
- K. Giles, J. P. Williams and I. Campuzano, Rapid Commun. Mass Spectrom., 2011, 25, 1559–1566 CrossRef CAS PubMed.
- Y. Zhong, S. J. Hyung and B. T. Ruotolo, Analyst, 2011, 136, 3534–3541 RSC.
- J. L. Benesch and B. T. Ruotolo, Curr. Opin. Struct. Biol., 2011, 21, 641–649 CrossRef CAS PubMed.
- S. J. Hyung and B. T. Ruotolo, Proteomics, 2012, 12, 1547–1564 CrossRef CAS PubMed.
- Y. Zhong, S. J. Hyung and B. T. Ruotolo, Expert Rev. Proteomics, 2012, 9, 47–58 CrossRef CAS PubMed.
- M. F. Jarrold, Phys. Chem. Chem. Phys., 2007, 9, 1659–1671 RSC.
- C. Atmanene, D. Chaix, Y. Bessin, N. Declerck, A. Van Dorsselaer and S. Sanglier-Cianferani, Anal. Chem., 2010, 82, 3597–3605 CrossRef CAS PubMed.
- C. Atmanene, S. Petiot-Becard, D. Zeyer, A. Van Dorsselaer, V. Vivat Hannah and S. Sanglier-Cianferani, Anal. Chem., 2012, 84, 4703–4710 CrossRef CAS PubMed.
- S. Niu, J. N. Rabuck and B. T. Ruotolo, Curr. Opin. Chem. Biol., 2013, 17, 809–817 CrossRef CAS PubMed.
- M. P. Nyon, T. Prentice, J. Day, J. Kirkpatrick, G. N. Sivalingam, G. Levy, I. Haq, J. A. Irving, D. A. Lomas, J. Christodoulou, B. Gooptu and K. Thalassinos, Protein Sci., 2015, 24, 1301–1312 CrossRef CAS PubMed.
- C. Giglione, M. Pierre and T. Meinnel, Mol. Microbiol., 2000, 36, 1197–1205 CrossRef CAS.
- C. T. Supuran, F. Carta and A. Scozzafava, Expert Opin. Ther. Pat., 2013, 23, 777–788 CrossRef CAS PubMed.
- Z. Q. Xu, M. T. Flavin and J. Flavin, Expert Opin. Invest. Drugs, 2014, 23, 163–182 CrossRef CAS PubMed.
- C. Giglione, A. Boularot and T. Meinnel, Cell. Mol. Life Sci., 2004, 61, 1455–1474 CrossRef CAS PubMed.
- C. Giglione, S. Fieulaine and T. Meinnel, Trends Biochem. Sci., 2009, 34, 417–426 CrossRef CAS PubMed.
- S. Ragusa, S. Blanquet and T. Meinnel, J. Mol. Biol., 1998, 280, 515–523 CrossRef CAS PubMed.
- C. Giglione, A. Serero, M. Pierre, B. Boisson and T. Meinnel, EMBO J., 2000, 19, 5916–5929 CrossRef CAS PubMed.
- A. Serero, C. Giglione and T. Meinnel, J. Mol. Biol., 2001, 314, 695–708 CrossRef CAS PubMed.
- S. Fieulaine, A. Boularot, I. Artaud, M. Desmadril, F. Dardel, T. Meinnel and C. Giglione, PLoS Biol., 2011, 9, e1001066 CAS.
- A. Boularot, C. Giglione, S. Petit, Y. Duroc, R. Alves de Sousa, V. Larue, T. Cresteil, F. Dardel, I. Artaud and T. Meinnel, J. Med. Chem., 2007, 50, 10–20 CrossRef CAS PubMed.
- D. E. Koshland, Proc. Natl. Acad. Sci. U. S. A., 1958, 44, 98–104 CrossRef CAS.
- H. Rogniaux, A. Van Dorsselaer, P. Barth, J. F. Biellmann, J. Barbanton, M. van Zandt, B. Chevrier, E. Howard, A. Mitschler, N. Potier, L. Urzhumtseva, D. Moras and A. Podjarny, J. Am. Soc. Mass Spectrom., 1999, 10, 635–647 CrossRef CAS.
- O. El-Kabbani, H. Rogniaux, P. Barth, R. P. Chung, E. V. Fletcher, A. Van Dorsselaer and A. Podjarny, Proteins, 2000, 41, 407–414 CrossRef CAS.
- J. M. Clements, R. P. Beckett, A. Brown, G. Catlin, M. Lobell, S. Palan, W. Thomas, M. Whittaker, S. Wood, S. Salama, P. J. Baker, H. F. Rodgers, V. Barynin, D. W. Rice and M. G. Hunter, Antimicrob. Agents Chemother., 2001, 45, 563–570 CrossRef CAS PubMed.
- J. P. Guilloteau, M. Mathieu, C. Giglione, V. Blanc, A. Dupuy, M. Chevrier, P. Gil, A. Famechon, T. Meinnel and V. Mikol, J. Mol. Biol., 2002, 320, 951–962 CrossRef CAS.
- D. E. Clemmer and M. F. Jarrold, J. Mass Spectrom., 1997, 32, 577–592 CrossRef CAS.
- C. A. Scarff, V. J. Patel, K. Thalassinos and J. H. Scrivens, J. Am. Soc. Mass Spectrom., 2009, 20, 625–631 CrossRef CAS PubMed.
- G. Vonhelden, M. T. Hsu, N. Gotts and M. T. Bowers, J. Phys. Chem., 1993, 97, 8182–8192 CrossRef CAS.
- T. Wyttenbach, M. Witt and M. T. Bowers, J. Am. Chem. Soc., 2000, 122, 3458–3464 CrossRef CAS.
- A. A. Shvartsburg, R. R. Hudgins, P. Dugourd and M. F. Jarrold, Chem. Soc. Rev., 2001, 30, 26–35 RSC.
- A. A. Shvartsburg, R. R. Hudgins, P. Dugourd and M. F. Jarrold, J. Phys. Chem. A, 1997, 101, 1684–1688 CrossRef CAS.
- C. Uetrecht, I. M. Barbu, G. K. Shoemaker, E. van Duijn and A. J. Heck, Nat. Chem., 2011, 3, 126–132 CrossRef CAS PubMed.
- C. Uetrecht, R. J. Rose, E. van Duijn, K. Lorenzen and A. J. Heck, Chem. Soc. Rev., 2010, 39, 1633–1655 RSC.
- J. T. Hopper and N. J. Oldham, J. Am. Soc. Mass Spectrom., 2009, 20, 1851–1858 CrossRef CAS PubMed.
- S. J. Hyung, C. V. Robinson and B. T. Ruotolo, Chem. Biol., 2009, 16, 382–390 CrossRef CAS PubMed.
- J. N. Rabuck, S. J. Hyung, K. S. Ko, C. C. Fox, M. B. Soellner and B. T. Ruotolo, Anal. Chem., 2013, 85, 6995–7002 CrossRef CAS PubMed.
- F. Debaene, E. Wagner-Rousset, O. Colas, D. Ayoub, N. Corvaia, A. Van Dorsselaer, A. Beck and S. Cianferani, Anal. Chem., 2013, 85, 9785–9792 CrossRef CAS PubMed.
- B. T. Ruotolo, J. L. Benesch, A. M. Sandercock, S. J. Hyung and C. V. Robinson, Nat. Protoc., 2008, 3, 1139–1152 CrossRef CAS PubMed.
- M. F. Bush, Z. Hall, K. Giles, J. Hoyes, C. V. Robinson and B. T. Ruotolo, Anal. Chem., 2010, 82, 9557–9565 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS.
- A. A. Shvartsburg and M. F. Jarrold, Chem. Phys. Lett., 1996, 261, 86–91 CrossRef CAS.
- M. F. Mesleh, J. M. Hunter, A. A. Shvartsburg, G. C. Schatz and M. F. Jarrold, J. Phys. Chem., 1996, 100, 16082–16086 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5an01311a |
| This journal is © The Royal Society of Chemistry 2015 |