
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insights into the consequences of co-polymerisation in the early stages of IAPP and Aβ peptide assembly from mass spectrometry
Lydia M.
Young
a,
Rachel A.
Mahood
a,
Janet C.
Saunders
a,
Ling-Hsien
Tu
b,
Daniel P.
Raleigh
bc,
Sheena E.
Radford
*a and
Alison E.
Ashcroft
*a
aAstbury Centre for Structural Molecular Biology, School of Molecular and Cellular Biology, University of Leeds, LS2 9JT, UK. E-mail: s.e.radford@leeds.ac.uk; a.e.ashcroft@leeds.ac.uk
bDepartment of Chemistry, Stony Brook University, Stony Brook, New York 11794-3400, USA
cResearch Department of Structural and Molecule Biology, University College London, Gower Street, London, WC1E 6BT, UK
First published on 14th July 2015
Abstract
The precise molecular mechanisms by which different peptides and proteins assemble into highly ordered amyloid deposits remain elusive. The fibrillation of human amylin (also known as islet amyloid polypeptide, hIAPP) and the amyloid-beta peptide (Aβ-40) are thought to be pathogenic factors in Type 2 diabetes mellitus (T2DM) and Alzheimer's disease (AD), respectively. Amyloid diseases may involve co-aggregation of different protein species, in addition to the self-assembly of single precursor sequences. Here we investigate the formation of heterogeneous pre-fibrillar, oligomeric species produced by the co-incubation of hIAPP and Aβ-40 using electrospray ionisation-ion mobility spectrometry-mass spectrometry (ESI-IMS-MS)-based methods. Conformational properties and gas-phase stabilities of amyloid oligomers formed from hIAPP or Aβ40 alone, and from a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of hIAPP and Aβ40 monomers, were determined and compared. We show that co-assembly of the two sequences results in hetero-oligomers with distinct properties and aggregation kinetics properties compared with the homo-oligomers present in solution. The observations may be of key significance to unravelling the mechanisms of amyloid formation in vivo and elucidating how different sequences and/or assembly conditions can result in different fibril structures and/or pathogenic outcomes.
:1 mixture of hIAPP and Aβ40 monomers, were determined and compared. We show that co-assembly of the two sequences results in hetero-oligomers with distinct properties and aggregation kinetics properties compared with the homo-oligomers present in solution. The observations may be of key significance to unravelling the mechanisms of amyloid formation in vivo and elucidating how different sequences and/or assembly conditions can result in different fibril structures and/or pathogenic outcomes.
Introduction
Amyloid formation by proteins and peptides comprised of diverse sequences and folds contributes to more than 50 human disorders.1 Two well-known examples are the 40- and 42-residue amyloid-β peptides (Aβ-40 and Aβ-42) and the 37-residue human islet amyloid polypeptide (hIAPP). These amyloid sequences are associated with Alzheimer's disease (AD) and Type 2 diabetes mellitus (T2DM), respectively.1 AD is the major age-dependent neurodegenerative disease and the leading cause of dementia, characterised by progressive memory deficit and neuronal loss.1,2 T2DM is a complex, chronic metabolic disorder characterised by hyperglycemia and is associated with macrovascular and microvascular complications.1,3,4 Extracellular neuronal amyloid plaques formed in AD consist mainly of aggregated Aβ peptide, whereas in T2DM extracellular pancreatic islet amyloid deposits are comprised mainly of aggregated hIAPP. The formation of islet amyloid by hIAPP is a key factor contributing to the loss of β-cell function in T2DM and to the failure of islet transplants.4–7Both the Aβ and hIAPP peptide sequences contain hydrophobic regions with a high tendency to self-associate under a wide range of conditions. hIAPP and Aβ40 exhibit an overall 25% amino acid identity and 47% similarity (Fig. 1), with critical regions, Aβ40 (26–32) and hIAPP (20–29) believed to be involved in the self-assembly of each peptide,8–12 being most similar. Several epidemiological studies have suggested a link between AD and T2DM,13,14 with T2DM patients reported to have a two-to three-fold increased risk for AD.15
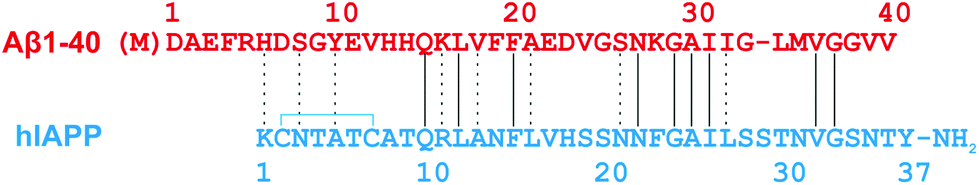 | ||
| Fig. 1 Sequence alignment of Aβ40 and hIAPP. Recombinant expression of Aβ40 results in an additional N-terminal methionine.16 The intramolecular disulfide bond in hIAPP is indicated by a blue line, and the amidated C-terminal is shown. Lines between the hIAPP and Aβ40 sequences indicate exact amino acid matches; dashes indicate chemical similarity. | ||
Although amyloid fibrils formed in vitro are commonly assembled from a single protein sequence, fibrils formed in vivo can contain more than one protein. Emerging evidence suggests that in addition to homo-polymerisation, cross-sequence interactions may also play a role in aggregation and pathogenicity.2,17–28 Reported examples of cross-amyloid interactions involving Aβ or hIAPP include the Aβ40–Aβ42,29 Aβ–tau,23 Aβ–α-synuclein,30,31 Aβ–transthyretin,20 Aβ–hIAPP,21 hIAPP–ratIAPP,19,32 hIAPP–partially processed pro-IAPP,33 and IAPP–insulin interactions.24,26,34,35
The Aβ40–hIAPP interaction, identified in vitro, has been suggested to have low nanomolar-affinity and to occur between pre-fibrillar Aβ40 and hIAPP species.2,26,36 The observed copolymerisation has been proposed to suppress cytotoxic homo-polymerisation and amyloidogenesis by both Aβ40 and hIAPP.26 In another study, two regions of Aβ40 (residues 11–21 and 23–37) with high binding affinity for hIAPP, and two analogous regions of hIAPP (residues 8–20 and 21–37) with corresponding affinity for Aβ40, have been identified.36
In this study we utilise electrospray ionisation-ion mobility spectrometry-mass spectrometry (ESI-IMS-MS) to examine the similarities and differences between the oligomers formed from Aβ40, hIAPP and a 1:1 mixture of Aβ40:hIAPP. ESI-IMS-MS has the unique capability of resolving complex, heterogeneous mixtures of species present in solution, including transiently populated states of intrinsically disordered proteins, without requiring their prior separation. By isolating individual ions within the mass spectrometer, information about mass, shape (collision cross-sectional area (CCS)), stability (using collision induced unfolding (CIU), collision induced dissociation (CID), surface induced dissociation (SID) and hydrogen-deuterium exchange (HDX)) and kinetics (using real time or subunit exchange experiments) can be obtained.37–40 Non-covalent assemblies, such as virus capsid intermediates,41 amyloid intermediates42 and membrane proteins,43,44 in addition to other protein assemblies, can be studied in detail using ESI-IMS-MS.
Here we use ESI-IMS-MS to observe and characterise hetero-oligomeric species containing monomer units of both Aβ40 and hIAPP formed early in amyloid assembly of these proteins. We compare the conformations, dynamics and relative gas-phase stabilities of the hetero- and homo-oligomers observed. The results reveal that unique oligomer conformations are formed as a consequence of co-polymerisation that have distinct stability and form amyloid at different rates compared with oligomers arising from a single peptide precursor. The findings highlight the further diversification of possible amyloid conformations that result from co-assembly of different disease-related amyloidogenic sequences.
Methods
hIAPP and Aβ40 preparation
hIAPP was synthesised using Fmoc chemistry, oxidised using dimethyl sulfoxide (DMSO) to form the disulfide bond linking residues Cys 2–Cys 7, and purified via HPLC.45 Hydrochloric acid was used as the counter ion in all HPLC purification steps.45 Aβ40 (containing an additional N-terminal methionine not present in wild-type Aβ40, produced by the cleavage of amyloid precursor protein), was expressed recombinantly in E. coli and purified as described elsewhere.35 The final stages of purification involved size exclusion chromatography (Superdex™ 75 GL 10/300 column, GE Healthcare, UK) with a volatile mobile phase (50 mM ammonium bicarbonate, pH 7.8) and peptide-containing fractions were lyophilised.Lyophilised hIAPP samples were dissolved in dimethyl sulfoxide (DMSO) at a final peptide concentration of 3.2 mM. After 24 h at 25 °C, stock solutions were diluted 100-fold into 200 mM ammonium acetate, pH 6.8, to a final peptide concentration of 32 μM for MS analysis. The final concentration of DMSO was 1% (v/v). Lyophilised Aβ40 was resolubilised in DMSO at 3.2 mM and diluted into 200 mM ammonium acetate, pH 6.8, 1% (v/v) DMSO at a final peptide concentration of 32 μM. The sample was centrifuged at 13000g (4 °C, 10 min) prior to MS analysis to remove any insoluble aggregates that may have formed. All samples were prepared in 96-well plates (Corning Costar 3915, Corning Life Sciences, Amsterdam, The Netherlands) at 25 °C without agitation, for infusion into the mass spectrometer via a Triversa NanoMate (Advion Biosciences, Ithaca, NY, USA). The mixed sample was treated in the same manner as the Aβ40 sample.
ESI-IMS-MS
A Synapt HDMS quadrupole-time-of-flight mass spectrometer (Waters Corpn., Manchester, UK), equipped with a Triversa NanoMate (Advion Biosciences, Ithaca, NY, USA) automated nano-ESI interface, was used for these analyses. The mass spectrometer, described in detail elsewhere46 has a travelling-wave IMS device situated between the quadrupole and the time-of-flight analysers.hIAPP and Aβ40 samples were analysed using positive mode nanoESI with a capillary voltage of 1.7 kV and a nitrogen nebulising gas pressure of 0.8 psi. The following instrumental parameters were used: cone voltage 30 V; source temperature 60 °C; backing pressure 1.6 mBar; ramped travelling wave height 7–20 V; travelling wave speed 300 m s−1; IMS cell pressure 0.55 mBar. Data were acquired over the range m/z 500–6000. Mass calibration was achieved using caesium iodide solution, prepared by dissolving the compound in 50% (v/v) water/isopropanol to a concentration of 2 mg mL−1. CCS measurements were estimated by use of a calibration obtained by analysis of denatured proteins (cytochrome c, ubiquitin, lysozyme) and peptides (tryptic digests of alcohol dehydrogenase (ADH) and cytochrome c) with known CCSs obtained elsewhere from drift tube ion mobility measurements.47,48
Collision Induced Dissociation (CID)-MS/MS experiments were performed using the quadrupole analyzer to select isobaric m/z ions representing the dimer 5+ ions and performing CID in the trap collision cell prior to the IMS device and time-of-flight analyzer. Increasing collision energy was applied to the trap collision cell in 10 V increments from 10–100 V until the oligomer ions were completely dissociated into monomer ions.
Data were processed by use of MassLynx v4.1 and Driftscope v2.4 software supplied with the mass spectrometer.
Fibril depolymerisation
A mixed sample containing a 1:1 molar ratio of hIAPP:Aβ40 was prepared by diluting 3.2 mM stock solutions of each peptide in DMSO, 100-fold, into 200 mM ammonium acetate, pH 6.8, to give a final concentration of each peptide of 16 μM in 1% (v/v) DMSO. After 7.5 h or 5 days of incubation at 25 °C without agitation in low binding tubes (Maxymum Recovery® TM tubes, Axygen, Corning Life Sciences, Amsterdam, The Netherlands), mixed samples were centrifuged in a Beckman ultracentrifuge at 300000g for 45 min. Fibrillar samples in the pellet were depolymerised by incubation in 100% (v/v) HFIP for 24 h. Samples were air-dried and then redissolved in 50:40:10 (v/v/v) acetonitrile/water/acetic acid, and fibril constituent peptides were identified by ESI-MS.
Thioflavin T fluorescence (ThT) assays
Samples were prepared in clear-bottomed, low volume, non-binding 96-well microplates (Corning Costar NBS™ 3881, Corning Life Sciences, Amsterdam, The Netherlands) sealed with clear sealing film and were incubated in a FLUOstar OPTIMA plate reader (BMG Labtech, Aylesbury, Bucks, UK) for 5 days at 25 °C, without agitation. Samples had a volume of 100 μL containing 100 μM ThT and 32 μM peptide in 200 mM ammonium acetate, pH 6.8 and a 1% (v/v) final concentration of DMSO. Fluorescence was excited at 440 nm and emission intensity was measured at 485 nm.Transmission electron microscopy (TEM)
The TEM images of each peptide solution were acquired after 5 days incubation at 25 °C in low binding tubes (Maxymum Recovery® TM tubes, Axygen, Corning Life Sciences, Amsterdam, The Netherlands), using a JEM-1400 (JEOL Ltd, Tokyo, Japan) transmission electron microscope. Carbon grids were prepared by irradiation under UV light for 30 min and stained with 4% (w/v) uranyl acetate solution as described previously.49Results
Formation of homo oligomers and fibrils from hIAPP and Aβ40
ESI-IMS-MS has been used in previous studies to probe the monomeric and oligomeric populations originating from Aβ40 and hIAPP.6,10,19,35,50–53 Consistent with these data, the ESI mass spectra of hIAPP (Fig. 2a i.) and Aβ40 (Fig. 2b i.) show dominant 2+/3+ (hIAPP) and 2+/3+/4+ (Aβ40) monomer charge state ions, respectively. For both peptides, small oligomeric components are also observed in the ESI-mass spectra. ESI-IMS-MS enables detection of hIAPP and Aβ40 dimer through to hexamer/pentamer inclusively, as observed in the Driftscope plots (Fig. 2a ii. and b ii., respectively).10,19 hIAPP and Aβ40 oligomers appear, and subsequently disappear, as aggregation proceeds (data not shown),19,35 resulting ultimately in the formation of long straight amyloid fibrils, as observed by TEM (Fig. 2a iii. and b iii).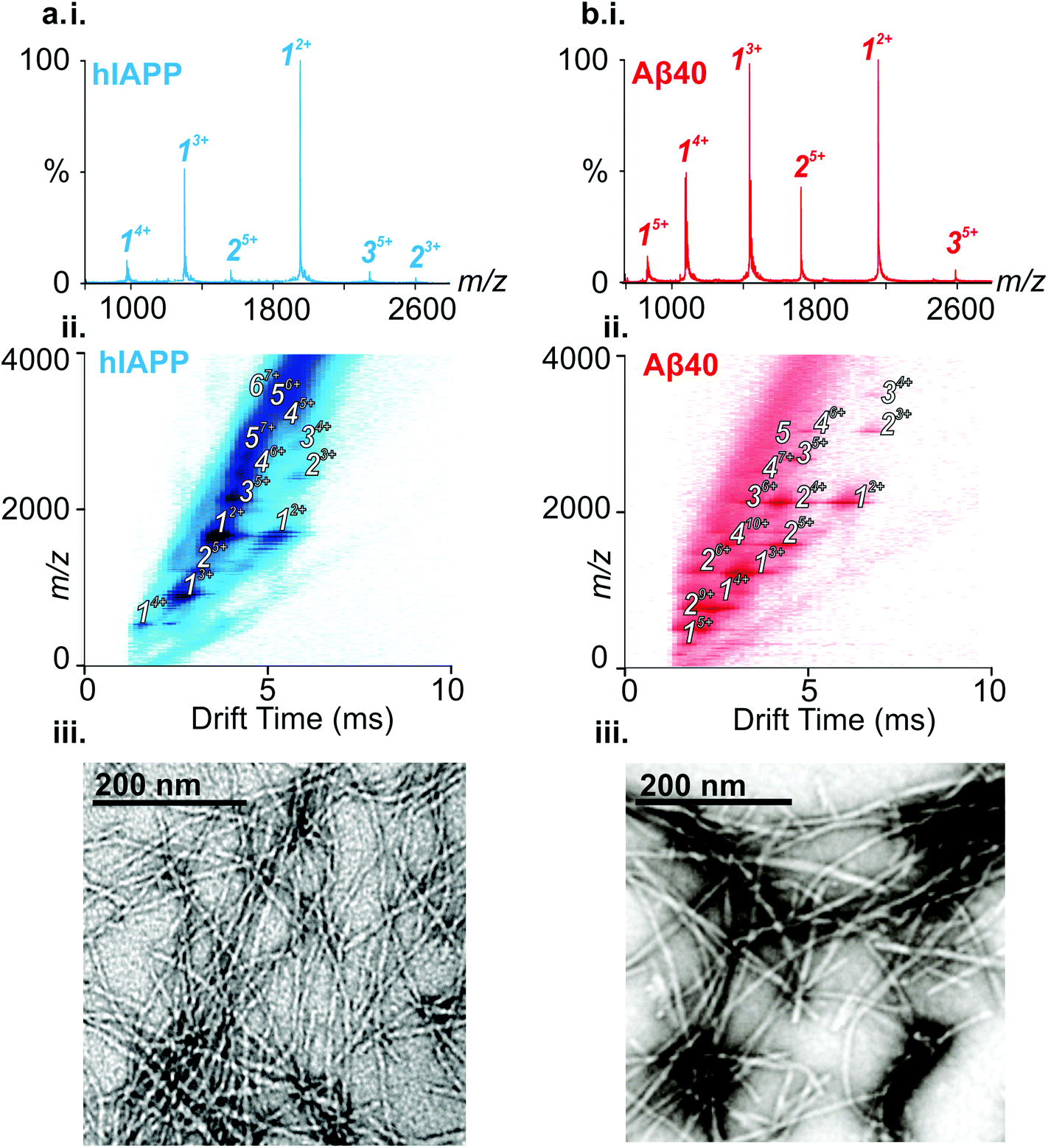 | ||
| Fig. 2 hIAPP and Aβ40 occupy monomer through higher order oligomer species en route to long-straight fibrils. (a) (i) ESI-MS mass spectrum of hIAPP. Numbers above peaks denote oligomer order, with the positive charge state of ions in superscript. (ii) ESI-IMS-MS Driftscope plot of hIAPP, showing monomer (1) through hexamer (6), present 2 min after diluting the monomer to a final peptide concentration of 32 μM in 200 mM ammonium acetate, pH 6.8, 1% DMSO (v/v). ESI-IMS-MS Driftscope plots show IMS drift time versus m/z versus intensity (z = square root scale). (iii) Negative stain TEM image of hIAPP fibrils after 5 days in 200 mM ammonium acetate buffer, pH 6.8, 1% DMSO (v/v) (25 °C, quiescent) (scale bar = 200 nm). (b) (i) ESI-MS mass spectrum of Aβ40. (ii) ESI-IMS-MS Driftscope plot of Aβ40, showing monomer (1) through pentamer (5), present 12 min after diluting the peptide to a monomer concentration of 32 μM in 200 mM ammonium acetate, pH 6.8. (iii) Negative stain TEM image of Aβ40 fibrils after 5 days in 200 mM ammonium acetate buffer, pH 6.8, 1% DMSO (v/v) (25 °C, quiescent) (scale bar = 200 nm). | ||
Formation of hetero-oligomers and hetero-fibrils from hIAPP and Aβ40
We next used ESI-IMS-MS to study the monomeric and oligomeric states present in a mixture (1:1 molar ratio) of hIAPP and Aβ40. The ESI-mass spectrum of a 1:1 mixture of hIAPP:Aβ40 (Fig. 3a) shows hIAPP and Aβ40 monomers and homo-oligomers occupying similar charge state distributions as those observed for each peptide when incubated in isolation (dominant monomer 2+/3+ and 2+/3+/4+ for hIAPP and Aβ40, respectively; Fig. 2 and 3). In addition to homo-oligomeric peaks (5+ dimers and 5+ trimers for both peptides), new peaks appear in the ESI-MS spectrum, with mass values corresponding to a mixture of hIAPP and Aβ40 monomer sub-units. These unique assemblies represent 1:1 hIAPP:Aβ40 dimer 5+ and 4+ ions as well as 2:1 and 1:2 hIAPP:Aβ40 trimer 5+ ions. Peak intensities observed for mixed assemblies are suggestive of a random mixing of the two sequences, i.e. three peaks are observed for dimer 5+ ions in a 1:2:1 ratio corresponding to hIAPP:hIAPP, hIAPP:Aβ40 and Aβ40:Aβ40 dimers. The Driftscope plot (Fig. 3b) gives a visual representation of the heterogeneous ensemble of homo- and hetero-oligomers formed in the mixed sample, each species having a unique drift time thus enabling CCS values to be assigned to every oligomer present via the use of a calibration approach.47,48 The observed homo-oligomers of hIAPP and Aβ40 and the mixed hetero-oligomers appear and subsequently disappear as protein aggregation proceeds. The lifetime of the oligomers corresponds with the lag time of fibril assembly (Fig. 4a), after which oligomers are no longer observed (data not shown). This is likely due to the decreased oligomer concentration as these species are incorporated into fibrils and/or that the large aggregates present within the sample perturb spraying and infusion into the mass spectrometer. Although higher order species disappear concomitantly with fibril formation, the CCS values and the charge states of the oligomers observed from any of the samples did not vary significantly over the time course of the experiment (data not shown).
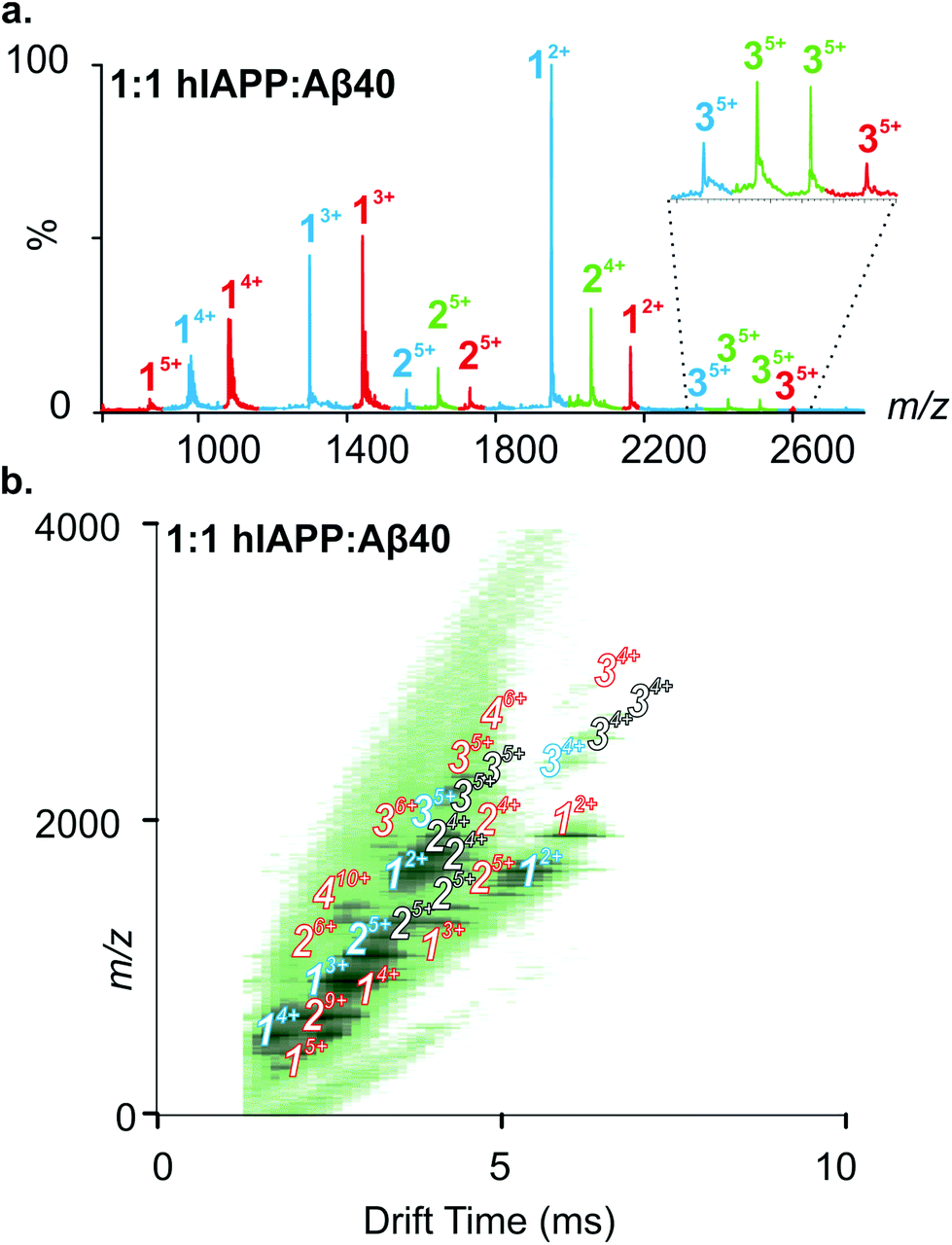 | ||
| Fig. 3 A 1:1 mixture of hIAPP and Aβ40 results in the population of homo- and hetero-oligomers. (a) ESI-MS mass spectrum of 1:1 hIAPP:Aβ40. Numbers above peaks denote oligomer order, with the positive charge state of ions in superscript. Peaks coloured red represent Aβ40-alone species, peaks coloured blue represent hIAPP-alone species and peaks coloured green represent hetero-oligomers with m/z corresponding to a mixture of hIAPP and Aβ40 monomer units. A zoom of m/z 2300–2650 is inset to highlight the presence of trimer 5+ ions. Two unique hetero-trimers are observed, one at a lower m/z corresponding to 2hIAPP:1Aβ40 trimer 5+ ions and one at a higher m/z corresponding to 1hIAPP:2Aβ40 trimer 5+ ions. (b) ESI-IMS-MS Driftscope plot of the 1:1 hIAPP:Aβ40 aggregation mixture showing all-hIAPP species (blue) monomer through trimer, all-Aβ40 species (red) monomer through tetramer, and hIAPP-Aβ40 mixed species (white) dimer through trimer. The species shown are present 12 min after diluting the monomers to a final total peptide concentration of 32 μM in 200 mM ammonium acetate, pH 6.8. | ||
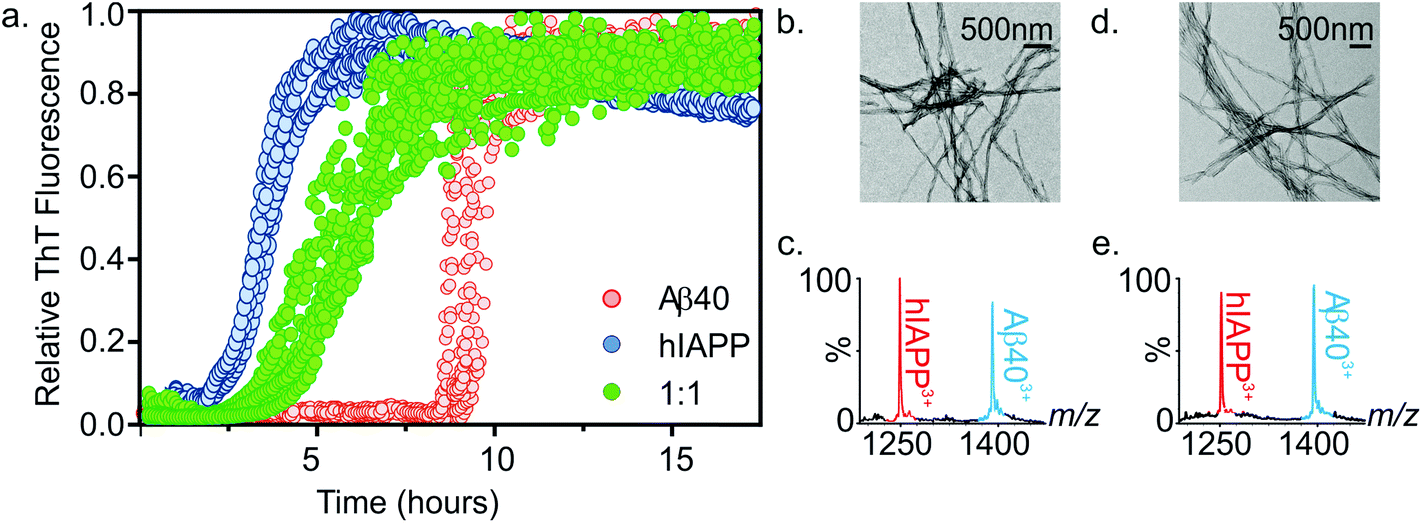 | ||
| Fig. 4 (a) ThT fluorescence intensity of hIAPP (blue), Aβ40 (red) and a 1:1 mixture of hIAPP:Aβ40 (green) indicating the growth of fibrils over time. All samples contained a final peptide concentration of 32 μM in 200 mM ammonium acetate buffer, pH 6.8, 1% DMSO. Fibril kinetics were monitored over a 17.5 h period (25 °C, quiescent). Note that final amplitudes of ThT signal were similar prior to normalization. (b) Negative stain TEM image of fibrils formed from a 1:1 mixture of hIAPP:Aβ40 after 7.5 h in 200 mM ammonium acetate buffer, pH 6.8 (25 °C, quiescent) (scale bar = 500 nm). (c) ESI mass spectrum of depolymerised fibrils at 7.5 h showing the presence of both hIAPP and Aβ40 monomer constituents in the pellet. (d) and (e) as (b) and (c) but analyzed after 5 days. | ||
hIAPP and Aβ40 co-assemble at an intermediate rate compared with the individual assembly rates
The kinetics of fibril formation when each peptide was present in a 1:1 mixture was compared with the rate of fibril formation of each peptide in isolation. Thioflavin T (ThT), a benzothiazole dye which displays an enhanced fluorescence upon non-covalent binding to amyloid fibrils,54,55 was used to monitor fibrillation kinetics in real-time. Under the conditions employed, both peptides form fibrils on rapid time scales (Fig. 4a). Consistent with previous studies that report hIAPP to be the more amyloidogenic of the two sequences,21,35 hIAPP exhibits a lag-phase of ∼2 hours while that of Aβ40 is ∼9 hours. When incubated together, co-polymerisation occurs, resulting in the formation of long, straight fibrils (Fig. 4b), with a lag-phase of ∼3.5 hours, intermediate between the lag-phases of each peptide in isolation. Strikingly, a single transition was observed in the mixed sample consistent with co-aggregation of the two sequences, with no evidence for independent assembly of either peptide. Both peptides form fibrillar aggregates in the mixture, with both hIAPP and Aβ40 monomer subunits present in the aggregate pellet obtained after 7.5 h (Fig. 4a and c), as analysed by ESI-MS after centrifugation and depolymerisation (see Methods) (Fig. 4c); TEM images reveal amyloid fibrils, but no amorphous aggregates (Fig. 4b). Note that Aβ40 does not form significant quantities of fibrils at this time when incubated alone. Similarly, both peptides are found in the pellet after 5 days incubation (Fig. 4e). These experiments do not report on the extent to which mixing occurs within each amyloid fibril. It is clear from these data that the presence of the more highly aggregation-prone sequence (hIAPP) has a profound effect on the rate of the less aggregation-prone sequence (Aβ40), the mixed sample having a lag-phase close to, but distinguishable from, that of hIAPP alone. This is interesting, given that previous reports have shown that Aβ40 fibrils will cross-seed hIAPP monomer, but hIAPP fibrils are inefficient at cross-seeding Aβ40 monomer.21 It is important to distinguish between the phenomena of cross-seeding and co-polymerisation: hetero-amyloid assemblies composed of multiple species can arise either through co-polymerisation of two pools of monomer as shown here or by cross-seeding, in which existing fibrils (seeds) of one species catalyse fibril formation from monomers of a different sequence. The discrepancy between our data and previously published cross-seeding experiments indicates that the determinants of cross-seeding and co-polymerisation of these two sequences are distinct.
ESI-IMS-MS reveals conformations of hetero-oligomers
Ion mobility arrival time distributions (ATDs) for individual charge state ions of each peptide species were extracted from the three-dimensional ESI-IMS-MS datasets. Fig. 5 shows a comparison of the ATDs of homo-oligomers of hIAPP and Aβ40 and their corresponding hetero-oligomers. Dimer 5+ ions are significantly populated by both hIAPP and Aβ40 alone (Fig. 2) and in the 1:1 mixture of hIAPP:Aβ40 (Fig. 3). For the hIAPP:hIAPP dimer 5+ ions (Fig. 5 a i.), a single peak is observed with a drift time of 3.2 ms, corresponding to a CCS of ∼1170 Å2. Similarly, for the Aβ40:Aβ40 dimer 5+ ions (Fig. 5 a ii.) a single peak is observed with a drift time of 4.2 ms, corresponding to a CCS of ∼1330 Å2. The ∼14% difference in the CCS of the two dimer 5+ ions is expected, given that there is a ∼14% difference in the molecular weight of each sequence and the fact that Aβ40 is three amino acid residues longer than hIAPP. There is less of a difference (∼5%) between the measured CCS values for the Aβ40:Aβ40:Aβ40 trimer 5+ and the hIAPP:hIAPP:hIAPP trimer 5+ ions (1470 and 1400 Å2, respectively). This could be explained by a conformational change occurring that results in relative compaction of the trimer compared with the dimer in both systems. Alternatively, the larger difference in CCS between homo-dimers of either peptide could result from a greater degree of Coulombic repulsion in the dimer with five charges, relative to a trimer with five charges. A similar observation can be seen for homo-assemblies, particularly that of Aβ40, with the trimer 5+ ions occupying narrower CCS ranges with respect to dimer 5+ ions (Fig. 5a ii. and b ii.).
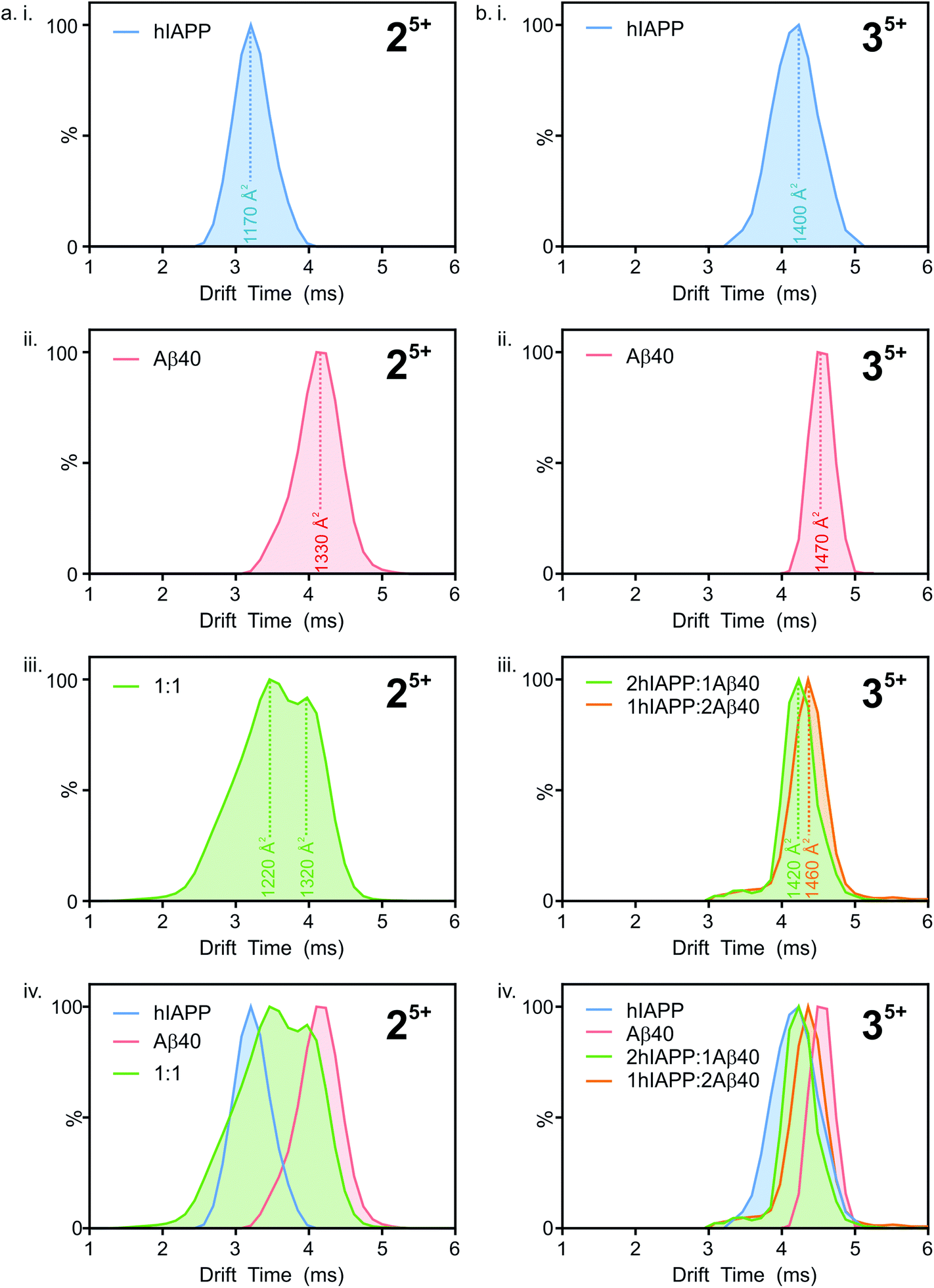 | ||
| Fig. 5 Extracted arrival time distributions (ATDs) for the most abundant oligomeric charge state ions (dimer 5+ and trimer 5+) observed within the ESI-IMS-MS spectra for hIAPP (blue), Aβ40 (red) and ions corresponding to heterodimers from a 1:1 mixture of hIAPP:Aβ40 (green). Estimated cross-sections are shown, with the drift times taken from the apex of the ATDs. (a) ATDs of dimer 5+ ions from hIAPP (i), Aβ40 (ii), 1:1 hIAPP:Aβ40 heterodimers (iii) and all ATDs shown overlaid (iv). (b) ATDs of trimer 5+ ions from hIAPP (i), Aβ40 (ii), heterotrimers from a 1:1 mixture of hIAPP:Aβ40 (iii) and all ATDs shown overlaid (iv). | ||
ATD profiles that deviate slightly from Gaussian are observed for both peptides and could be indicative of the presence of multiple conformers that are rapidly interconverting on the ESI-IMS-MS timescale. For the purposes of this study, however, we focus on the major conformers present that give distinguishable peaks in the extracted ATDs. In the mixed sample, the hIAPP:Aβ40 dimer 5+ (Fig. 5a iii.) exhibits two distinguishable conformers at 3.5 and 4.0 ms. These drift times correspond to mixed dimers of CCS ∼1220 and 1320 Å2, respectively. The drift times of the two major conformers populated by the mixed dimer 5+ ions are distinct from those of each of the homo-dimers. One has a drift time maximum which is closer to that of the Aβ dimer, while the other is closer to the hIAPP dimer. The results observed are intriguing given that if the two peptides were to mix to form a dimer with a unique conformation, a single peak with a drift time intermediate between that of each homo-oligomer may result. Alternatively a unique species with a different conformation and hence different CCS would result. Instead, the mixed species partition into two populations of dimers which are distinct from either parent homo-dimer. Neither of the hetero-dimer conformers has a drift time (peak top) which is identical to that of either homo-oligomer, suggesting that species with unique conformations are populated in the mixed sample. Given the width of the ATDs, however, it is likely that within the dynamic ensemble of hetero-dimer species populated, some ions may have drift times, and hence conformational properties, similar to those of the homo-dimers.
A similar observation can be made for the trimer 5+ ions from the hIAPP, Aβ40 and mixed samples. hIAPP and Aβ40 homo-trimer 5+ ions populate ions with drift times 4.1 ms and 4.5 ms corresponding to CCSs of ∼1400 and ∼1470 Å2, respectively (Fig. 4b i. and ii.). Two unique hetero-trimers exist, comprised of 2:1 and 1:2 hIAPP:Aβ40 monomer subunits. These ions each appear to populate single conformations (Fig. 5 b iii.) with calculated CCSs of ∼1420 and ∼1460 Å2, respectively. The conformations of the mixed trimer ions (in CCS) are in between the conformations of the hIAPP homo-trimer and the Aβ40 homo-trimer (Fig. 5b iv.).
The mixed sample, therefore, is comprised of homo-oligomers of hIAPP, homo-oligomers of Aβ40, hetero-oligomers of hIAPP:Aβ40 (that contain species that are hIAPP-like in CCS) and hetero-oligomers of hIAPP:Aβ40 (that contain species that are Aβ40-like in CCS). The homo-oligomers formed in the mixed sample are indistinguishable in CCS from the homo-oligomers formed when each peptide is incubated in isolation, suggesting that the presence of hetero-dimers does not alter the structure of homo-dimers.
In summary, the results presented demonstrate that the mixed oligomers observed here are capable of populating conformational states similar to, but unique from, those occupied by each of their constituent peptides when incubated in isolation.
CID-MS/MS reveals differences in gas-phase stability between hIAPP, Aβ40 and mixed oligomers
Next, the gas-phase stabilities of the different oligomers of hIAPP and Aβ40 were probed to investigate whether their susceptibility to gas-phase dissociation can be related to their different rates of amyloid formation and can help to rationalise the consequences of mixing the peptides. Given that the oligomers observed by ESI-IMS-MS are not only lowly populated (<10% of total ion intensity), but also co-populated with each other and the monomeric species, they are not amenable to solution-phase stability assays. Although solution-phase stability cannot be directly inferred from gas-phase stability,56,57 the latter has been implemented successfully in the interrogation of the stability of both protein/ligand58 and protein/protein complexes.17,59 Accordingly, CID-MS/MS was utilised to provide a direct comparison of the gas-phase stabilities of the dimers formed from the two peptides in isolation and within a mixture. In this experiment, ions of specific m/z were selected by the quadrupole analyser and fragmented in the trap collision cell immediately prior to IMS separation and time-of-flight analysis. The data revealed that hIAPP homo-dimers dissociate at lower collision voltages than Aβ40 homo-dimers in the gas-phase. hIAPP dimers (5+ charge state ions) were found to be fully dissociated to two monomer ions (2+ and 3+ charge states) at a lower energy (10 V) than that required to dissociate the equivalent Aβ40 dimers: the Aβ40 homo-dimers begin to dissociate at 20 V but can be observed as intact dimers until a voltage of 50 V is applied (Fig. 6a i. and ii.). This difference in gas-phase stability of the dimer 5+ ions of hIAPP and Aβ40 may be related to the difference in fibrillation kinetics observed for the two sequences (Fig. 4) i.e. hIAPP dissociates more readily and therefore may be able to access amyloidogenic conformations more readily than Aβ40. This proposition relies on the assumption that gas-phase stability is related to the ability of the protein to dissociate and/or unfold and re-form/assemble into higher-order, “on-pathway” species. If, on the contrary, this dimer was an “on-pathway” species, we would expect decreased stability to result in slower relative aggregation rates given that dissociation would impede amyloid assembly.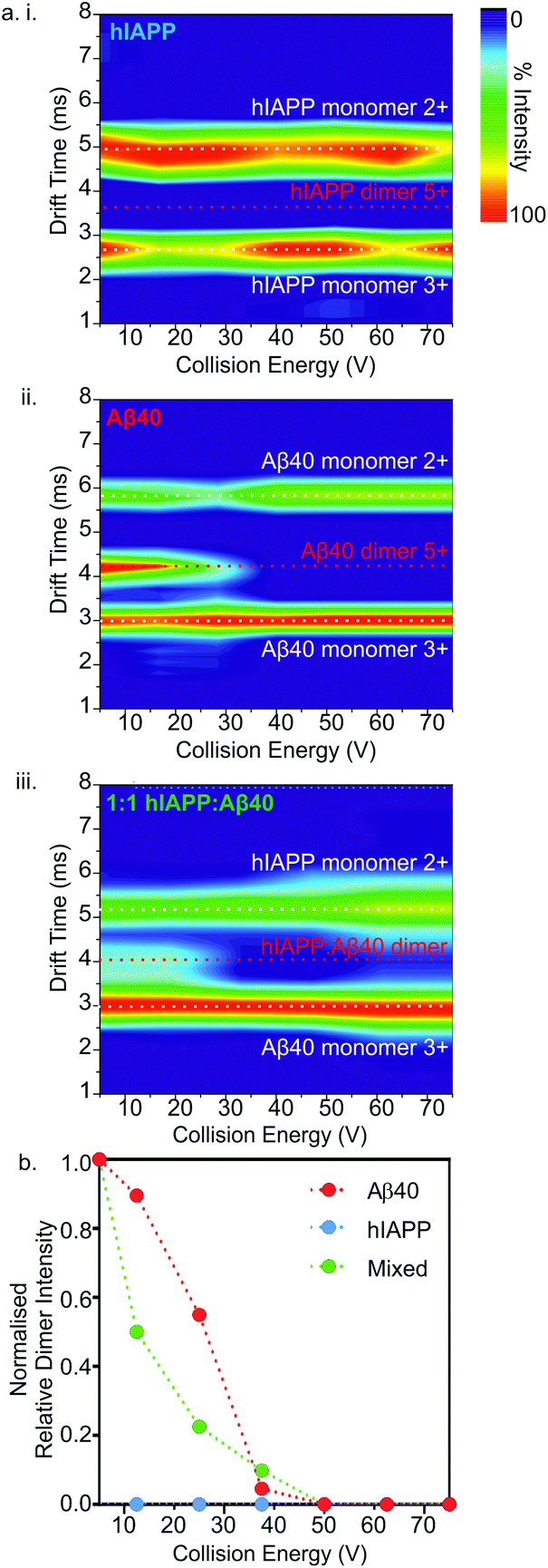 | ||
| Fig. 6 Collision induced dissociation (CID)-MS/MS of homo- and hetero-dimers. (a) CID heat maps showing relative peak intensities of mass-selected dimer 5+ ions and dissociated monomers upon activation for hIAPP alone (i), Aβ40 alone (ii) and a 1:1 mixture of hIAPP:Aβ40 (n.b. the predominant monomer charge state ions of each peptide that dissociates from the hetero-dimers are shown) (iii). (b) CID-MS/MS of hIAPP (blue), Aβ40 (red) and hIAPP:Aβ40 (green) dimers (32 μM peptide, 200 mM ammonium acetate buffer, pH 6.8). Intensity of the 5+ dimer ions of each sample, relative to total ion intensity in the spectrum, is plotted versus increasing ion-accelerating voltage into the trap T-wave collision cell), normalized to the ion intensity at 5 V collision energy. | ||
Interestingly, the mixed dimer 5+ ions, formed from hIAPP and Aβ40 monomer subunits, exhibited a gas-phase stability intermediate between those of homo-oligomers of hIAPP and Aβ40 of the same order, being less stable than Aβ40 oligomers, but more stable than hIAPP oligomers (Fig. 6a iii.) and giving rise primarily to Aβ40 monomer (3+ charge state ions) and hIAPP monomer (2+ charge state ions). Ions corresponding to homo- and hetero-trimers were not observed with sufficient intensity to perform stability analysis by MS/MS with confidence. The intermediate stability of hetero-dimers is consistent with the mixed assemblies occupying new conformations measured by their CCS (Fig. 5) and forming fibrils with aggregation kinetics different from both of their parent sequences (Fig. 4a). Taken together, the data suggest that MS-based methods can provide direct insights into the conformational properties of oligomers during fibrillation that can be related to the rate of aggregation.
Discussion
Here we have investigated the effects of co-incubating two disease-related amyloidogenic sequences on the heterogeneous array of oligomeric structures which assemble during fibril formation. Conformational properties and gas-phase stabilities of amyloid oligomers formed from hIAPP or Aβ40 alone and from a 1:1 mixture of hIAPP and Aβ40 monomers were compared. Co-assembly of the two sequences were observed and hetero-oligomers with conformational properties distinct from their homo-oligomeric counterparts have been characterised. As well as having fibrillation rates intermediate between that of the homo-assembly, the hetero-oligomeric species observed have conformations and gas-phase stabilities intermediate between those of their homo-assembly counterparts, as judged by ESI-IMS-MS and ESI-MS-CID-IMS-MS. The observations made could be important in aiding our quest to unravel the mechanisms of amyloid formation and the origins of its heterogeneous assembly pathways.
Given the similarity in length and sequence of hIAPP and Aβ40 (Fig. 1), it is perhaps not surprising that their two sequences co-assemble.17,60 Indeed, previous studies of both ΔN6 and hβ2microglobulin (pH 6.2),18 and hIAPP and rat-IAPP,19,32 using ESI-MS and ESI-IMS-MS, have demonstrated that pairs of proteins that possess fundamentally different abilities to form fibrils are able to co-polymerise into amyloid.18,19,32 Co-polymerisation results ultimately in a greater degree of polymorphism, with the hetero-oligomers and fibrils exhibiting unique conformational and thermodynamic properties compared with their homo-counterparts, thus expanding the repertoire of amyloid species populated in terms of both structure and stability.17,18 In addition to co-assembly from two distinct monomer pools, cross-seeding is a common cause of co-polymerisation of amyloid sequences. This phenomenon occurs when existing fibrils (known as ‘seeds’) of one precursor sequence catalyse fibrillation from monomer pools of a different sequence, via templating of the seed's structure. Seeded fibrils form at an increased rate, compared with their unseeded counterparts, and can be structurally distinct from fibrils formed de novo.21,61 Interestingly, whilst hIAPP and Aβ40 co-assemble early during fibrillation (at least in dimeric and trimeric forms) and Aβ40 fibrils seed hIAPP assembly, hIAPP fibrils have been reported not to seed Aβ40 assembly.35 Templating the cross-β structure of amyloid is thus very different to the repertoire of protein–protein interactions in pre-amyloid states.
In vivo, co-polymerisation of different protein precursors may be relevant to amyloid disease. Amyloid plaques are highly heterogeneous, being comprised of monomers with different truncations (e.g. hβ2microglobulin and ΔN6; hIAPP and pro-hIAPP processing intermediates62), mutations (e.g. wild-type Aβ and Aβ E22G), relative compositions (e.g. the ratio of Aβ40:Aβ42), post-translational modifications (e.g. phosphorylated/nitrosylated α-synuclein), as well associated co-factors (e.g. GAGS, SAP).17,18,61,63,64 In AD, N-terminally truncated, pyroglutamated forms of Aβ co-polymerise with Aβ42, resulting in oligomers that are more toxic than homo-oligomers formed by either peptide alone.65 Additionally the ratio of Aβ40:42 has been shown to be crucial in determining the location and associated toxicity of amyloid deposits.66 There is also new evidence that Aβ43, a peptide that is more neurotoxic than Aβ42, can co-polymerise with other Aβ peptides and accelerate AD pathology.67 Conversely, hetero-assemblies have been reported that are capable of blocking and/or reversing amyloidosis. A conformationally constrained analogue of hIAPP, for example, designed to mimic a non-amyloidogenic conformation, can bind to oligomers of Aβ and this hetero-association inhibits Aβ self-assembly,2 while assembly of hβ2microglobulin is accelerated by ΔN6 but mouse β2microglobulin inhibits ΔN6 assembly, reminiscent of strains in prion disease.68 In either case, the consequences of co-polymerisation are significant, and distinct from the outcomes of polymerisation of a single protein sequence.
Under the conditions employed here, consistent with previous studies,32,69 hIAPP fibrillates with a shorter lag time compared with that of Aβ40. Similarly, when hIAPP and Aβ42 are mixed at equimolar ratios, fibril formation and membrane permeabilization occurs at a rate intermediate between that observed for hIAPP or Aβ42 alone.69 Membrane permeabilization has been proposed to play a role in amyloid induced toxicity and hIAPP-Aβ42 hetero-aggregates adsorb, aggregate, and permeabilise membranes significantly more slowly than pure hIAPP, but at a much faster rate than observed for pure Aβ42.69 In addition there is evidence that Aβ fibrils can cross seed hIAPP in a transgenic mouse model.35 These data, combined with the results presented here, are suggestive of unique and/or intermediate structures being occupied in the mixed samples that have significant effects on the progress of fibril formation in vitro and may have biological consequences in vivo.
Conclusions
The ESI-IMS-MS, ThT and TEM studies described here demonstrate that mixing hIAPP and Aβ40 monomers does not prevent oligomerisation and/or fibril formation but, in fact, can enhance the rate of fibril formation by Aβ40 alone, and retard the rate of hIAPP assembly. Hetero-oligomers formed during fibrillation have unique conformations and gas-phase stabilities with respect to their homo-polymeric counterparts and thus extend the repertoire of amyloid species formed. Using the unique separative features of ESI-IMS-MS to characterise transient components individually from within highly heterogeneous mixtures, we reveal here that hetero-dimers and hetero-trimers of hIAPP-Aβ40 have unique conformations compared with the structures of homo-oligomers present within the mixtures, possibly determining the outcome of the course of amyloid assembly. Unravelling the process of co-polymerisation further could pave the way to understanding the fundamental mechanisms of amyloidosis, and how the population of hetero-polymeric species affects the rate, stability, toxicity and biological consequences of amyloid deposition.Acknowledgements
L. M. Y. is funded by a Biotechnology and Biological Sciences Research Council (BBSRC) CASE studentship (Grant Number BB/I015361/1) sponsored by Micromass UK Ltd/Waters Corpn., Manchester, UK. R. A. M. is funded by a BBSRC studentship (Grant Number BB/F01614X/1). J. C. S. is funded by a BBSRC CASE studentship (Grant Number BB/H014713/1) sponsored by Avacta Analytical PLC, Wetherby, UK. L.-H., T, and D. P. R are funded by a US National Institutes of Health grant GM078114. The Synapt HDMS mass spectrometer was purchased with funds from the Biotechnology and Biological Sciences Research Council through its Research Equipment Initiative scheme (BB/E012558/1). We thank Dominic Walsh (Brigham & Women's Hospital, Boston, USA) and Sara Linse (Lund University, Sweden) for provision of Aβ40 vector. We also acknowledge all members of the Ashcroft, Radford and Raleigh groups for helpful discussions.References
- J. D. Sipe, M. D. Benson, J. N. Buxbaum, S.-i. Ikeda, G. Merlini, M. J. M. Saraiva and P. Westermark, Amyloid, 2014, 21, 221–224 CrossRef PubMed.
- L.-M. Yan, A. Velkova, M. Tatarek-Nossol, E. Andreetto and A. Kapurniotu, Angew. Chem., Int. Ed., 2007, 46, 1246–1252 CrossRef CAS PubMed.
- P. Westermark, Ups. J. Med. Sci., 2011, 116, 81–89 CrossRef PubMed.
- P. Westermark, A. Andersson and G. T. Westermark, Physiol. Rev., 2011, 91, 795–826 CrossRef CAS PubMed.
- P. A. Halban, K. S. Polonsky, D. W. Bowden, M. A. Hawkins, C. Ling, K. J. Mather, A. C. Powers, C. J. Rhodes, L. Sussel and G. C. Weir, J. Clin. Endocrinol. Metab., 2014, 99, 1983–1992 CrossRef CAS PubMed.
- G. T. Westermark, P. Westermark, C. Berne and O. Korsgren, New Eng. J. Med., 2008, 359, 977–979 CrossRef CAS PubMed.
- K. J. Potter, A. Abedini, P. Marek, A. M. Klimek, S. Butterworth, M. Driscoll, R. Baker, M. R. Nilsson, G. L. Warnock, J. Oberholzer, S. Bertera, M. Trucco, G. S. Korbutt, P. E. Fraser, D. P. Raleigh and C. B. Verchere, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4305–4310 CrossRef CAS PubMed.
- K. Tenidis, M. Waldner, J. Bernhagen, W. Fischle, M. Bergmann, M. Weber, M. L. Merkle, W. Voelter, H. Brunner and A. Kapurniotu, J. Mol. Biol., 2000, 295, 1055–1071 CrossRef CAS PubMed.
- A. T. Petkova, Y. Ishii, J. J. Balbach, O. N. Antzutkin, R. D. Leapman, F. Delaglio and R. Tycko, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16742–16747 CrossRef CAS PubMed.
- L. Young, H. Ndlovu, T. Knapman, S. Harris, S. Radford and A. Ashcroft, Int. J. Ion Mobil. Spectrom., 2013, 16, 29–39 CrossRef CAS.
- P. Westermark, U. Engström, K. H. Johnson, G. T. Westermark and C. Betsholtz, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 5036–5040 CrossRef CAS.
- L. E. Buchanan, E. B. Dunkelberger, H. Q. Tran, P. N. Cheng, C. C. Chiu, P. Cao, D. P. Raleigh, J. J. de Pablo, J. S. Nowick and M. T. Zanni, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 19285–19290 CrossRef CAS PubMed.
- Y. Yang and W. Song, Neuroscience, 2013, 250, 140–150 CrossRef CAS PubMed.
- J. Tang, Y. Pei and G. Zhou, Exp. Gerontol., 2013, 48, 744–750 CrossRef CAS PubMed.
- D. Kopf and L. Frolich, J. Alzheimers Dis., 2009, 16, 677–685 Search PubMed.
- D. M. Walsh, E. Thulin, A. M. Minogue, N. Gustavsson, E. Pang, D. B. Teplow and S. Linse, FEBS J., 2009, 276, 1266–1281 CrossRef CAS PubMed.
- C. J. Sarell, P. G. Stockley and S. E. Radford, Prion, 2013, 7, 359–368 CrossRef CAS PubMed.
- C. J. Sarell, L. A. Woods, Y. Su, G. T. Debelouchina, A. E. Ashcroft, R. G. Griffin, P. G. Stockley and S. E. Radford, J. Biol. Chem., 2013, 288, 7327–7337 CrossRef CAS PubMed.
- L. M. Young, P. Cao, D. P. Raleigh, A. E. Ashcroft and S. E. Radford, J. Am. Chem. Soc., 2014, 136, 660–670 CrossRef CAS PubMed.
- J. N. Buxbaum, Z. Ye, N. Reixach, L. Friske, C. Levy, P. Das, T. Golde, E. Masliah, A. R. Roberts and T. Bartfai, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2681–2686 CrossRef CAS PubMed.
- B. O'Nuallain, A. D. Williams, P. Westermark and R. Wetzel, J. Biol. Chem., 2004, 279, 17490–17499 CrossRef PubMed.
- J. J. Wiltzius, S. A. Sievers, M. R. Sawaya and D. Eisenberg, Protein Sci., 2009, 18, 1521–1530 CrossRef CAS PubMed.
- J. P. Guo, T. Arai, J. Miklossy and P. L. McGeer, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 1953–1958 CrossRef CAS PubMed.
- P. Westermark, Z. C. Li, G. T. Westermark, A. Leckstrom and D. F. Steiner, FEBS Lett., 1996, 379, 203–206 CrossRef CAS.
- B. I. Giasson, M. S. Forman, M. Higuchi, L. I. Golbe, C. L. Graves, P. T. Kotzbauer, J. Q. Trojanowski and V. M. Lee, Science, 2003, 300, 636–640 CrossRef CAS PubMed.
- A. Velkova, M. Tatarek-Nossol, E. Andreetto and A. Kapurniotu, Angew. Chem., Int. Ed., 2008, 47, 7114–7118 CrossRef CAS PubMed.
- S. Gilead, H. Wolfenson and E. Gazit, Angew. Chem., Int. Ed., 2006, 45, 6476–6480 CrossRef CAS PubMed.
- J. Lauren, D. A. Gimbel, H. B. Nygaard, J. W. Gilbert and S. M. Strittmatter, Nature, 2009, 457, 1128–1132 CrossRef CAS PubMed.
- K. Pauwels, T. L. Williams, K. L. Morris, W. Jonckheere, A. Vandersteen, G. Kelly, J. Schymkowitz, F. Rousseau, A. Pastore, L. C. Serpell and K. Broersen, J. Biol. Chem., 2012, 287, 5650–5660 CrossRef CAS PubMed.
- I. F. Tsigelny, L. Crews, P. Desplats, G. M. Shaked, Y. Sharikov, H. Mizuno, B. Spencer, E. Rockenstein, M. Trejo, O. Platoshyn, J. X. Yuan and E. Masliah, PLoS One, 2008, 3, e3135 Search PubMed.
- K. Ono, R. Takahashi, T. Ikeda and M. Yamada, J. Neurochem., 2012, 122, 883–890 CrossRef CAS PubMed.
- C. T. Middleton, P. Marek, P. Cao, C. C. Chiu, S. Singh, A. M. Woys, J. J. de Pablo, D. P. Raleigh and M. T. Zanni, Nat. Chem., 2012, 4, 355–360 CrossRef CAS PubMed.
- F. Meng, A. Abedini, B. Song and D. P. Raleigh, Biochem., 2007, 46, 12091–12099 CrossRef CAS PubMed.
- A. C. Susa, C. Wu, S. L. Bernstein, N. F. Dupuis, H. Wang, D. P. Raleigh, J.-E. Shea and M. T. Bowers, J. Am. Chem. Soc., 2014, 136, 12912–12919 CrossRef CAS PubMed.
- M. E. Oskarsson, J. F. Paulsson, S. W. Schultz, M. Ingelsson, P. Westermark and G. T. Westermark, Am. J. Pathol., 2015, 185, 834–846 CrossRef CAS PubMed.
- E. Andreetto, L. M. Yan, M. Tatarek-Nossol, A. Velkova, R. Frank and A. Kapurniotu, Angew. Chem., Int. Ed., 2010, 49, 3081–3085 CrossRef CAS PubMed.
- S. Mehmood, T. M. Allison and C. V. Robinson, Annu. Rev. Phys. Chem., 2015, 66, 453–474 CrossRef CAS PubMed.
- A. Konijnenberg, A. Butterer and F. Sobott, Biochim. Biophys. Acta, 2013, 1834, 1239–1256 CrossRef CAS PubMed.
- E. Boeri Erba and C. Petosa, Protein Sci., 2015 DOI:10.1002/pro.2661.
- L. A. Woods, S. E. Radford and A. E. Ashcroft, Biochimica et Biophysica Acta, Gen. Sub., 2013, 1834, 1257–1268 CrossRef CAS PubMed.
- C. Uetrecht, C. Versluis, N. R. Watts, W. H. Roos, G. J. L. Wuite, P. T. Wingfield, A. C. Steven and A. J. R. Heck, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9216–9220 CrossRef CAS PubMed.
- D. P. Smith, S. E. Radford and A. E. Ashcroft, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6794–6798 CrossRef CAS PubMed.
- M. Landreh and C. V. Robinson, J. Physiol., 2015, 593, 355–362 CrossRef CAS PubMed.
- A. N. Calabrese, T. G. Watkinson, P. J. Henderson, S. E. Radford and A. E. Ashcroft, Anal. Chem., 2015, 87, 1118–1126 CrossRef CAS PubMed.
- L. H. Tu, A. L. Serrano, M. T. Zanni and D. P. Raleigh, Biophys. J., 2014, 106, 1520–1527 CrossRef CAS PubMed.
- K. Giles, S. D. Pringle, K. R. Worthington, D. Little, J. L. Wildgoose and R. H. Bateman, Rapid Commun. Mass Spectrom., 2004, 18, 2401–2414 CrossRef CAS PubMed.
- D. P. Smith, T. W. Knapman, I. Campuzano, R. W. Malham, J. T. Berryman, S. E. Radford and A. E. Ashcroft, Eur. J. Mass Spectrom., 2009, 15, 113–130 CrossRef CAS PubMed.
- S. J. Valentine, A. E. Counterman and D. E. Clemmer, J. Am. Soc. Mass Spectrom., 1999, 10, 1188–1211 CrossRef CAS.
- G. W. Platt, K. E. Routledge, S. W. Homans and S. E. Radford, J. Mol. Biol., 2008, 378, 251–263 CrossRef CAS PubMed.
- N. F. Dupuis, C. Wu, J. E. Shea and M. T. Bowers, J. Am. Chem. Soc., 2009, 131, 18283–18292 CrossRef CAS PubMed.
- S. L. Bernstein, N. F. Dupuis, N. D. Lazo, T. Wyttenbach, M. M. Condron, G. Bitan, D. B. Teplow, J. E. Shea, B. T. Ruotolo, C. V. Robinson and M. T. Bowers, Nat. Chem., 2009, 1, 326–331 CrossRef CAS PubMed.
- N. F. Dupuis, C. Wu, J.-E. Shea and M. T. Bowers, J. Am. Chem. Soc., 2011, 133, 7240–7243 CrossRef CAS PubMed.
- M. M. Gessel, S. Bernstein, M. Kemper, D. B. Teplow and M. T. Bowers, ACS Chem. Neurosci., 2012, 3, 909–918 CrossRef CAS PubMed.
- P. Ladewig, Nature, 1945, 156, 81–82 CrossRef PubMed.
- M. Biancalana and S. Koide, Biochimica et Biophysica Acta, Gen. Sub., 2010, 1804, 1405–1412 CrossRef CAS PubMed.
- J. Marcoux and C. V. Robinson, Structure, 2013, 21, 1541–1550 CrossRef CAS PubMed.
- A. Dyachenko, M. Goldflam, M. Vilaseca and E. Giralt, Biopolymers, 2010, 94, 689–700 CrossRef CAS PubMed.
- A. Abedini, F. Meng and D. P. Raleigh, J. Am. Chem. Soc., 2007, 129, 11300–11301 CrossRef CAS PubMed.
- S. Sakagashira, H. J. Hiddinga, K. Tateishi, T. Sanke, T. Hanabusa, K. Nanjo and N. L. Eberhardt, Am. J. Pathol., 2000, 157, 2101–2109 CrossRef CAS.
- M. R. Krebs, L. A. Morozova-Roche, K. Daniel, C. V. Robinson and C. M. Dobson, Protein Sci., 2004, 13, 1933–1938 CrossRef CAS PubMed.
- C. Iannuzzi, G. Irace and I. Sirangelo, Molecules, 2015, 20, 2510–2528 CrossRef PubMed.
- J. F. Paulsson and G. T. Westermark, Diabetes, 2005, 54, 2117–2125 CrossRef CAS.
- C. A. De Carufel, P. T. Nguyen, S. Sahnouni and S. Bourgault, Biopolymers, 2013, 100, 645–655 CrossRef CAS PubMed.
- L. Stefanis, Cold Spring Harbor Perspect. Med., 2012, 2, a:009399 Search PubMed.
- J. M. Nussbaum, S. Schilling, H. Cynis, A. Silva, E. Swanson, T. Wangsanut, K. Tayler, B. Wiltgen, A. Hatami, R. Ronicke, K. Reymann, B. Hutter-Paier, A. Alexandru, W. Jagla, S. Graubner, C. G. Glabe, H. U. Demuth and G. S. Bloom, Nature, 2012, 485, 651–655 CrossRef CAS PubMed.
- D. Eisenberg and M. Jucker, Cell, 2012, 148, 1188–1203 CrossRef CAS PubMed.
- T. Saito, T. Suemoto, N. Brouwers, K. Sleegers, S. Funamoto, N. Mihira, Y. Matsuba, K. Yamada, P. Nilsson, J. Takano, M. Nishimura, N. Iwata, C. Van Broeckhoven, Y. Ihara and T. C. Saido, Nat. Neurosci., 2011, 14, 1023–1032 CrossRef CAS PubMed.
- T. K. Karamanos, A. P. Kalverda, G. S. Thompson and S. E. Radford, Mol. Cell, 2014, 55, 214–226 CrossRef CAS PubMed.
- L. Khemtémourian, J. Killian, J. Höppener and M. Engel, Exp. Diabetes Res., 2008, 2008, 1–9 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2015 |