
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Flow induced dispersion analysis rapidly quantifies proteins in human plasma samples†
Nicklas N.
Poulsen
,
Nina Z.
Andersen
,
Jesper
Østergaard‡
,
Guisheng
Zhuang
,
Nickolaj J.
Petersen
and
Henrik
Jensen‡
*
Department of Pharmacy, University of Copenhagen, Universitetsparken 2, 2100 Copenhagen, Denmark. E-mail: henrik.jensen@sund.ku.dk
First published on 27th May 2015
Abstract
Rapid and sensitive quantification of protein based biomarkers and drugs is a substantial challenge in diagnostics and biopharmaceutical drug development. Current technologies, such as ELISA, are characterized by being slow (hours), requiring relatively large amounts of sample and being subject to cumbersome and expensive assay development. In this work a new approach for quantification based on changes in diffusivity is presented. The apparent diffusivity of an indicator molecule interacting with the protein of interest is determined by Taylor Dispersion Analysis (TDA) in a hydrodynamic flow system. In the presence of the analyte the apparent diffusivity of the indicator changes due to complexation. This change in diffusivity is used to quantify the analyte. This approach, termed Flow Induced Dispersion Analysis (FIDA), is characterized by being fast (minutes), selective (quantification is possible in a blood plasma matrix), fully automated, and being subject to a simple assay development. FIDA is demonstrated for quantification of the protein Human Serum Albumin (HSA) in human plasma as well as for quantification of an antibody against HSA. The sensitivity of the FIDA assay depends on the indicator-analyte dissociation constant which in favourable cases is in the sub-nanomolar to picomolar range for antibody–antigen interactions.
Introduction
The detection and quantification of proteins represent a considerable challenge in diagnostics, disease treatment and drug development. Especially the evolving fields of personalized medicines and therapeutic drug monitoring rely on general methodologies for rapid decentralized quantification of protein based biomarkers.1–3 Most immunological assays, such as e.g. Enzyme-linked Immunosorbent Assays, ELISA, involve surface chemistries which require substantial development and optimisation for each new application.4 Furthermore, such assays usually involve several steps including analyte capture, washing, secondary antibody incubation and detection, leading to a relatively long analysis time and assays prone to operator errors. Consequently, there is substantial interest in alternatives to traditional bio-assays such as ELISA.5In this work we demonstrate a simple one-step procedure for quantification of proteins. It has previously been shown that Taylor Dispersion Analysis (TDA) is an effective method for determination of diffusivities from dispersion in a hydrodynamic flow.6–10 The present work builds on these studies as it aims to relate changes in the dispersion of an indicator molecule to protein concentrations. The apparent size of the indicator molecule (i.e. the ligand interacting with the protein of interest) is relatively small in the absence of the protein, but its apparent size increases with the protein concentration due to complex formation. Here we demonstrate that the apparent increase in size of the indicator is an accurate approach for protein quantification. This approach, termed Flow Induced Dispersion Analysis (FIDA), does not require separation/isolation of an affinity complex, is not dependent on a secondary antibody interaction, it is fast (minutes compared to hours for ELISA), and easy to automate. The method may be used for any protein (analyte) to which an indicator molecule (affinity ligand) binding with high affinity and specificity exist.
Experimental section
Experimental details, including materials and procedures are provided in the ESI.† Briefly, the FIDA experiments are conducted using standard fused silica capillaries (50 μm inner diameter, 360 μm outer diameter) and standard apparatus for capillary electrophoresis (Agilent 3DCE instrument, Agilent Technologies, Waldbronn, Germany) employing laser induced fluorescence detection (ZETALIF Evolution, Picometrics). A 488 nm Melles Griot Diode laser was used for fluorophore excitation. The mobilisation pressure were either 50 mbar (provided by the CE system) or 250 mbar obtained by coupling an external nitrogen source to the CE apparatus. These pressures corresponded to linear flow rates of 0.6 and 3 mm s−1 respectively.An overview of the automated workflow is shown in Fig. 1. Initially the capillary is rinsed to remove contaminants prior to analysis. Second, the capillary is filled with analyte. Third, a small well defined indicator zone is introduced (typically around 10 nL) and mobilized by a pressure driven flow of the sample. The signal corresponding to the indicator is recorded at the detection window located at 48 cm. From the indicator peak shape, the apparent indicator diffusivity is obtained.
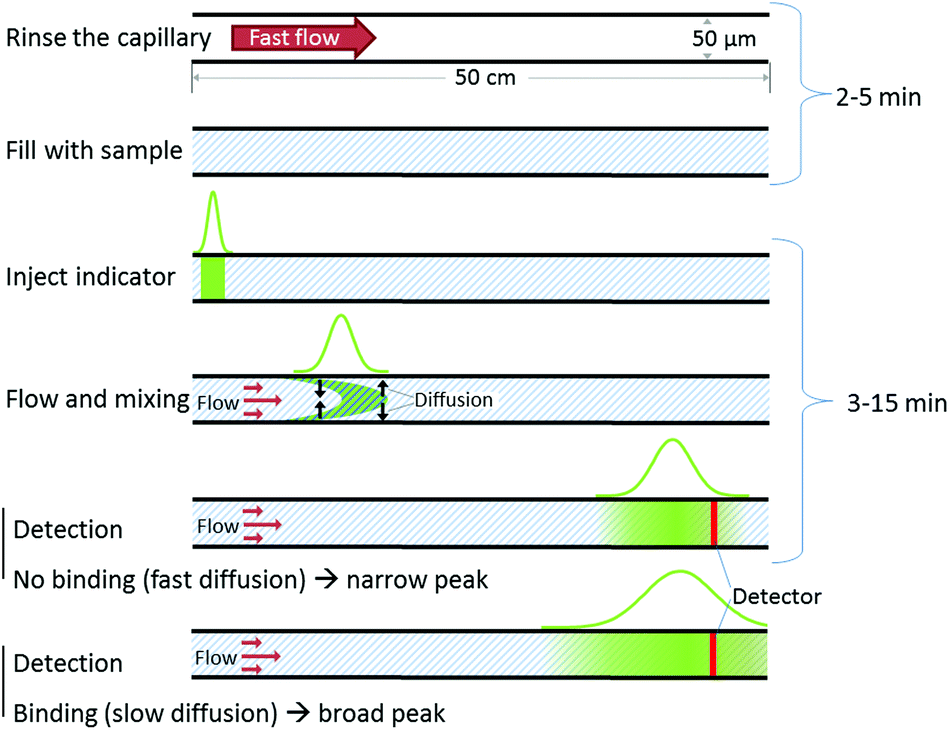 | ||
| Fig. 1 Schematic presentation of the workflow in a FIDA experiment. After rinsing the capillary is filled with analyte. Then a small indicator zone is introduced (typically around 10 nL) and moved to the detector by a pressure driven flow of sample. During the flow the indicator is mixed with the sample. The indicator peak variance is linked to the apparent diffusivity, which in turn is dependent on the degree of binding. | ||
Results and discussion
In a FIDA experiment, the binding equilibrium between the indicator (I) and the analyte (A) is probed:Indicator (I) + Analyte (A) = Complex (IA)
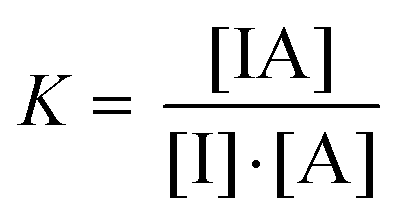 | (1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding stoichiometry the unbound fraction of the indicator (x) may be calculated from eqn (2):
:1 binding stoichiometry the unbound fraction of the indicator (x) may be calculated from eqn (2):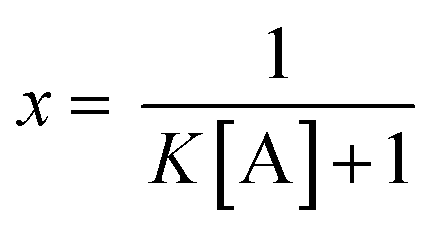 | (2) |
If the analyte is in a large excess compared to the indicator molecule, the free concentration may be approximated by the formal analyte concentration (cA).
When on-and-off binding kinetics is fast the apparent diffusivity of the indicator molecule (Dapp) is a linear function of the unbound fraction of the indicator,10
| Dapp = DIx + DIA(1 − x) | (3) |
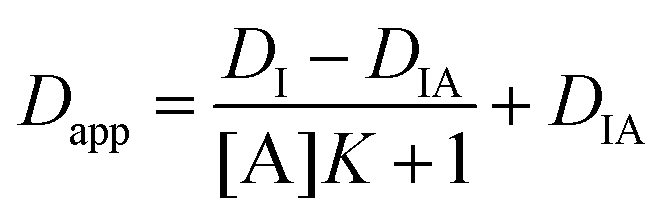 | (4) |
Diffusion coefficients may be determined by Taylor Dispersion Analysis (TDA) in capillaries with small inner diameter. This principle has been exploited for measurement of diffusion coefficients7–9 as well as for assessment of non-covalent binding constants of small molecules.10–12
In a FIDA experiment, the indicator zone is dispersed due to the parabolic flow profile and radial diffusion. As the indicator zone is dispersed it is mixed with the analyte and binding occurs. The indicator peak shape is monitored at the capillary outlet and fitted to a Gaussian peak shape. From the indicator peak variance (σ2), capillary radius (r) and the peak appearance time (tR) the apparent indicator diffusivity (Dapp) can be calculated using eqn (5).6
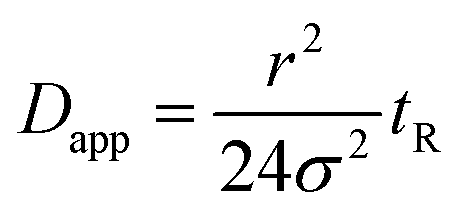 | (5) |
Details on the conditions for eqn (5) to be valid are provided in the ESI.† The binding isotherm shown in eqn (4) relates the apparent indicator diffusivity to the analyte concentration and can thus be used as a standard curve for quantifying the analyte.
The FIDA methodology is first demonstrated by quantifying the protein Human Serum Albumin (HSA). In addition to being a model system, a low HSA concentration is associated with a poor recovery in critical illness.13 Fluorescein was chosen as the indicator molecule because it is known to bind to HSA.14 Fluorescein is a small molecule (332 g mol−1) and therefore a considerable change in apparent diffusivity is seen upon binding to HSA resulting in a significant broadening of the fluorescein peak as shown in Fig. 2 (insert). Furthermore, fluorescein is a fluorescent molecule allowing sensitive and selective fluorescence detection. The high HSA concentrations lead to an increase in the background fluorescence, but this does not complicate the data treatment since it is simply seen as a baseline offset. Following initial experiments, a fluorescein concentration of 50 nM and an injection volume 7 nL were selected. Experimental details and data treatment are provided in the ESI.† The indicator apparent diffusivity as a function of the concentration of the analyte HSA in buffer provides the standard curve shown in Fig. 2. Pressures of 50 and 250 mbar resulting in analysis times of 14 and 3 min, respectively, were applied. The standard curves were found to be well described by eqn (4) and similar diffusion coefficients where obtained at both pressures. Thus it is possible to perform a measurement in 3 min. It should, however, in general be carefully evaluated that there is sufficient time for diffusion, mixing and binding. Using the data obtained at 50 mbar, the diffusivity of unbound fluorescein was determined to be 4.3 × 10−10 m2 s−1, close to the value of 4.25 × 10−10 m2 s−1 reported by Culbertson et al.15 The diffusivity of the fluorescein-HSA complex estimated to 6.3 × 10−11 m2 s−1 is close to 6.41 × 10−11 m2 s−1 previously found for HSA.16 The association equilibrium binding constant was calculated to 2.8 × 104 M−1 at 25 °C assuming 1:1 binding stoichiometry. As expected this value is higher than the previously reported value of 6.0 × 103 M−1 at 37 °C.14
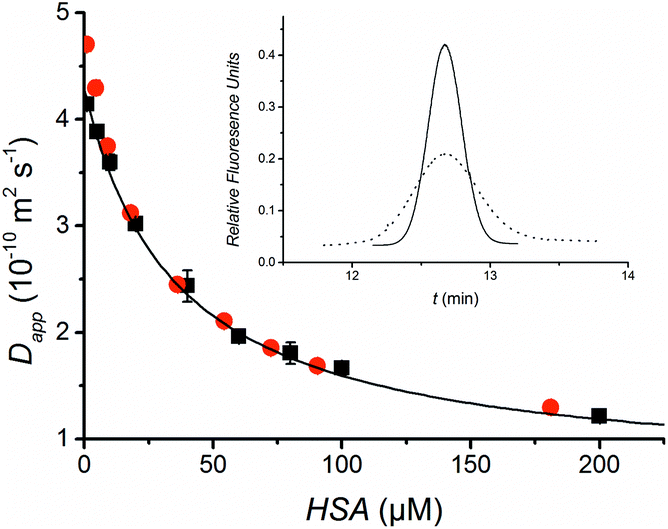 | ||
| Fig. 2 The standard curve for HSA is obtained by plotting the apparent diffusivity of the indicator peak against the HSA concentration at 50 mbar (black) and 250 mbar (red) applied pressures. The data points obtained at 50 mbar have been fitted to the binding isotherm given in eqn (4). Insert: Peak profiles (Taylorgrams) of 50 nM fluorescein indicator in buffer (solid line) and 200 μM HSA solution (dotted line). The time for the red curve has been slightly offset in order to overlay the two curves and HSA background fluorescence has been subtracted to allow easy comparison of the signals. | ||
The standard curve shown in Fig. 2 was used to quantify HSA in diluted (2%) human plasma. The colorimetric Bromo-Cresol Purple assay (BCP) was selected as a comparative method as it is widely used for quantifying HSA.17 Three different plasma samples were analysed using FIDA and BCP and comparable results were achieved as seen in Table 1. The selectivity of the assay is linked to the specificity of the non-covalent binding to the protein. Obviously, fluorescein and bromo-cresol purple may be expected to bind to other proteins, which could give rise to false results especially at high plasma concentrations. Matrix effects have previously been reported for the BCP assay.18
| Plasma sample | HSA (g l−1) | |
|---|---|---|
| FIDAa | BCPb | |
| a The uncertainty of the FIDA assay is based on independent measurement of apparent diffusivities (triplicates). b The uncertainty of the BCP assay is based on triplicate absorbance measurements of the plasma sample. The relative standard deviation of the BCP assay was previously reported to be 1.6%–2.3%.18 | ||
| 1 | 33 (±0.6%) | 32 (±0.2%) |
| 2 | 30 (±5.8%) | 27 (±0.5%) |
| 3 | 32 (±6.8%) | 32 (±0.2%) |
Possible matrix effects in the FIDA assay were studied in a series of standard addition experiments shown in the ESI (Fig. S2†). The standard addition curve and the standard curve are similar indicating very limited matrix effect from plasma in the FIDA assay.
As briefly mentioned above, non-specific binding will affect the dispersion of the indicator and, hence, affect results. In practice a strong specific binding between indicator and analyte is optimal. Therefore FIDA is well suited for antibody based quantification as most antibody–antigen interactions are characterized by high affinity and specificity. The ability of FIDA to quantify antibodies was investigated using Anti-HSA (an antibody against HSA) as the analyte and HSA covalently bound to the fluorescent probe ATTO488A as the indicator (F-HSA). The standard curve is shown in Fig. 3. The diffusivity of the indicator in the absence of antibody is 8.6 × 10−11 m2 s−1. This value is larger than 6.4 × 10−11 m2 s−1 previously found for HSA in this work and reported in the literature,14 indicating that unreacted fluorescent dye has not been completely removed from the tagged F-HSA. Experimentally, this manifests itself as an apparent higher diffusivity for the indicator molecule. This also explains the determined apparent antibody complex diffusivity of 5.7 × 10−11 m2 s−1 which is larger than the reported value of 4.4 × 10−11 m2 s−1.19 Still, a clear F-HSA-antibody binding is observed at antibody concentrations as low as 1 nM as shown in Fig. 3, which shows that a small fraction of non-covalently bound fluorescent tag does not hamper the FIDA assay. This point is rather important as complete removal of non-reacted or non-covalently bound fluorescent tag is often challenging.
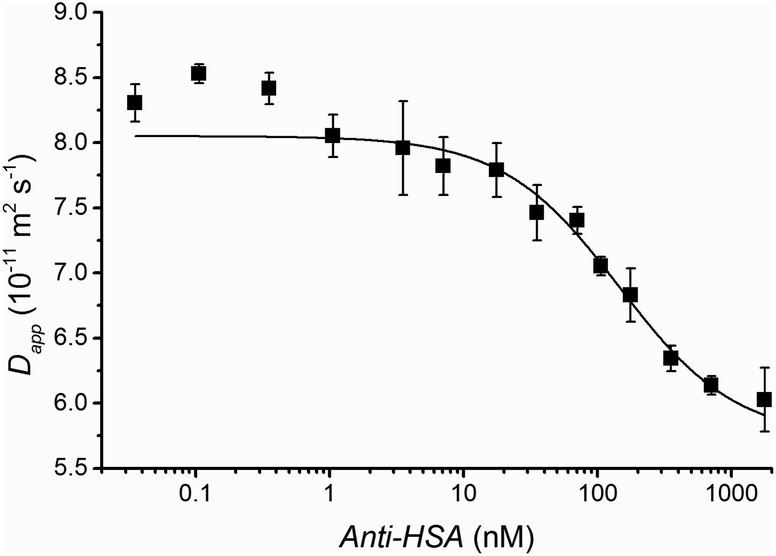 | ||
| Fig. 3 Apparent diffusivity of the F-HSA indicator plotted as a function of the concentration of Anti-HSA. All measurements are triplicates and standard deviation is shown. The solid black line shows the fit obtained with eqn (4). A 7 nL volume of 20 nM fluorescently tagged HSA (F-HSA) was injected into a 50 μm ID capillary filled with various concentration of Anti-HSA. The analysis time was less than 12 min at 50 mBar. | ||
Two binding events are evident in Fig. 3. A small fraction of the indicator (HSA) is bound with a strong affinity below 1 nM Anti-HSA, whereas the main binding event occurs at concentrations above 10 nM. This heterogenic binding may be due to covalently bound fluorescence tags hampering the epitope recognition for part of the HSA molecules or due to a small fraction of antibodies binding with higher affinity.20 The dynamic range for this assay is between 10 nM and 1 μM. The dissociation constant was estimated to be 1 × 10−7 M according to eqn (4) assuming a 1:1 interaction.
The binding curve in Fig. 3 may be used as a standard curve for quantifying Anti-HSA. This point was tested for independently prepared anti-HSA samples, where the antibody was successfully quantified in buffer solution. The accuracy was approximately 10% (see ESI†).
The dynamic range of the FIDA assay can be adjusted by changing the indicator concentration. Theoretical standard curves, based on the dissociation constant of 1 × 10−7 M found for HSA-anti HSA system, are shown in Fig. 4a. The four curves correspond to different indicator concentrations. The curves have been calculated assuming fast binding kinetics, 1:1 binding, and that the Taylor conditions are fulfilled (ESI†). During the analysis the indicator is diluted as it is dispersed over a longer part of the capillary. In practice this causes the binding curve to be shifted to lower analyte concentration, but this effect was not taken into account in the theoretical curves. The calculated curves thus merely describe how dynamic range and sensitivity can be adjusted in a practical situation rather than providing an exact match to experiments.
 | ||
| Fig. 4 (a) Theoretical binding curves describing the relationship between the apparent indicator diffusivity and analyte concentration for indicator concentrations 0.1, 1, 10 and 100 times the dissociation constant (black, green, blue and purple curve respectively). The diffusivity of the unbound indicator is defined as 1.0 × 10−10 m2 s−1 and for the complex it is defined as 0.50 × 10−10 m2 s−1. (b) The analyte concentration needed to bind 10%, 50% and 90% of the indicator (pink, black and blue) is plotted against the indicator concentration. The dissociation constant is marked by a vertical red line. The dissociation constant for the HSA-anti HSA interaction of 1 × 10−7 M was used for the calculations in both (a) and (b). | ||
It can be seen in Fig. 4a that lowering the indicator concentration will shift the dynamic range to smaller concentrations until a concentration corresponding to the dissociation equilibrium constant is reached. This effect is also visible from Fig. 4b, where the analyte concentration needed to bind 10%, 50% and 90% of the indicator is plotted against indicator concentration. It is apparent that decreasing analyte concentrations are needed to obtain the same degree of binding at lower indicator concentrations. However, when decreasing the indicator concentration below the dissociation constant (shown as a vertical red line) the effect quickly wears off and any further reduction of the indicator concentration does not lead to a change in the analyte concentration needed to bind the analyte. If the dynamic range is defined as being between 10% and 90% binding of the indicator it can be seen that the dynamic range is a factor of 10 when the indicator concentration is larger than the dissociation constant and a factor of one hundred when the indicator concentration is smaller than the dissociation constant (Fig. 4b).
The limit of detection for FIDA will in general be determined by the binding constant for the indicator-ligand interaction, the limit of detection for the indicator (in the order of picomolar for many fluorophores using the present instrumentation), the difference in diffusivity between the unbound indicator and the indicator-analyte complex, and the standard deviation for the determination of the peak variance. In the case of a strong selective interaction it should thus be feasible to reach sub-nano to picomolar sensitivities.
Conclusions
The present work demonstrates a new general approach for rapid protein quantification. The relative simplicity of the methodology and high tolerance to matrix effects is likely to make assays development easy; the main requirement is availability of a ligand (indicator molecule) which binds to the analyte with high affinity and specificity. Indicator molecules will often be available as antibodies (or antibody fragments). The FIDA protocol is straight forward to automate. It is further a significant advantage that only one interaction is required and a secondary antibody is not needed as in ELISA assays. Another important point concerns the fact that the affinity interaction takes place in solution, therefore problems associated with non-specific surface adsorption are minimized. Essentially, the FIDA methodology is a 1-channel assay which is also suited for microfluidic systems.21 FIDA may thus ultimately be implemented in microfluidic based point-of-care devises, for e.g. quantification of bio-markers.Acknowledgements
Financial support from the Danish Council for Independent Research, Technology and Production Science (grant: 11-106647) is gratefully acknowledged.References
- I. Cascorbi, Eur. J. Clin. Pharmacol., 2010, 66, 749–754 CrossRef PubMed.
- J. M. Perkel, Science, 2009, 324, 815–817 CrossRef.
- L. J. Lesko, Clin. Pharmacol. Ther., 2007, 81, 807–816 CrossRef CAS PubMed.
- R. A. Vijayendran and D. E. Leckband, Anal. Chem., 2001, 73, 471–480 CrossRef CAS.
- J. Comley, Drug Discovery World, 2012, 23–45 Search PubMed.
- G. Taylor, Proc. R. Soc. London, A, 1953, 219, 186–203 CrossRef CAS.
- M. S. Bello, R. Rezzonico and P. G. Righetti, Science, 1994, 266, 773–776 CAS.
- H. Cottet, J. P. Biron and M. Martin, Anal. Chem., 2007, 79, 9066–9073 CrossRef CAS PubMed.
- U. Sharma, N. J. Gleason and J. D. Carbeck, Anal. Chem., 2005, 77, 806–813 CrossRef CAS PubMed.
- H. Jensen and J. Østergaard, J. Am. Chem. Soc., 2010, 132, 4070–4071 CrossRef CAS PubMed.
- A. Bielejewska, A. Bylina, K. Duszczyk, M. Fialkowski and R. Holyst, Anal. Chem., 2010, 82, 5463–5469 CrossRef CAS PubMed.
- H. Jensen and J. Østergaard, Analyte Quantification using Flow Induced Dispersion Analysis, US provisional Application, Serial number 61/309,384.
- K. N. Apelgren, J. L. Rombeau, P. L. Twomey and R. A. Miller, Crit. Care Med., 1982, 10, 305–307 CrossRef CAS PubMed.
- J. T. Penniston, Exp. Eye Res., 1982, 34, 435–443 CrossRef CAS.
- C. T. Culbertson, S. C. Jacobson and J. M. Ramsey, Talanta, 2002, 56, 365–373 CrossRef CAS.
- J. Østergaard and H. Jensen, Anal. Chem., 2009, 81, 8644–8648 CrossRef PubMed.
- B. T. Doumas and T. Peters, Clin. Chem., 2009, 55, 583–584 CAS.
- H. A. Assink, B. G. Blijenberg, G. J. M. Boerma and B. Leijnse, J. Clin. Chem. Clin. Bio., 1984, 22, 685–692 CAS.
- B. Pokric and Z. Pucar, Anal. Biochem., 1979, 93, 103–114 CrossRef CAS.
- A. S. Muerhoff, K. Rupprecht, Q. Ruan, B. Zeck, C. Ramsay, C. Zhao and S. M. Desai, J. Immunol. Methods, 2009, 345, 60–69 CrossRef CAS PubMed.
- K. F. Lei, J. Lab. Autom., 2012, 17, 330–347 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, supplementary data and theoretical considerations. See DOI: 10.1039/c5an00697j |
| ‡ Henrik Jensen and Jesper Østergaard are co-founders of FIDA-Tech Aps, a university spin-out exploring opportunities to commercialize FIDA. |
| This journal is © The Royal Society of Chemistry 2015 |