
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Facile and controllable synthesis of hydroxyapatite/graphene hybrid materials with enhanced sensing performance towards ammonia†
Qing
Zhang
a,
Yong
Liu
*a,
Ying
Zhang
b,
Huixia
Li
a,
Yanni
Tan
a,
Lanlan
Luo
a,
Junhao
Duan
a,
Kaiyang
Li
a and
Craig E.
Banks
*c
aState Key Laboratory of Powder Metallurgy, Central South University, Changsha 410083, PR China. E-mail: yonliu@csu.edu.cn; Fax: +86-731-88836939; Tel: +86-731-88836939
bCollege of Science, Huazhong Agricultural University, Wuhan 430070, PR China
cFaculty of Science and Engineering, School of Science and the Environment, Division of Chemistry and Environmental Science, Manchester Metropolitan University, Chester Street, Manchester M1 5GD, UK. E-mail: c.banks@mmu.ac.uk; Web: http://www.craigbanksresearch.com Fax: +44 (0)1612476831; Tel: +44 (0)1612471196
First published on 27th May 2015
Abstract
In this work, needle-like and micro-spherical agglomerates of nanocrystalline hydroxyapatite (HA) were successfully assembled on the surface of graphene sheets with the aid of dopamine having two roles, as a template and a reductant for graphite oxide during the process of self-polymerization. The crystalline structure and micromorphology of HA can be conveniently regulated by controlling the mineralization route either with a precipitation (cHA/GR) or biomimetic methodology (bHA/GR). Both the composites exhibit improvements of ∼150% and ∼250% in sensitivity towards the sensing of ammonia at room temperature, compared with that of bare graphene. The combination of the multi-adsorption capability of HA and the electric conductivity of graphene is proposed to be the major reason for the observed enhancements. Gas sensing tests demonstrated that the HA/GR composites exhibit excellent selectivity, high sensitivity and repeatable stability towards the analytical sensing of ammonia.
1. Introduction
Environmental pollution caused by combustion from vehicles, agricultural sector and industrial leakages of toxic chemicals and gases is one of the most serious problems facing our society.1–3 Amongst various environmental pollutants, ammonia is one of the most widely existing toxic chemicals resulting from various common sources, such as intensive farming, automobile exhaust gases, air-conditioners and the chemical industry. Ammonia is irritating to the respiratory system, skin and eyes,4,5 with short-term exposure to a high ammonia concentration leading to fatal or severe long-term respiratory system damage and lung disorders.Currently, ammonia sensors are widely used in environmental analysis, automotive monitoring and the chemical industry to provide with a safe and pleasant environment. In order to meet the requirements of these applications, semi-conducting metal oxides6,7 and conducting polymers8 have been extensively investigated and utilized to construct commercial sensing devices. However, each kind of sensing material has respective drawbacks. Specifically, a metal oxide based sensor has high sensitivity and low detection limit, but its selectivity is not satisfactory, while conducting polymers have excellent selectivity and high sensitivity, but poor regeneration property. Additionally, all these materials need to operate at a relatively high temperature.9 Nowadays, nanotechnology is intensively explored in fabricating gas sensing materials to improve the comprehensive performance and indeed some significant progress has been made,10 but the cost and the power consumption are very high. Therefore, there is still an urgent need to develop new ammonia sensors which are reliable, inexpensive, with lower operating temperatures and can provide useful analytical outputs.
Since 2004 graphene, a two dimensional monolayer of sp2 carbon in a honeycomb-like network, has attracted tremendous scientific and technological interest due to its intriguing physical, chemical and mechanical properties.11 Moreover, based on its unique microstructures and high quality of the crystal lattice, graphene has been considered as an outstanding candidate for gas sensing applications owing to its high sensitivity and low electronic noise.12 Novoselov's group firstly demonstrated that graphene based sensors are capable of detecting individual gas molecules (e.g., NH3, CO, NO2, H2O, and ethanol) through gas-induced changes in resistivity.13 Thereafter, researchers have been endeavoring in improving the sensitivity, stability, and selectivity of graphene based gas sensors through decoration with nanoparticles, such as metals,14 metal oxides,15,16 organic crystals,17 conductive polymers,18 and metal–organic frameworks.19 Although there are some achievements, its application in ammonia sensing is restricted by its unsatisfactory sensitivity and drift of the sensing signals due to weak retention forces caused by the small size of ammonia particles (0.3 nm)20 and its low adsorption heat upon graphene (very close to the heat of vaporization).21,22 To address these issues, Camille Petit et al. functionalized graphene with copper based metal–organic frameworks, which exhibit a porous structure with functional groups providing a substantial number of adsorption sites for ammonia.23
Hydroxyapatite (Ca10(PO4)6(OH)2, HA) is extensively applied as a medical implant material for its similar composition and structure to the natural bone mineral apatite.24,25 In addition, it has various other applications, such as in chromatography,26 biosensors,27 catalysts28 and ion-exchange.29 HA has also attracted wide attention for its applications in gas sensing30 and fuel cells31 related to its porous nature and ionic conductivity. It has a lattice of hydroxyl ions (OH−) located at the center of Ca2+ triangles along the c-axis of a hexagonal unit cell.32 An ionic conducting mechanism in HA has been proposed and accepted by researchers. The transfer occurs through an OH− ion interacting with the double bonded oxygen of –PO4 groups, because the distance between two adjacent OH− ions is too large (0.344 nm).32 Moreover, the surface of HA possesses several P–OH groups, which act as adsorption sites for various molecules, such as H2O, CH3OH, and CH3I.33,34 The nanocrystalline and porous nature of HA powder plays a vital role in the adsorption of a large number of chemical gases.35,36 However, for pure HA based gas sensors, the response is low and the operating temperature is relatively high, in comparison with that of semiconductor based gas sensors. Some studies have tried to improve the gas sensing property of HA by swift heavy ion irradiation and doping of Si, Fe and Co,37–40 but the enhancement is relatively limited. Actually, HA is a kind of one-dimensional ionic conductor and the conductivity highly relies on the concentration of protons and vacancies.35 Its gas sensing property is restricted by both the poor conductivity and contact between HA particles. Assembling HA nanoparticles onto a graphene matrix can potentially integrate an effective electron transfer medium with rich reactive adsorption sites from both graphene and HA.
In recent years, fabrication of hydroxyapatite and graphene hybrid materials has attracted more and more attention. Zhu et al. fabricated graphene/hydroxyapatite composites via spark plasma sintering and found that their mechanical properties and in vitro biocompatibility were enhanced.41 However, this method requires high temperature, and the hierarchical morphologies and structures inside the composites are difficult to control. Compared to this method, biomineralization is a competitive way in synthesizing graphene/hydroxyapatite. In this method, various biomolecules are utilized to functionalize graphene and introduce oxygenated species, such as hydroxyl groups, sulfate groups, and phosphate groups,42–47 which can provide nucleation sites for the formation of HA. Polydopamine, a mimic of the specialized adhesive foot protein, Mefp-5 (Mytilus edulis foot protein-5),48 exhibits an adhesive versatility with various metal ions owing to its abundant catechol moieties. It is suitable for the formation of hydroxyapatite by co-precipitation of calcium and phosphate ions.49 Park et al. reported a mussel-inspired and polydopamine-assisted method for the mineralization of hydroxyapatite on scaffold materials.50 Simultaneously, graphene oxide can be reduced to graphene with dopamine.44 Therefore, in this work, hydroxyapatite/graphene (HA/GR) composite materials were synthesized through chemical precipitation (cHA/GR) and biomimetic (bHA/GR) methodologies, with the aid of dopamine. The gas sensing properties towards gaseous ammonia were investigated.
2. Results
2.1 Microstructure characterization
After the reduction and mineralization process (see the Experimental section), the obtained composites have two distinguished morphologies. The cHA/GR composites prepared by the chemical precipitation methodology exhibit needle-like HA crystals which appear to be closely attached to graphene flakes. Fig. 1a shows that HA crystals are 60–120 nm in length and 5 nm in diameter. Following the immersion in 1.5 × SBF (see the Experimental section) for 15 days, HA micro-spheres are formed with a diameter of 3μm, which appear to be wrapped by the graphene network, as shown in Fig. 1d and f. It can be clearly seen that HA agglomerates show interconnected and porous structures. | ||
| Fig. 1 TEM images, SAED patterns and HRTEM images of hydroxyapatite–graphene composites synthesized by the chemical precipitation method (a, b, c) and biomimetic method (d, e, f). SEM image of bHA/GR (top right of Fig. 1f). | ||
The TEM results reveal the oriented nucleation and epitaxial growth of the assembled hydroxyapatite in both cHA/GR and bHA/GR composites. The selected area electron diffraction (SAED) patterns are shown in Fig. 1b and e. The diffraction dots show (100) and (110) planes of GO and the diffraction rings correspond to HA hexagonal phases. HRTEM images at the HA/GR interfaces disclose the crystallographic orientation (Fig. 1c and f). HA grows along the (211) plane, which has a lattice spacing of 0.283 nm. There is a similar research revealing the interface structure of HA/GR composites, which shows that the (300) plane of HA superimposed on the surface of graphene sheets, and (002) plane preferentially matches with the cross-section of graphene sheets.51 In this work, at the cross-section of graphene flakes, the multilayered walls are exposed to an interspacing of 0.356 nm, and form an angle of 26° with the (211) plane (Fig. 1c). For hexagonal HA crystals, the angle between the (300) and (211) planes is 25°. Therefore, it is more likely that the (300) plane of HA is parallel to the surface of graphene walls, and the open ends of graphene multi-sheets form interfaces with the (002) plane of HA crystals. These results are in good accordance with the findings on the combination pattern of graphene and hydroxyapatite crystals reported before,51 and the formation mechanisms will be discussed later in this article.
2.2 Phase constitutions
The composition and phase structures of HA/GR composites are shown in Fig. 2. Graphite oxide (GO) has a peak centered at 2θ = 9.3°, corresponding to the (002) plane. After chemical reaction with dopamine, graphite oxide is reduced to graphene (fGR) with a characteristic peak at a 2θ of 24.4°. The composite materials, both cHA/GR and bHA/GR, show the main characteristic peaks of the hexagonal HA phase (JCPDS card no. 09-0432).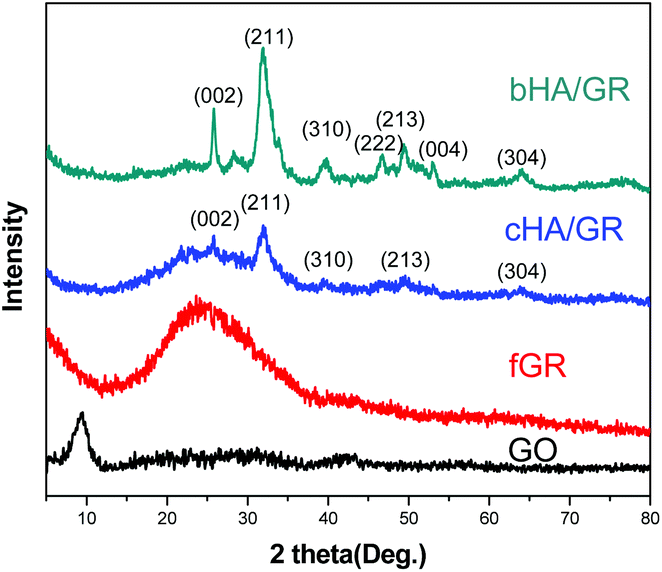 | ||
| Fig. 2 XRD patterns of GO, fGR, cHA/GR, and bHA/GR composites. | ||
Broad diffraction peaks in both patterns demonstrate that HA particles are not well crystallized. Apparently, HA in bHA/GR has sharper peaks, suggesting it has a higher crystallinity than that in cHA/GR. Additionally, the peak at about 25° corresponding to the (002) plane of GR can be distinguished in the patterns of cHA/GR, while this peak disappears in bHA/GR, because the intensity of peaks corresponding to HA in bHA/GR is so high that the diffraction pattern of GR is covered.
In order to confirm the reductant function and investigate the mineralization of HA on graphene, Raman spectroscopy, FTIR and XPS were performed in collaboration and supplementary with each other (find more details in the ESI†). The appearance of peaks corresponding to (PO4)3− in Raman and FTIR spectra suggests the successful deposition of HA (Fig. S1 and S2†). Quantitative analysis by XPS shows that HA deposited on fGR is calcium-deficient and much closer to biological apatite found in humans (Fig. S3a–d†). Moreover, XPS analysis shows that the relative content of the oxygen atom in fGR declines obviously compared with that of GO, which provides strong evidence for the reduction of GO by dopamine (Fig. S3e and f†).
2.3 Gas sensing property of HA/GR composites
Gas sensing performances of the obtained composites were further explored. It is found, as shown in Fig. 3a and c, that HA/GR exhibits responses to various volatile gases at room temperature, including acetone, methanol, ethanol, ammonia, toluene, heptane and tetrahydrofuran (see Fig. 3a). Response sensitivities (denoted by S, see the Experimental section) are 35.7%, 41.6%, 38.4%, 69.5%, 36.5%, 49.6% and 35.5%, respectively. Considering that the composites exhibit the highest response to ammonia with good selectivity (expressed by the ratio of the sensitivities between two specific gases, 1–1.96 in this work), the gas sensing performance to ammonia was investigated further.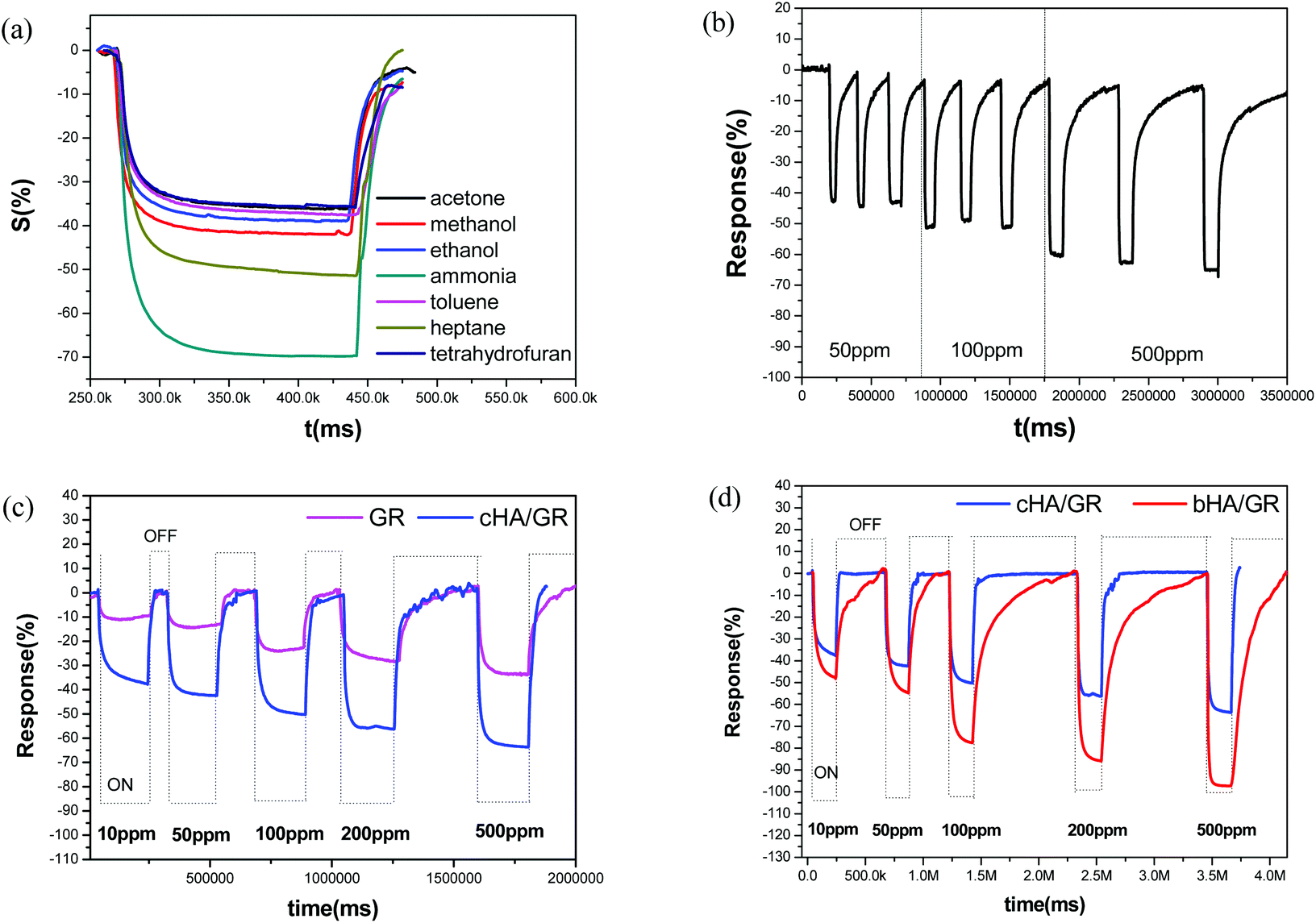 | ||
| Fig. 3 Response–recovery curves of the cHA/GR composite based sensor in various gases with a concentration of 500 ppm (a), cyclic stability of sensing property of cHA/GR to NH3 with different concentrations (b), response–recovery curve of fGR and cHA/GR based sensors to NH3 with different concentrations (c), the comparison of sensing behaviors of cHA/GR and bHA/GR composite based sensors to NH3 with different concentrations (d). All the above sensing behaviors were investigated at room temperature. | ||
Fig. 3b depicts response–recovery curves of fGR and cHA/GR based sensors towards ammonia in the detection range of 10–500 ppm recorded at room temperature. It can be clearly observed that the cHA/GR composite based sensor shows a much larger response magnitude at each level of ammonia concentration, compared with fGR based sensors, with a 1.9–2.7 times enhancement in sensitivity. The stability of the cHA/GR based sensor is also investigated, as shown in Fig. 3c. The continual cycles of response–recovery curves are measured by repeated exposure and removal of ammonia gas. The variances of sensitivities (denoted by v, expressed by the standard deviation of the sensitivities in the same concentration level) are 0.0509, 0.0585 and 0.0732 in 50 ppm, 100 ppm and 500 ppm ammonia, respectively. It is proved that the obtained composites have excellent sensing stability towards ammonia.
The composite bHA/GR is also applied in ammonia sensing and compared with that of cHA/GR. As shown in Fig. 3d, it is evident that the response sensitivities of the bHA/GR based sensor are much higher than that of cHA/GR sensors at each ammonia concentration level. The response time is close, but the recovery time of the bHA/GR based sensor is significantly prolonged. The differences of gas sensing behaviours of the composites obtained by precipitation and biomimetic methodologies are mainly attributed to the differences of morphologies and microstructures.
Moreover, the comparison of sensing performances between HA/GR composites and other carbon materials based sensors in recent studies has been made as shown in Table 1. It is fascinating that HA/GR composites exhibit obvious improvements in performance over the other carbon based materials in recent reports, with both higher sensitivity and lower operation temperature, suggesting the hybrid materials have great potential in a range of applications.
| NH3 sensing materials | Response to 50 ppm NH3 (%) | Temperature range |
|---|---|---|
| a RT: Room temperature. | ||
| fGR (this work) | 14 | RTa |
| cHA/GR (this work) | 42 | RT |
| bHA/GR (this work) | 54 | RT |
| Graphene/Au nanoparticles | 852 | RT |
| Graphene foam | 653 | RT (Joule heating to 400 K is needed during desorption) |
| Bare graphene | 21% to 100 ppm NH354 | RT |
| Carbon nanotube/Pt | 1.255 | 120 °C |
3. Discussion
3.1 In situ synthetic reaction mechanism
Based on the above results, possible reaction mechanisms are proposed for the formation of cHA/GR and bHA/GR composites. As illustrated in Fig. 4, GO can be simultaneously reduced and functionalized by dopamine. It is reported that dopamine shows self-polymerization behavior in the presence of a Tris-HCl buffer solution, thus it can partially reduce the oxygenated species in GO. In this process, the negatively charged functional groups interact with polydopamine (PDA) through hydrogen bonding or electrostatic attraction. Polydopamine is a mimic of the specialized adhesive foot protein, Mefp-5 (Mytilus edulis foot protein-5), in which the catechol moiety strongly binds to various metal ions. Therefore, the PDA layer on fGR can provide more efficient nucleation sites for HA mineralization.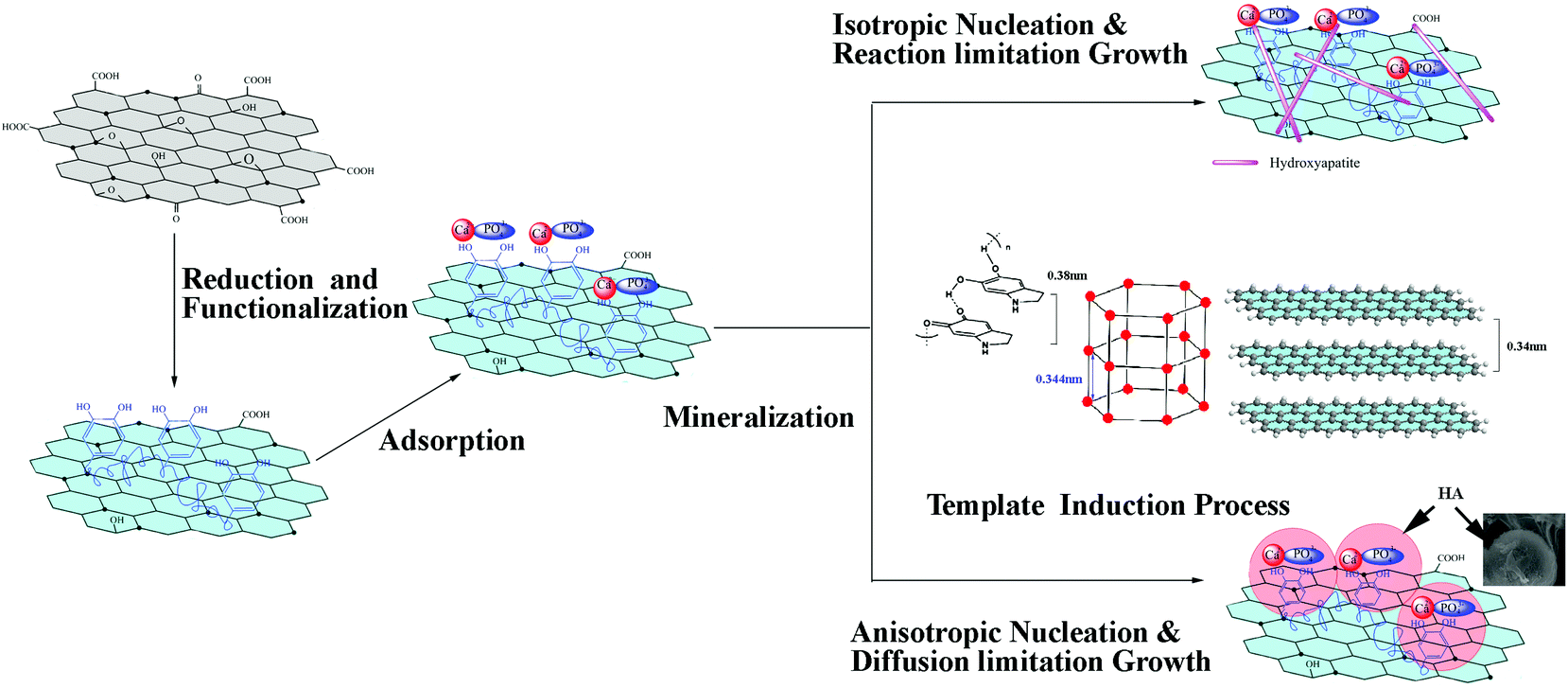 | ||
| Fig. 4 Schematic illustration of the mechanism for the orientated nucleation and epitaxial growth of hydroxyapatite in situ on graphene functionalized with polydopamine, and the morphology control during the mineralization in the chemical precipitation method and the biomimetic method. | ||
It is worth noting that both the special structures of GR and polydopamine exhibit significant influences on the microstructures of HA crystals. It is reported that polydopamine forms stacked and layered structures, which are held together by strong non-covalent forces, including charge transfer, π-stacking, and hydrogen bonding, with a d-spacing (0.38 nm) consistent with that observed in other π-stacked materials.56 Graphene is a two dimensional material with a layered structure. The multilayered walls are exposed with an inter-wall spacing of ∼0.34 nm while the lattice spacing of the (002) plane of HA is 0.344 nm. The layered structure of polydopamine and GR induce orientated nucleation and epitaxial growth of HA. As illustrated in Fig. 4, the (002) plane of HA takes priority to match with the cross-section of both graphene sheets and polydopamine layers, as the lattice mismatches (δ) are respectively 0.01 and 0.1, which are both lower than the incoherence limit of 0.25. Thus a good interfacial bonding can be achieved by the small lattice mismatch, when the (002) plane forms relatively stronger interfaces with the open ends, and the (300) plane matches with the surface of graphene and the polydopamine layer.
There is a classical crystal growth model previously proposed,57 which is suitable to explain the forming mechanism of HA particles with different morphologies. According to the model, growth units diffuse and collide on the growing surface followed by a reaction. The collision frequency is determined by the diffusion rate of growth units, while the reaction rate is determined by thermodynamic conditions, which is associated with the heat transfer rate in aggregation. In chemical precipitation, as Ca(NO3)2 is added into the fGR solution, Ca2+ cations will be attracted and anchored onto hydroxyl groups through electrostatic interactions, and function as nuclei for the crystallization and the growth of HA particles. Ca2+ can in situ react with phosphate ions via electrovalent bonds to form HA nano-particles. In the process, the solution is under vigorous stirring and fGR is homogeneously dispersed, then isotropic nucleation and reaction controlled growth occur. The particles undergo multiple collisions before getting attached, leading to the formation of a compacted structure, a needle-like morphology, while in the biomimetic method SBF is a metastable solution containing calcium and phosphate ions, which are already supersaturated with respect to the apatite. The strong ionic strength prevents the homogeneous dispersion of fGR, and induces anisotropic nucleation. Additionally, the solution is kept still, so diffusion becomes the controlling step in which particles adhere once they encounter each other, leading to an open structure as shown in Fig. 1d.
In this work, it is demonstrated that HA nano-structures can be formed by two in situ precipitation methods assisted by dopamine. Dopamine can reduce and functionalize GO, and introduce more reactive sites and defects for the nucleation and growth of HA nano-particles. The formation of different HA agglomerates is owing to the differences of the microchemical environment and the crystal growth process.
3.2 Mechanism of gas sensing
The fGR behaves like an n-type semiconductor sensor, i.e. the resistance decreases in the presence of ammonia, and electrons dominate in conductivity. After deposition with HA crystals, active adsorption and reaction sites increase. HA acts as anchor to capture ammonia molecules, while GR plays the role of an efficient electronic transmission medium as well as gas sensing. Therefore, there is a good combination of the adsorption capability of HA and the conductivity of GR, and it is exactly the reason for the significant enhancement of the gas sensing sensitivity of the composites. However, there are obvious noises during the desorption process of cHA/GR, because of the existence of grain boundary barriers between HA crystals and GR. Considering the fact that the response sensitivity is obviously enhanced while the response and recovery time is not prolonged, it can be concluded that cHA/GR hybrid materials have more excellent gas sensing behavior than fGR.It is interesting that the gas sensing sensitivity of bHA/GR (with a specific surface area of 161.67 m2 g−1) is higher than that of cHA/GR, which is with a much lower specific surface area of 20.40 m2 g−1. The reason is that bHA/GR has much finer crystals, and the nano-porous structure can facilitate the adsorption and the diffusion of gas. Additionally there are more active reaction sites on the open structure of bHA/GR than in the compacted structure of cHA/GR. The bHA/GR based sensor shows an elongated recovery time, compared with a cHA/GR based sensor, because the adsorbed gas must diffuse out from the interconnected channels of the HA agglomerates, thus it requires much time.
Fig. 5 displays a plausible model for the gas sensing mechanism of HA/GR composites towards ammonia. As there are various adsorption sites on the surface of HA crystals, HA can adsorb molecules with positively or negatively charged groups, and can capture gas molecules effectively. To explain the gas sensing property of HA, it is vitally important to understand the conductivity mechanism of HA. Protons are charge carriers in HA crystals at room temperature, rather than calcium cations and phosphate ions. HA crystals display P63/m hexagonal symmetry, and consist of an adaptable framework, penetrated by channels along the c-axis in which protons can migrate. Protons as the charge carriers migrate from OH− to the adjacent PO43−. Under an ammonia atmosphere, the HA/GR composite can capture NH3 molecules onto its surface. NH3 acts as a proton donor, and produces the intermediate NH2−, which reacts with the chemisorbed oxygen ions, such as O2−, O− and O2−:
| NH3 ⇌ NH2− + H+ |
| NH2− + O−(O2−/O2−) ⇌ NOxn− + H2O |
 | ||
| Fig. 5 Scheme of the mechanism of the gas sensing property of HA/graphene to NH3. | ||
In this process, HA crystals act as proton acceptor, and the conductivity decreases due to the increment of charge carriers. Additionally, NOxn− can be incorporated in the lattice of HA crystals. It is more likely that NOxn− will replace OH− on the surface or in the channel of HA crystals, and introduce a slight change of the framework structure. If there is no graphene, carriers must cross interface potential barriers, and increase the electrical resistance of the HA. However, GR, which has a good conductivity, plays the role of an efficient carrier transfer medium. In this work, polydopamine functionalized graphene shows n-type semiconductor properties, that is electrons dominate the conductivity. Under a reducing atmosphere, graphene takes electrons from NH3 molecules and the resistance decreases. Although graphene has a very large specific surface area, there are not enough active reaction sites towards ammonia, so the gas sensitivity of pure graphene is limited. After the mineralization, HA can provide various adsorption and active sites, and graphene acts as a bridge between HA crystals, and hence charge carriers can transfer more efficiently. It is the combination of the adsorption capability of HA and the conductivity of graphene that leads to the observed excellent gas sensing property of HA/GR composite materials.
4. Conclusions
HA crystals with different morphologies and structures were assembled upon graphene successfully with the aid of dopamine. The layered structures of graphene and polydopamine induced orientated nucleation and epitaxial growth along the (211) plane of HA. It is proposed that a reaction-controlled growth process occurs in the chemical precipitation method, leading to needle-like HA crystals, while in the biomimetic method, anisotropic nucleation and diffusion-limited growth occur, thus forming microspherical agglomerates. The composites are effective in gas sensing. It was demonstrated that the HA/GR composite has much better sensing performance towards ammonia at room temperature due to the synergy of the adsorption capabilities of HA and the electric conductivity of graphene. For cHA/GR and bHA/GR composites, different gas sensing performances are mainly attributed to the differences in structures and morphologies. bHA/GR composites with much finer crystals and nano-porous structures show a much higher response (improved analytical sensitivity) to ammonia than that of cHA/GR; therefore, the gas sensing properties can be readily adjusted by controlled syntheses of HA/GR composites.5. Experimental
5.1 Materials
Graphene oxide platelets were purchased from Nanjing Xian Feng Nanotechnology Co., P.R. China. Dopamine-hydrochloride (98.5%), Ca(NO3)2·4H2O, and (NH4)2·HPO4 were purchased from Alfa Aesar. All other chemicals were of analytical reagent grade, and used as received unless otherwise noted. Deionized water (DI water, ULUP-1-20 T) was used as solvent in all experiments.5.2 Synthesize of functionalized graphene based HA composites
Based on an in situ chemical precipitation reaction, the preparation details are listed as follows:Preparation and functionalization of graphene oxide (GO) solution: raw GO platelets were dissolved in DI water by ultrasonic dispersion (10 ml of a 1 mg ml−1 solution). Polydopamine (PDA) coating was performed by mixing the GO suspension (1 mg ml−1) with a buffer solution (10 ml of 2 mg ml−1 of dopamine-hydrochloride in 10 mM Tris buffer, pH 8.5) at room temperature. The solution was subjected to centrifugation, and washed with DI water.
In situ synthesis of HA nanoparticles: The concentration of functionalized graphene (fGR) produced by centrifugation was dissolved in 10 mL DI water utilizing ultrasonic dispersion, and 0.0005 mol Ca(NO3)2·4H2O was then added. Then 10 mL of 0.03 mol L−1 (NH4)2·HPO4 solution was added dropwise into the mixture at a pH value of 11. The suspensions were subjected to centrifugation, and washed with DI water and alcohol, respectively. The precipitates were dried at 60 °C. The composite prepared by this method is named cHA/GR.
Biomineralization of HA nanoparticles: The concentrate of fGR produced by centrifugation was dispersed in 1.5 × SBF (matrix to simulate body fluids, Na+, 213.0 mM; K+, 7.5 mM; Mg2+, 2.25 mM; Ca2+, 3.75 mM; Cl−, 221.7 mM; HCO3−, 6.3 mM; HPO42−, 1.5 mM; and SO42−, 0.75 mM) solution by ultrasonic dispersion. The solution was incubated in a WAVE bioreactor at 37 °C for 14 days. The precipitates were washed and dried at 60 °C. The composite prepared by this method is named as bHA/GR.
5.3 Materials characterization
The crystalline phases of the composites were verified by X-ray diffraction (XRD) using a Rigaku D/max 2500 diffractometer with CuKα radiation (λ = 1.54178 Å) at 40 kV. The diffraction angles (2θ) were from 5 to 90° in steps of 0.02°. The morphologies of the composites were characterized by using a scanning electron microscope (SEM, NOVA NANOSEM 230). Transmission electron microscopy (TEM) was performed on a JEOL JEM-2100F at 200 kV. Samples were prepared by placing a drop of a dilute alcohol dispersion of the products on the surface of a copper grid. Raman spectroscopy was performed at 514.5 nm by using a Raman Imaging Microscope System (Renishaw RM1000) in a backscattering geometry. Fourier transform infrared spectroscopy (Thermo/USA Nicolet Nexus 670) was performed to identify the functional groups of the composites. The spectra were recorded from 400 cm−1 to 4000 cm−1. X-ray photoelectron spectroscopy (XPS, Perkin-Elmer, model PHI 5400) was employed to identify the composition and elemental states of the obtained powders. The binding energies were calibrated by the C 1s hydrocarbon peaks. The quantitative analyses and the curve fitting were conducted by the Casa XPS software package. The specific surface area was calculated by the Brunauer–Emmett–Teller (BET, Quantachrome Monosorb) method.5.4 Gas sensing properties of HA/GR composites
Ammonia was chosen as the target gas to study the gas-sensing properties. The HA/GR was mixed with polyvinyl alcohol to form a paste, and then coated on the outside surface of an alumina tube. A pair of gold electrodes was installed at each end of the ceramic tube. Each electrode was connected with two Pt wires. A Ni–Cr heating alloy coil was inserted into the tube to form an indirectly-heated gas sensor. The structure of the sensor is shown in Fig. 6. The electrical properties of the sensor were measured by using an intelligent gas sensing system (CGS-8, China).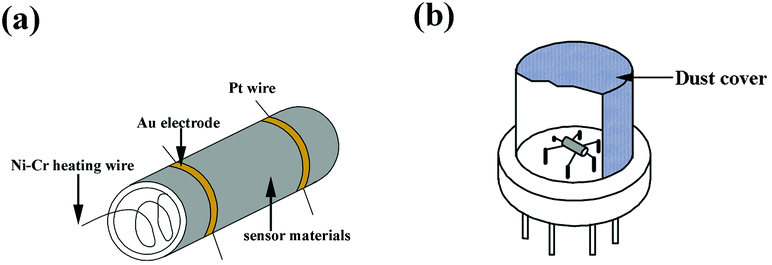 | ||
| Fig. 6 The assembly of sensing materials on the ceramic tube (a), and the gas sensing device (b). | ||
The response of the sensors was calculated using the equation:
| S = (Rg − Ra)/Rg × 100 |
Acknowledgements
This work was financially supported by the National Science Foundation of China (no. 51272289).Notes and references
- G. Dar, A. Umar, S. A. Zaidi, S. Baskoutas, S. Hwang, M. Abaker, A. Al-Hajry and S. Al-Sayari, Talanta, 2012, 89, 155–161 CrossRef CAS PubMed.
- G. Korotcenkov, Sens. Actuators, B, 2005, 107, 209–232 CrossRef CAS PubMed.
- A. Sutti, C. Baratto, G. Calestani, C. Dionigi, M. Ferroni, G. Faglia and G. Sberveglieri, Sens. Actuators, B, 2008, 130, 567–573 CrossRef CAS PubMed.
- L. G. Close, F. I. Catlin and A. M. Cohn, Arch. Otolaryngol., 1980, 106, 151–158 CrossRef CAS.
- C. Leung and C. Foo, Ann. Acad. Med., 1992, 21, 624–629 CAS.
- C. S. Rout, M. Hegde, A. Govindaraj and C. Rao, Nanotechnology, 2007, 18, 205504 CrossRef.
- G. Wang, Y. Ji, X. Huang, X. Yang, P.-I. Gouma and M. Dudley, J. Phys. Chem. B, 2006, 110, 23777–23782 CrossRef CAS PubMed.
- V. Chabukswar, S. Pethkar and A. A. Athawale, Sens. Actuators, B, 2001, 77, 657–663 CrossRef CAS.
- B. Timmer, W. Olthuis and A. v. d. Berg, Sens. Actuators, B, 2005, 107, 666–677 CrossRef CAS PubMed.
- Z. Fan, D. Wang, P.-C. Chang, W.-Y. Tseng and J. G. Lu, Appl. Phys. Lett., 2004, 85, 5923–5925 CrossRef CAS PubMed.
- M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2009, 110, 132–145 CrossRef PubMed.
- X. Huang, N. Hu, R. Gao, Y. Yu, Y. Wang, Z. Yang, E. S.-W. Kong, H. Wei and Y. Zhang, J. Mater. Chem., 2012, 22, 22488–22495 RSC.
- F. Schedin, A. Geim, S. Morozov, E. Hill, P. Blake, M. Katsnelson and K. Novoselov, Nat. Mater., 2007, 6, 652–655 CrossRef CAS PubMed.
- X. Huang, Z. Yin, S. Wu, X. Qi, Q. He, Q. Zhang, Q. Yan, F. Boey and H. Zhang, Small, 2011, 7, 1876–1902 CrossRef CAS PubMed.
- Y. Su, S. Li, D. Wu, F. Zhang, H. Liang, P. Gao, C. Cheng and X. Feng, ACS Nano, 2012, 6, 8349–8356 CrossRef CAS PubMed.
- Z. Zhang, R. Zou, G. Song, L. Yu, Z. Chen and J. Hu, J. Mater. Chem., 2011, 21, 17360–17365 RSC.
- S. Wang, B. M. Goh, K. K. Manga, Q. Bao, P. Yang and K. P. Loh, ACS Nano, 2010, 4, 6180–6186 CrossRef CAS PubMed.
- H. Kim, A. A. Abdala and C. W. Macosko, Macromolecules, 2010, 43, 6515–6530 CrossRef CAS.
- C. Petit and T. J. Bandosz, Adv. Mater., 2009, 21, 4753–4757 CAS.
- J. Thompson, Phys. Rev. A, 1971, 4, 802 CrossRef.
- W. Spencer, C. Amberg and R. Beebe, J. Phys. Chem., 1958, 62, 719–723 CrossRef CAS.
- R. Overstreet and W. Giauque, J. Am. Chem. Soc., 1937, 59, 254–259 CrossRef CAS.
- C. Petit, B. Mendoza and T. J. Bandosz, Langmuir, 2010, 26, 15302–15309 CrossRef CAS PubMed.
- T.-M. G. Chu, D. G. Orton, S. J. Hollister, S. E. Feinberg and J. W. Halloran, Biomaterials, 2002, 23, 1283–1293 CrossRef CAS.
- P. Habibovic, F. Barrere, C. A. Blitterswijk, K. Groot and P. Layrolle, J. Am. Ceram. Soc., 2002, 85, 517–522 CrossRef CAS PubMed.
- Y. Murakami, K. Sugo, T. Yoshitake, M. Hirano and T. Okuyama, Sep. Purif. Technol., 2013, 103, 161–166 CrossRef CAS PubMed.
- S. P. Wang, Y. Lei, Y. Zhang, J. Tang, G. L. Shen and R. Q. Yu, Anal. Biochem., 2010, 398, 191–197 CrossRef CAS PubMed.
- D. L. Zhang, H. W. Zhao, X. Y. Zhao, Y. M. Liu, H. Chen and X. J. Li, Prog. Chem., 2011, 23, 687–694 CAS.
- A. Yasukawa, T. Yokoyama, K. Kandori and T. Ishikawa, Colloids Surf., A, 2008, 317, 123–128 CrossRef CAS PubMed.
- R. S. Khairnar, R. U. Mene, S. G. Munde and M. P. Mahabole, 4th Nanoscience and Nanotechnology Symposium (Nns2011): An International Symposium, 2011, 1415.
- K. Muroyama, H. Munakata and K. Kanamura, Electrochemistry, 2007, 75, 807–812 CrossRef CAS PubMed.
- K. Yamashita, K. Kitagaki and T. Umegaki, J. Am. Ceram. Soc., 1995, 78, 1191–1197 CrossRef CAS PubMed.
- E. Fujii, K. Kawabata, K. Ando, K. Tsuru, S. Hayakawa and A. Osaka, J. Ceram. Soc. Jpn., 2006, 114, 769–773 CrossRef CAS.
- H. Tanaka, M. Chikazawa, K. Kandori and T. Ishikawa, Phys. Chem. Chem. Phys., 2000, 2, 2647–2650 RSC.
- M. Nagai and T. Nishino, Solid State Ionics, 1988, 28, 1456–1461 CrossRef.
- H. Owada, K. Yamashita, T. Umegaki, T. Kanazawa and M. Nagai, Solid State Ionics, 1989, 35, 401–404 CrossRef CAS.
- R. U. Mene, M. P. Mahabole and R. S. Khairnar, Radiat. Phys. Chem., 2011, 80, 682–687 CrossRef CAS PubMed.
- R. U. Mene, M. P. Mahabole, K. C. Mohite and R. S. Khairnar, Mater. Res. Bull., 2014, 50, 227–234 CrossRef CAS PubMed.
- R. U. Mene, M. P. Mahabole, K. C. Mohite and R. S. Khairnar, J. Alloys Compd., 2014, 584, 487–493 CrossRef CAS PubMed.
- R. U. Mene, M. P. Mahabole, R. Sharma and R. S. Khairnar, Vacuum, 2011, 86, 66–71 CrossRef CAS PubMed.
- J. Zhu, H. M. Wong, K. W. K. Yeung and S. C. Tjong, Adv. Eng. Mater., 2011, 13, 336–341 CrossRef CAS PubMed.
- Z. J. Fan, J. Q. Wang, Z. F. Wang, Z. P. Li, Y. N. Qiu, H. G. Wang, Y. Xu, L. Y. Niu, P. W. Gong and S. R. Yang, J. Phys. Chem. C, 2013, 117, 10375–10382 CAS.
- M. Li, Y. Wang, Q. Liu, Q. Li, Y. Cheng, Y. Zheng, T. Xi and S. Wei, J. Mater. Chem. B, 2013, 1, 475–484 RSC.
- H. Liu, P. Xi, G. Xie, Y. Shi, F. Hou, L. Huang, F. Chen, Z. Zeng, C. Shao and J. Wang, J. Phys. Chem. C, 2012, 116, 3334–3341 CAS.
- H. Y. Liu, J. Cheng, F. J. Chen, D. C. Bai, C. W. Shao, J. Wang, P. X. Xi and Z. Z. Zeng, Nanoscale, 2014, 6, 5315–5322 RSC.
- H. Y. Liu, J. Cheng, F. J. Chen, F. P. Hou, D. C. Bai, P. X. Xi and Z. Z. Zeng, ACS Appl. Mater. Interfaces, 2014, 6, 3132–3140 CAS.
- J. Zhao, Z. C. Zhang, Z. W. Yu, Z. N. He, S. S. Yang and H. Y. Jiang, Appl. Surf. Sci., 2014, 289, 89–96 CrossRef CAS PubMed.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- W. M. Chirdon, W. J. O'Brien and R. E. Robertson, J. Biomed. Mater. Res., Part B, 2003, 66, 532–538 CrossRef PubMed.
- J. Ryu, S. H. Ku, H. Lee and C. B. Park, Adv. Funct. Mater., 2010, 20, 2132–2139 CrossRef CAS PubMed.
- Y. Liu, J. Huang and H. Li, J. Mater. Chem. B, 2013, 1, 1826–1834 RSC.
- M. Gautam and A. H. Jayatissa, Solid-State Electron., 2012, 78, 159–165 CrossRef CAS PubMed.
- F. Yavari, Z. Chen, A. V. Thomas, W. Ren, H.-M. Cheng and N. Koratkar, Sci. rep., 2011, 1 Search PubMed.
- V. Dua, S. P. Surwade, S. Ammu, S. R. Agnihotra, S. Jain, K. E. Roberts, S. Park, R. S. Ruoff and S. K. Manohar, Angew. Chem., Int. Ed., 2010, 49, 2154–2157 CrossRef CAS PubMed.
- R. Rossi, M. Alvisi, G. Cassano, R. Pentassuglia, D. Dimaio, D. Suriano, E. Serra, E. Piscopiello, V. Pfister and M. Penza, in Sensors and Microsystems, Springer, 2012, pp. 115–119 Search PubMed.
- D. R. Dreyer, D. J. Miller, B. D. Freeman, D. R. Paul and C. W. Bielawski, Langmuir, 2012, 28, 6428–6435 CrossRef CAS PubMed.
- V. A. Bogoyavlenskiy and N. A. Chernova, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2000, 61, 1629 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5an00622h |
| This journal is © The Royal Society of Chemistry 2015 |