
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient intracellular delivery and improved biocompatibility of colloidal silver nanoparticles towards intracellular SERS immuno-sensing†
Vinay
Bhardwaj
,
Supriya
Srinivasan
and
Anthony J.
McGoron
*
10555 West Flagler Street, Department of Biomedical Engineering, Florida International University, Miami, FL, USA. E-mail: mcgorona@fiu.edu; Fax: +1-305-348-6954; Tel: +1-305-348-1352
First published on 29th April 2015
Abstract
High throughput intracellular delivery strategies, electroporation, passive and TATHA2 facilitated diffusion of colloidal silver nanoparticles (AgNPs) are investigated for cellular toxicity and uptake using state-of-art analytical techniques. The TATHA2 facilitated approach efficiently delivered high payload with no toxicity, pre-requisites for intracellular applications of plasmonic metal nanoparticles (PMNPs) in sensing and therapeutics.
Nanoparticles (NPs), and in particular noble metal nanoparticles (NMNPs) with plasmonic properties, e.g. gold (Au) and silver (Ag) are versatile agents with numerous diagnostic and therapeutic applications.1–6 However, the problem of poor cell uptake of NPs, including NMNPs, has limited their real-world in situ and in vivo applications.5 The most used practices to achieve cell uptake can be categorized into three intracellular delivery strategies: passive diffusion, facilitated diffusion and active delivery.6 The NPs can diffuse across cell membranes based on their physiochemical properties (passive diffusion) or they can be functionalized to mediate diffusion (facilitated diffusion).7–10 Increasing cell permeability to create localized membrane pores using electrical (electroporation) or mechanical (microinjection) force (active delivery) to allow improved diffusion is another strategy to increase cell uptake of NPs.11 Passive diffusion is usually very slow and technically fails to deliver high payload, which is a critical demand for intracellular applications, primarily sensing. Label-free cell-based biosensors (LF-CBB) have promising important applications in environmental monitoring, biosecurity and rapid diagnostics. The LF-CBB offers several important advantages over the conventional label-based cell-free assays, including polymerase chain reaction (PCR) and enzyme-linked immuno-sorbent assay (ELISA).12,13 Most noteworthy, the label-free approach allows direct detection of analyte, without any chance of uncertainty introduced by the dye/label conjugate. A cell-based biosensor (CBB) offers two major advantages over a cell-free detection. First, a CBB allows detection in live cells, measuring bioavailability of analyte and the functional response of cells to an analyte. Second, CBB enables dynamic monitoring as compared to end-point detection in cell-free assays, PCR and ELISA.
The scattering properties of plasmonic metal NPs become more dominant than absorbance when the diameter is greater than 40 nanometers (nm), the most suitable size for scattering based sensor techniques, including surface-enhanced Raman spectroscopy (SERS).14 Spherical NPs of ∼50 nm diameter have higher cell uptake than other shapes and sizes as well as maximum SERS enhancement.8,15 Interestingly, AgNPs exhibit higher scattering and absorption than AuNPs, which makes AgNPs a better choice over AuNPs for SERS sensing, near-infrared imaging and photothermal basis of therapeutic applications.16,17 Progress in SERS has enabled label-free/label-based detection in cell-free/cell-based systems from single cells to tissues, and entire organisms, including humans.18–21 The SERS immuno-sensing outperforms traditional ELISA technique in allowing RISE (Rapid, Inexpensive, Simple and Effective) detection, which are pre-requisites to develop a detect-to-protect class of biosensor, especially in the event of chemical/biological warfare and natural/industrial disasters.22–28 However, there is no CBB using SERS immuno-sensor technology (SIST), which allows intracellular detection of specific biomarker proteins.
The Lincoln Laboratory at Massachusetts Institute of Technology (MIT), U.S.A. has developed one-of-its-kind CBB technology, CANARY (Cellular Analysis and Notification of Antigen Risks and Yields), which is truly a pioneer in detect-to-protect biosensors.29 CANARY allows an optimum combination of speed and sensitivity required to provide warning signals in response to biological warfare agents (BWAs), less than 50 pathogen particles in less than 3 minutes. A portable CBB device based on CANARY, called PANTHER (for PAthogen Notification for THreatening Environmental Releases), allowing rapid detection of several pathogens (biological-toxins) is licensed by Innovative Biosensors Inc., U.S.A. Unfortunately, the use of mammalian cells in the CANARY design limits its shelf-life to 2 days at room temperature (RT), which can be increased to 2 weeks by using genetic engineering to over-express protective genes in the CANARY cells.30 The poor shelf-life of the mammalian CBB technologies is an inherent limitation and unfavorable attribute for in-field environmental surveillance of toxins.
Our group focuses on the development of a portable CBB-SIST to meet the aforementioned need of a biosensor for the environmental surveillance of chemical-toxins. We use yeast, a single-celled eukaryotic organism, which is the choice of sensor organisms in commercial portable CBB assays/sensors with high shelf-life, a requirement for environmental monitoring.31–33 Current, commercial CBB assays, primarily bioreporters, are limited by the use of genetically modified organisms (GMOs), the requirement of labels, long incubation time and the need for a special skill-set.33 We recently reported the rapid and label-free colloidal AgNPs-based SERS immuno-sensor that has an edge over ELISA for cell-free, end-point detection of RAD54 and HSP70 biomarkers expressed by yeast in response to environmental-toxins.22 The efficient delivery of AgNPs into yeast is a pre-requisite to develop the proposed portable CBB-SIST for dynamic monitoring of chemical-toxins in the environment. Herein, we report our investigation of strategies to efficiently deliver colloidal AgNPs into yeast. Yeasts have an extra barrier (rigid cell wall) compared to animal cells, which provide robustness, but in turn challenge the intracellular delivery of cargo.34 Several delivery strategies have been reported to increase cell uptake in intact yeast: electroporation, bombardment using microprojectiles, microinjection, etc.35–37 However, almost all are intended to deliver DNA (transformation) and most of these remain proof-of-concept delivery techniques. There are numerous, but contrary reports on the toxicity of NMNPs, primarily AgNPs.38–41 There are a few reports on the rapid and targeted delivery of AgNPs using electroporation and microinjection for intracellular SERS sensing in animal cells.42–45 However, little effort was put into investigating the cellular damage of NMNPs. The inherent heating and leaching effect of NMNPs are primary mechanisms of toxicity.17,40 Additionally, the qualitative and quantitative uptake of AgNPs was not comprehensively studied and the uptake was not characterized using state-of-the-art techniques. Vo-Dinh and co-workers have used the TAT-mediated approach to efficiently deliver gold nanostars for intracellular SERS applications21 but the entrapment of TAT-functionalized NPs in endosomes is a major concern.46,47 TATHA2, a fusogenic viral peptide with cell permeability (TAT) as well as the endosome rupture release (HA2) properties facilitates rapid and high cell uptake of NPs by pinocytosis.46–49 The TATHA2-mediated delivery has primarily been investigated for intracellular targeting of the nucleus for applications in gene expression46,47 and not for intracellular immuno-sensing, which requires preferentially uniform intracellular distribution of NPs, as the proteins are ubiquitously distributive inside cells. Although, S. Kumar and co-workers have demonstrated TATHA2-mediated delivery for intracellular protein detection, they used AuNPs.48,49 Indeed, in our knowledge we are the first group to investigate TATHA2-mediated intracellular delivery of AgNPs and the first group to study TATHA2-mediated delivery for intracellular SERS immuno-sensing. We did a comprehensive study to investigate AgNPs uptake in yeast by using three delivery strategies, which are most suitable for our proposed CBB-SIST: passive diffusion, TATHA2 facilitated diffusion and electroporation.
We synthesized the colloidal AgNPs using a simple citrate reduction method and characterized them by UV-Vis spectrophotometry, dynamic light scattering (DLS) and transmission electron microscopy (TEM). Then, the effects of the three delivery strategies; active, passive and facilitated were studied to characterize their cell toxicity and uptake in yeast. The toxicity of AgNPs and the physical damage by the delivery strategies was investigated by cell viability, growth inhibition assays and scanning electron microscopy (SEM). The cell uptake was measured by inductively coupled plasma mass spectrometry (ICPMS) to quantify silver content and in situ TEM to observe AgNPs in ultrathin sections of yeast. The AgNPs adsorbed on the cell surface were removed using a mild iodine/potassium iodide (I2/KI) etching solution to enable the measurement of only the internalized particles.9 The selective removal and cytotoxic effect of the etching solution was tested by SEM and cell viability assays, respectively. The toxicity and uptake of AgNPs in yeast was performed in minimal medium (DI water buffered to pH 8.5) to avoid the confounding effect of serum proteins on cell uptake, a concern when using complete culture medium.8,38–41 Uptake studies were normalized to incubation of 105 AgNPs per cell and assessed for AgNPs surface localization (adsorbed on cell surface) and volume localization (intracellular or fully penetrating) before and after etching, respectively, for in situ TEM.
Colloidal AgNPs were prepared by a single step facile synthesis using the conventional citrate reduction method with some modifications, as reported in our previous work.22 The yellow-greenish colloidal solution containing almost spherical, mono-dispersed particles with average diameter ∼60 nm and maximum absorption at 436 nm wavelength were prepared (Fig. 1). The AgNP colloids had high negative surface charge (−40 millivolts), contributed by loosely attached citrate (COO)33− and nitrate ions (NO3−). The AgNP colloids were used without any modifications for free diffusion (passive delivery) and electroporation (active delivery) in yeast. The AgNPs were functionalized with TATHA2 to mediate/facilitate diffusion (facilitated delivery). TATHA2 was conjugated to AgNPs through biotinylation and was quantified using fluorescein isothiocyanate (FITC) labeled biotin monoclonal antibody (MAb) to estimate the number of TATHA2 molecules conjugated to single AgNP.49 See ESI† for more details on Experimental section.
 | ||
| Fig. 1 Physical characterization of AgNPs: TEM image (scale bar 0.5 μm) and inset, top right, shows an individual spherical AgNP (scale bar 20 nm). The lower insets show the UV-Vis absorption maximum at 436 nm (left) and DLS histogram average size 60 nm (right). | ||
The scheme of the TATHA2 conjugation process and the chemical characterization using Raman micro-spectroscopy is presented (Fig. 2). The change in the chemical specific marker peaks/bands indicate the successful conjugation of peptides to AgNPs. The loosely bound nitrate groups (NO3−) on bare-AgNPs are easily displaced by other active functional groups indicated by a decrease/disappearance of the NO3 peak (Fig. 2#1, marker peak ∼1060 cm−1).25 The conjugation between the AgNPs and MMT via SH group (linker) is evident by metal–S–C–C bending, including metal–S (∼360 cm−1) and υ(C–S)g at 614 cm−1, gauche conformation, dominant in a monolayer arrangement (Fig. 2#2.a). The decrease in the marker band of the linker at 1280 cm−1, with subsequent peptide conjugation, is in agreement with another report.26 The torsion of metal–S–C–C results in trans conformation, υ(C–S)t at 713 cm−1, a dominant form in bilayers, observed after subsequent peptide binding, anti-biotin MAb (Fig. 2#2.b) and biotin-TATHA2 (Fig. 2#3). The biotinylation, linkage between biotin-TATHA2 and anti-biotin MAb is validated by the biotin marker band (713 cm−1), which overlap with υ(C–S)t but shows increased peak intensity. Additionally, the changes in tryptophan (significant increase in 1390 cm−1 and decrease in 1342 and 1590 cm−1) support the tryptophan sensitive interaction between protein-biotin conjugates (indole N1–H bond). An average of 22 TATHA2 molecules was conjugated to single AgNP. The physiochemical properties of colloidal AgNPs designed to investigate three intracellular delivery strategies are summarized in the ESI (Table S1†). The significant change in surface charge of the AgNPs after TATHA2 functionalization is primarily attributed to the replacement of the loose anionic cap by the cationic groups, TAT moiety. The concentration of AgNPs after synthesis, estimated by ICPMS was 70 ppm or 0.07 mg ml−1, roughly equiv. to 7 × 1010 AgNPs ml−1. The yield of TATHA2-AgNPs was quite high, 80%. The high yield is attributed to the conjugation strategy, the linker-Ab was conjugated to the AgNPs rather than a general procedure of first conjugating the linker to the AgNPs and then to the Ab, which involves extra washing steps using centrifugation.22,24–26 Washing using centrifugation not only decreases the yield but also leads to leaching or dissolution of the AgNPs into free ions, one of the primary reasons for AgNPs toxicity.38–40
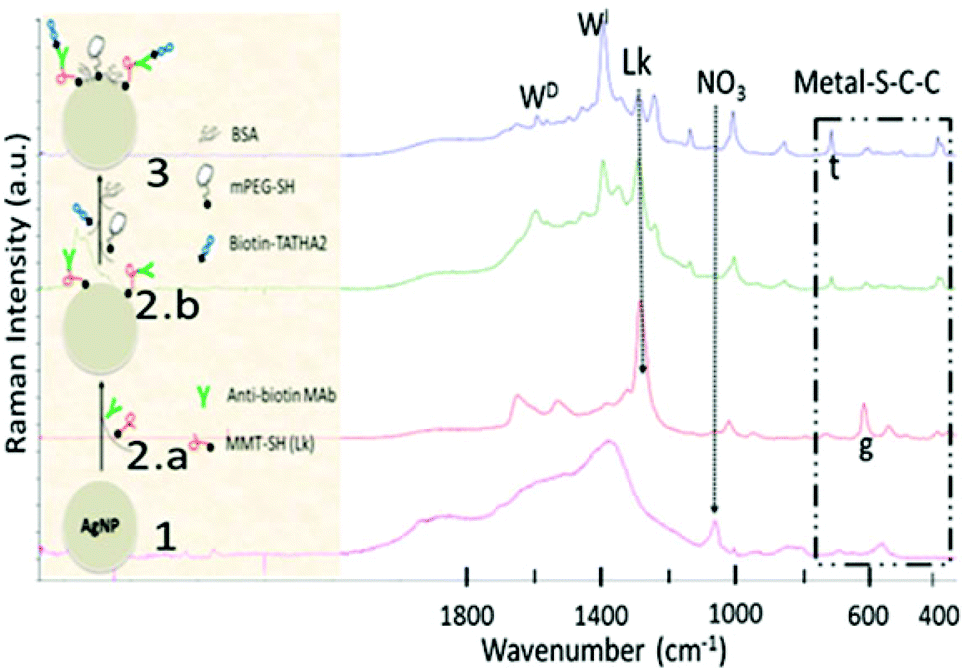 | ||
| Fig. 2 Conjugation of TATHA2 to AgNPs: Schematic of the conjugation process (left) and the Raman micro-spectroscopic characterization of the conjugation steps (right). The characteristic spectra of the conjugation steps are denoted as follows 1: AgNPs, 2.a: AgNPs-MMT, 2.b: AgNPs-MMT-MAb, and 3: AgNPs-MMT-MAb-TATHA2 (TATHA2-functionalized-AgNPs). g and t are gauche and trans conformers of υ(C–S) or metal–S–C–C bending, arrows indicates decrease in NO3 (nitrate) and Lk (Linker MMT) peaks, WI and WD are increase and decrease in tryptophan peaks, respectively. | ||
To test the effect of electroporation to deliver AgNPs in yeast, we used a BIO RAD MicroPulser Electroporation Apparatus designed primarily for transformation, DNA delivery in bacteria and yeast. We tested the pre-programmed company specific settings for the electroporation of yeast, cuvette gap (d) = 0.2 cm and voltage (V) = 1.5 kV, allowing electric field strength (E) = 1.5/0.2 = 7.5 kV cm−1 (E is V/d). We also tested the parameters optimized by Yu and coworkers, the only group to deliver AgNPs into animal cells using electroporation for intracellular SERS sensing; E = 0.875 kV cm−1 using four consecutive pulses.42,43 Also, we tested the lowest possible dose and time achievable by the instrument (E = 0.5 kV cm−1 for 1 ms, single pulse) for electroporation of AgNPs into yeast. We noticed, yeast cells undergo severe physical damage and toxicity even at the lowest electroporation dose (Fig. 3, I.C & II.C), inconsistent with Yu and coworkers observation in animal cells.42,43 No damage and toxicity to yeast was observed by electroporation in the absence of AgNPs (Fig. 3, I.A & II.A). Heat generation from NMNPs (AgNPs has ∼10 fold more heat potential than AuNPs) in the presence of electric field and/or laser is a characteristic physical phenomenon.17 We observed a damaging effect of TEM (200 kV) to individual AgNP as well as the effect of near-infrared (785 nm) Raman laser (100 mW) to AgNPs aggregates (concentrated 10 fold, 10 × 70 = 700 ppm) in presence of YPD media (Fig. S1 and S2†), consistent with other reports.50,51 It is essential to optimize key parameters of electroporation; electric pulse strength (E), pulse exposure (time × number) and temperature,44 and SERS measurement; concentration of AgNPs to analyte and time of incubation,24 excitation laser's wavelength, power output, spot size (objective) and exposure (time × number of exposures)51 for efficient intracellular delivery and detection, respectively. The induction of stress-proteins by electroporation, microinjection and other similar active delivery methods deter their applications for intracellular SERS immuno-sensing.45
 | ||
| Fig. 3 Cellular damage (I) and toxicity (II & III) of AgNPs delivery strategies: Panel I and II show the effect of electroporation by SEM (damaging effect) and agar plating (toxicity effect), respectively. A: no AgNPs, B: no electroporation and C: both, electroporation in presence of AgNPs, cell damage indicated by arrows. Panel III shows the growth inhibition effect (toxicity) of BR- (bare) and TH- (TATHA2) AgNPs compared to CT (control), CH (chitosan) and TH. *: Significant difference relative to CT, **: significant difference between silver treated groups; Ag+, BR- and TH-AgNPs, 100 ppm for 12 hours. Significant difference is P < 0.05 and SEM scale bar is 1 μm. | ||
The cytotoxicity of bare-AgNPs and TATHA2-AgNPs after lyophilization was tested on yeast using a growth inhibition assay. Roughly 60 nm NPs of chitosan (CH), a natural polymer with well characterized stability, biocompatibility and biomedical applications, primarily for drug delivery, were used as negative control in the toxicity study. The effect of the colloidal solution in which AgNPs were suspended before lyophilization (suspending solution) and the silver ions (Ag+) were also included to test the effect of the dissolution of AgNPs on cytotoxicity.38–40 The test agents were incubated with yeast to do the dose and time dependent toxicity study: 0.001, 0.01 and 0.1 mg ml−1 equiv. to 1, 10 and 100 ppm each for 3, 6 and 12 hours. A dose and time dependent growth inhibition was observed, cell viability >85% and 95% after exposure to 10 and 1 ppm AgNPs, respectively (data not reported). At the highest dose and time, 100 ppm for 12 hours, the AgNPs exhibit >70% cell-viability and Ag+ resulted in almost 100% cell killing (Fig. 3, III).
TATHA2, chitosan NPs (negative control) with ∼60 nm diameter and suspending solution were non-toxic. Among silver treated groups, Ag+ showed a significant difference in cell viability/toxicity, but no significant difference was observed between the cells exposed to bare vs. TATHA2-functionalized-AgNPs. Similar AgNPs-specific non-toxicity to microbes has been reported.40 Also, we support their claim that thiol-PEG coating around AgNPs is able to block Ag+ dissolution under special test environment, cell growth inhibition studies under anaerobic conditions and minimal media. In agreement with previous reports on AgNPs toxicity, we can deduce two major points. First, the dissolution of AgNPs to Ag+ is the major cause of toxicity.40 Second, the degree of cell uptake of AgNPs (bioavailable dose) influences the AgNPs cell toxicity.41 The significant difference in bioavailability of AgNPs delivered via passive and facilitated diffusion is definitely another critical factor controlling toxicity, as discussed in the following section.
To study the cell uptake of AgNPs via passive and facilitated delivery, we incubated the cells with particles for 3, 6 and 12 hours. The number of AgNPs taken up by the cells was quantified using ICPMS to measure silver content after dissolution of AgNPs into Ag+. The measured silver content was converted back to number of AgNPs to report uptake data in AgNPs per cell. The AgNPs were selectively removed from the cell surfaces using a mild etching procedure reported elsewhere, I2/KI (0.34/2 mM) for 5 minutes, to estimate the number of particles internalized by the cells.9 The etching parameters were tested (by SEM imaging) to validate the effective removal of AgNPs from the yeast surface (Fig. S3†). The procedure resulted in >95% cell viability. ICPMS quantitative results were verified by a UV-Vis spectrophotometer to quantify AgNPs, using AgNPs characteristic absorbance. To further validate the cellular uptake (adsorption + internalization) of AgNPs via the two strategies, we observed the AgNPs in ultrathin (≤50 nm) sections of yeast using TEM. TATHA2 facilitated delivery resulted in rapid (within 3 hours) and high internalization (∼15 fold) of TATHA2-AgNPs, compared to bare-AgNPs (Fig. 4A). The several fold difference in internalization of AgNPs by the two strategies seems to be due to the difference in endocytic pathways. TATHA2-mediated intracellular delivery is lipid raft-dependent macropinocytosis, a rapid receptor-independent form of endocytosis as compared to receptor-mediated endocytosis,46,47 which is a slow mechanism typical of charge-driven cellular uptake of NPs.8–10,41
 | ||
| Fig. 4 Cell uptake of AgNPs in yeast via passive and TATHA2 facilitated diffusion. Upper panel (A) shows the kinetics of cell uptake of AgNPs quantified by ICPMS (sample size, n = 3). Total uptake is adsorption + internalization. BR: bare and TH: TATHA2-functionalized. The lower panel (in situ TEM) shows the intracellular distribution of AgNPs into yeast after surface etching, 3 hours of TATHA2 facilitated diffusion (B) and 12 hours of passive diffusion (C), AgNPs aggregates appear as dark spots. Endocytic pathway: endocytic vesicle (EV), endosome (E) and late endosome (LE); nucleus (N) and mitochondrion (M). Scale bar is 0.5 μm. | ||
In our observation, passively diffusing bare-AgNPs reaches internalization saturation by 6 hours, while the internalization curve for the TATHA2-AgNPs shows significant increase even at 12 hours (Fig. 4A). The rapid and preferentially uniform intracellular distribution of TATHA2-AgNPs, without any apparent compartmentalization was observed within 3 hours (Fig. 4B). However, the bare-AgNPs were primarily found adsorbed to the cell surface, with little/no internalization. Observations of a few other sections gave similar information, where the passively diffusing bare-AgNPs were preferentially found entrapped in endosomes (Fig. 4C). The high cellular uptake/internalization of AgNPs by TATHA2 mediated diffusion over passive diffusion realistically exposes the cells to much higher doses of AgNPs (∼15 fold difference in bioavailable dose). The degree of cell uptake of AgNPs and AuNPs, bioavailable dose directly impacts the cell toxicity.41
Conclusions
We have tested the three most widely accepted cellular delivery strategies; active/electroporation, free/passive and TATHA2 mediated/facilitated diffusion to efficiently deliver AgNPs into yeast. Although, yeast tolerate a very high electroporation dose (E ≥ 7.5 kV cm−1 for two consecutive pulses of 1 and 4 ms), they undergo severe cell damage even at lower doses (E ≤ 0.5 kV cm−1 for 1 ms pulse) in the presence of AgNPs, incubation normalized to 105 AgNPs per cell. The conventional chemical synthesis and storage of colloidal AgNPs, and exposure of AgNPs to cells was modified to minimize the oxidation and dissolution of AgNPs to Ag+, mitigating AgNPs-specific toxicity and increasing their biocompatibility (cell viability >70% at 100 ppm).40 Free diffusion of bare-AgNPs resulted in poor uptake, internalization saturation and endosome entrapment. The TATHA2-AgNPs on the other hand have rapid and high internalization potential resulting in a several fold increase in cell uptake and preferentially uniform intracellular distribution, a requirement for detection of ubiquitously distributive proteins. Our findings suggest that TATHA2 facilitated delivery of AgNPs in yeast is a better delivery strategy over active/electroporation and passive/free diffusion for intracellular SERS sensing towards development of first CBB-SIST. Although, we studied AgNPs delivery in yeast in context to intracellular SERS sensing, our contribution will directly benefit the scientific community exploring delivery of other noble, plasmonic metal NPs, such as gold, platinum, palladium, cadmium, copper, zinc etc. in microbial and mammalian cells. Our study will have wider implications in the development of cell-based technologies for sensing and therapeutics.Acknowledgements
We thank Dr Barry P. Rosen at the Herbert Wertheim College of Medicine and Dr Walter M. Goldberg in the Department of Biological Sciences at Florida International University (FIU), U.S.A. for their support in the electroporation and ultramicrotomy experiments. We also thank the Advanced Materials Engineering Research Institute (AMERI) and the Trace Evidence Analysis Facility (TEAF) at FIU for TEM and ICPMS, respectively. We acknowledge the Department of Defense (DOD) U.S.A. for financial support (Award#W81XWH-10-1-0732). Publication of this article was funded in part by FIU Open Access Publishing Fund.Notes and references
- M. Cametti and Z. Dzolic, Chem. Commun., 2014, 50, 8273 RSC.
- T. K. Sau, A. L. Rogach, F. Jackel, T. A. Klar and J. Feldmann, Adv. Mater., 2010, 22, 105 Search PubMed.
- G. Doria, J. Conde, B. Veigas, L. Giestas, C. Almeida, M. Assuncao and J. Rosa, Sensors, 2012, 12, 1657 CrossRef CAS PubMed.
- J. Conde, G. Doria and P. Baptista, J. Drug Delivery, 2012, 2012, 1 CrossRef PubMed.
- B. Yameen, W. I. Choi, C. Vilos, A. Swami, J. Shi and O. C. Farokhzad, J. Controlled Release, 2014, 190, 485 CrossRef CAS PubMed.
- R. Levy, U. Shaheen, Y. Cesbron and V. See, Nano Rev., 2010, 1, 4889 Search PubMed.
- A. Verma and F. Stellaci, Small, 2010, 6, 12 CrossRef CAS PubMed.
- B. D. Chithrani, A. A. Ghazani and W. C. W. Chan, Nano Lett., 2006, 6, 662 CrossRef CAS PubMed.
- E. C. Cho, L. Au, Q. Zhang and Y. Xia, Small, 2010, 6, 517 CrossRef CAS PubMed.
- Z. J. Zhu, P. S. Ghosh, O. R. Miranda, R. W. Vachet and V. M. Rotello, J. Am. Chem. Soc., 2008, 130, 14139 CrossRef CAS PubMed.
- P. Verderio, S. Awakumova, G. Alessio, M. Bellini, M. Colombo, E. Galbiati, S. Mazzuchhelli, J. P. Avila, B. Santini and D. Prosperi, Adv. Healthcare Mater., 2014, 3, 957 CrossRef CAS PubMed.
- J. M. Atienza, N. Yu, S. L. Kirstein, B. Xi, X. Wang, X. Xu and Y. A. Abassi, Assay Drug Dev. Technol., 2006, 4, 597 CrossRef CAS PubMed.
- Y. Feng, Int. J. Electrochem., 2011, 2011, 16 Search PubMed.
- D. Boyer, P. Tamarat, A. Maali, B. Lounis and M. Orrit, Science, 2002, 297, 1160 CrossRef CAS PubMed.
- K. G. Stamplecoskie and J. C. Scaiano, J. Phys. Chem. C, 2011, 115, 1403 CAS.
- K. S. Lee and M. A. El-Sayed, J. Phys. Chem. B, 2006, 110, 19220 CrossRef CAS PubMed.
- A. O. Govorov and H. H. Richrdson, Nano Today, 2007, 2, 30 CrossRef.
- S. Wachsmann-Hogiu, T. Weeks and T. Huser, Curr. Opin. Biotechnol., 2009, 20, 63 CrossRef CAS PubMed.
- J. Kneipp, H. Kneipp and K. Kneipp, Chem. Soc. Rev., 2008, 37, 1052 RSC.
- M. Vendrell, K. K. Maiti, K. Dhaliwal and Y.-T. Chang, Curr. Trends Biotechnol., 2013, 31, 249 CrossRef CAS PubMed.
- A. M. Fales, H. Yuan and T. Vo-Dinh, Mol. Pharmaceutics, 2013, 10, 2291 CrossRef CAS PubMed.
- V. Bhardwaj, S. Srinivasan and A. J. McGororn, J. Biosens. Bioelectron., 2013, 4, 7 Search PubMed.
- M. Lee, S. Lee, J. H. Lee, H. W. Lim, G. H. Seong, E. K. Lee, S. L. Chang, C. H. Oh and J. Choo, Biosens. Bioelectron., 2011, 26, 2135 CrossRef CAS PubMed.
- S. Pang, T. P. Labuza and L. He, Analyst, 2014, 139, 1895 RSC.
- L. He, T. Rodda, C. L. Haynes, T. Deschaines, T. Strother, F. Diez-Gonzalez and T. P. Labuza, Anal. Chem., 2011, 83, 1510 CrossRef CAS PubMed.
- H. Li, J. Sun and B. M. Cullum, Nano Biotechnol., 2006, 2, 17 CAS.
- H. Chon, S. Lee, S. Y. Yoon, S. L. Chang, D. W. Lim and J. Choo, Chem. Commun., 2011, 47, 12515 RSC.
- K. Brown, Science, 2004, 305, 1228 CrossRef CAS PubMed.
- T. H. Rider, M. S. Petrovick, F. E. Nargi, J. D. Harper, E. D. Schwoebel, R. H. Mathews, D. J. Blanchard, L. T. Bortolin, A. M. Young, J. Chen and M. A. Hollis, Science, 2003, 301, 213 CrossRef CAS PubMed.
- M. S. Petrovick, F. E. Nargi, T. Towle, K. Hogan, M. Bohane, D. J. Wright, T. H. MacRae, M. Potts and R. F. Helm, Biotechnol. Bioeng., 2010, 106, 474 CAS.
- K. H. Baronian, Biosens. Bioelectron., 2004, 15, 953 CrossRef PubMed.
- P. A. Cahill, A. W. Knight, N. Billinton, M. G. Barker, L. Walsh, P. O. Keenan, C. V. Williams, D. J. Tweats and R. M. Walmsley, Mutagenesis, 2004, 19, 105 CrossRef CAS.
- H. Harms, M. C. Wells and J. R. van der Meer, Appl. Microbiol. Biotechnol., 2006, 70, 273 CrossRef CAS PubMed.
- H. Zlotnik, M. P. Fernandez, B. Bowers and E. Cabib, J. Bacteriol., 1984, 159, 1018 CAS.
- H. Hashimoto, H. Morikawa, Y. Yamada and A. Kimura, Appl. Microbiol. Biotechnol., 1985, 21, 336 CrossRef CAS.
- S. A. Johnston, P. Q. Anziano, K. Shark, J. C. Sanford and R. A. Butow, Science, 1988, 240, 1538 CAS.
- D. Riveline and P. Nurse, Nat. Methods, 2009, 6, 513 CrossRef CAS PubMed.
- O. Bondarenko, K. Juganson, K. Kasemets, M. Mortimer and A. Kahru, Arch. Toxicol., 2013, 87, 1181 CrossRef CAS PubMed.
- C. Levard, E. M. Hotze, G. V. Lowry and G. E. Brown, Environ. Sci. Technol., 2012, 46, 6900 CrossRef CAS PubMed.
- Z. M. Xiu, Q.-bo Zhang, H. L. Puppala, V. L. Colvin and P. J. J. Alvarez, Nano Lett., 2012, 12, 4271 CrossRef CAS PubMed.
- H. J. Yen, S. H. Hsu and C. L. Tsai, Small, 2009, 5, 1553 CrossRef CAS PubMed.
- Y. Yu, J. Lin, Y. Wu, S. Feng, Y. Li, Z. Huang, R. Chen and H. Zheng, Spectroscopy, 2011, 25, 13 CrossRef CAS.
- Y. Yu, J. Lin, Z. Huang, G. Xi, D. Lin, Y. Chen, R. Chen and H. Zeng, J. Phys.: Conf. Ser., 2011, 277, 012045 CrossRef.
- J. Lin, R. Chen, S. Feng, Y. Li, Z. Huang, S. Xie, Y. Yu, M. Cheng and H. Zeng, Biosens. Bioelectron., 2009, 25, 388 CrossRef CAS PubMed.
- E. A. Vitol, Z. Orynbayeva, M. J. Bouchard, J. A. Clifford, G. Friedman and Y. Gogotsi, ACS Nano, 2009, 3, 3529 CrossRef CAS PubMed.
- J. S. Wadia, R. V. Stan and S. F. Dowdy, Nat. Med., 2004, 10, 310 CrossRef CAS PubMed.
- S. F. Ye, M. M. Tian, T. X. Wang, L. Ren, D. Wang, L. H. Shen and T. Shang, Nanomedicine, 2012, 8, 833 CrossRef CAS PubMed.
- S. Kumar, N. Harrison, R. R. Kortum and K. Sokolov, Nano Lett., 2007, 7, 1338 CrossRef CAS PubMed.
- S. Kumar, J. Aaron and K. Sokolov, Nat. Protoc., 2008, 3, 314 CrossRef CAS PubMed.
- A. Takami, H. Kurita and S. Koda, J. Phys. Chem. B, 1999, 103, 1226 CrossRef CAS.
- J. Zheng, S. Pang, T. P. Labuza and L. He, Talanta, 2014, 129, 79 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details; synthesis and TATHA2-functionalization of AgNPs, characterization of AgNPs and evaluation of intracellular delivery strategies for cellular toxicity and uptake. Table S1, physiochemical properties of AgNPs delivered by three strategies; Fig. S1 and S2, physical damage by Raman laser and TEM in presence of AgNPs, respectively; Fig. S3, SEM image to show selective removal of AgNPs from cell surface. See DOI: 10.1039/c5an00435g |
| This journal is © The Royal Society of Chemistry 2015 |