
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multiplexed femtomolar quantitation of human cytokines in a fluoropolymer microcapillary film†
Ana P.
Castanheira
a,
Ana I.
Barbosa
b,
Alexander D.
Edwards
ac and
Nuno M.
Reis
*ab
aCapillary Film Technology Ltd, Billingshurst, West Sussex RH14 9SJ, UK
bDepartment of Chemical Engineering, Loughborough University, Leicestershire LE11 3TU, UK. E-mail: n.m.reis@lboro.ac.uk; Fax: +44 (0)1509 223 923; Tel: +44 (0)1509 222 505
cReading School of Pharmacy, University of Reading, Reading RG6 6AD, UK. E-mail: a.d.edwards@reading.ac.uk; Fax: +44 (0)118 931 4404; Tel: +44 (0)118 378 4253
First published on 18th June 2015
Abstract
Sensitive quantitation of multiple cytokines can provide important diagnostic information during infection, inflammation and immunopathology. In this study sensitive immunoassay detection of human cytokines IL-1β, IL-6, IL-12p70 and TNFα is shown for singleplex and multiplex formats using a novel miniaturized ELISA platform. The platform uses a disposable plastic multi-syringe aspirator (MSA) integrating 8 disposable fluoropolymer microfluidic test strips, each containing an array of ten 200 μm mean i.d. microcapillaries coated with a set of monoclonal antibodies. Each MSA device thus performs 10 tests on 8 samples, delivering 80 measurements. Unprecedented levels of sensitivity were obtained with the novel fluoropolymer microfluidic material and simple colorimetric detection in a flatbed scanner. The limit of detection for singleplex detection ranged from 2.0 to 15.0 pg ml−1, i.e. 35 and 713 femtomolar for singleplex cytokine detection, and the intra- and inter-assay coefficient of variation (CV) remained within 10%. In addition, a triplex immunoassay was developed for measuring IL-1β, IL-12p70 and TNFα simultaneously from a given sample in the pg ml−1 range. These assays permit high sensitivity measurement with rapid <15 min assay or detection from undiluted blood serum. The portability, speed and low-cost of this system are highly suited to point-of-care testing and field diagnostics applications.
Introduction
Cytokines are important immune signaling proteins produced following infection, tissue injury or during immunological disease.1,2 Characteristic expression patterns have been identified both locally in tissues and in various clinical samples (e.g. blood, serum, urine) that are associated with particular infections or immune and inflammatory diseases.1,3 Although cytokine measurement is nowadays a routine task in research laboratories, the ability to quantify rapidly multiple human cytokines simultaneously, down to pg ml−1 range, in a disposable miniaturized platform has the potential of providing a rapid identification and treatment of acute conditions such as sepsis4,5 and accurate point-of-care (POC) discrimination6–10 between bacterial and viral infection based on few key cytokines such as IL-6, TNFα, IL-10 combined with other biomarkers like C reactive protein (CRP) and procalcitonin (PCT).11A major challenge to the routine measurement of cytokine levels in conventional clinical samples (i.e. blood or urine) is that cytokines are highly potent and typically exert their action in a local tissue environment. For this reason, systemic levels are normally very low, typically in the range of 1–10 pg ml−1 or even lower, and highly elevated levels are usually only found in clinical samples such as blood only either transiently or during severe disease. Consequently, diagnostic tools must be capable of accurately quantifying multiple biomarkers from biological samples at very low concentrations. Although rapid near-patient measurement is desirable, current methods for sensitive quantitation of individual cytokines remain limited to the laboratory setting.1 The gold standard technique for cytokine quantitation is the enzyme-linked immunosorbant assay (ELISA) performed in a microtiter plate (MTP) which can meet the required high sensitivity and low assay variance, using enzyme amplification to provide measureable signals. This method typically measures only one cytokine per well whilst requiring a skilled laboratory operator to perform and analyse the test, and requires laboratory based detection systems such as plate readers. MTP based sandwich immunoassays require long incubation times, multiple washing steps and large reagent and sample volumes.12
To adapt the MTP format for measuring more than one cytokine from each sample, a technique termed multiplexing, fluorescence-based immunoassays platforms such as the Luminex® system were developed. These systems are capable of quantifying a set of cytokines using fluorescence detection on antibody-coated polystyrene microspheres, reporting sensitivities of 5.7 pg ml−1 for IL-6 and 5.2 pg ml−1 for IL-12.13 This effective laboratory method is particularly suited to high-throughput biomarker screening and identification given the capacity for very large numbers of simultaneous analytes (e.g. 20–40 per sample). Quinn et al.1 developed a related multiplexed cytokine assay utilizing VeraCode technology that uses holographically encoded silica cylinders to perform fluorescence immunoassays, and a two-laser scanning CCD imager to identify individual beads and quantify fluorescence signal. The sensitivity reported for all cytokines analyzed (IL-1b, TNFα, IL-2, IL-5, IL-6, IL-12 and IL-10) was 0.5 pg ml−1, except for IL-4 that presented a limit of detection of 14 pg ml−1. However, both Luminex and VeraCode technologies require long incubation times, complex fluid handling steps and costly and complex software to analyze the fluorescence signal.1 Consequently, these techniques have application limited to the central diagnostic laboratory setting.
An alternative approach for multiplex quantitative cytokine measurement is the use of microarrays. Wang et al.14 described a chip capable of profiling human cytokine expression of breast tumor cells and in cervical tumor samples, which was fabricated by printing 43 cytokine and chemokine antibodies into a microfluidic chip using a specialized polyacrylamide formulation. The authors reported assay sensitivities below 20 pg ml−1 for five out of six cytokines studied. Although capable of detecting a large set of cytokines, the assay protocol remains complex requiring skilled personnel and currently presents high manufacturing costs.
To overcome current limitations of laboratory diagnostic systems, a range of microfluidic devices were recently developed and applied to the detection and quantitation of biomarkers. Microfluidic technologies can reduce sample and reagent volumes, whilst greatly shortening incubation times. Many microfluidic assays can be measured using inexpensive optoelectronic components developed for consumer electronics products such as smartphones. The major barrier to market penetration of microfluidic immunoassays is the current high fabrication cost and limited scale of manufacture for microfluidic devices,15 which is combined with the difficulty in achieving the high analytical sensitivity required for measurement of cytokines in clinical samples (typically in the range of picograms per milliliter in serum, in contrast to nanograms to micrograms per milliliter for many other biomarkers) without the use of more sophisticated signal detection equipment. A microfluidic study reported multiple cytokine measurement in a miniaturized device with a biochip array to analyze circulating human cytokines.3 Monoclonal capture antibodies were bound in predefined positions on an activated biochip surface, providing discrete test regions.3 Chemiluminescence was used to measure analyte, with a reported sensitivity from 0.12 pg ml−1 for IL-6 to 2.12 pg ml−1 for IL-4, with twelve cytokines analysed in total. However, the biochip still requires sophisticated automated fluid processing and a dedicated benchtop reader, and remains laboratory based.
Colorimetric ELISA exploits simple, inexpensive and well established enzyme assay chemistry with simpler detection than fluorescent or chemiluminescent detection. For this reason, colorimetric readouts are ideal for POC testing applications. Indeed, colorimetric detection in miniaturized immunoassay formats has proved successful for detection of biomarkers present at higher concentrations.16,17 However, achieving sufficient sensitivity to measure analytes in the pg ml−1 range using colorimetric readout has proved highly challenging in portable microfluidic systems. In this paper we present a sensitive colorimetric sandwich assay with limits of detection (LoDs) <2 pg ml−1 using conventional ELISA reagents in miniaturized fluoropolymer multiplex test strips. This sensitivity is comparable to that achieved with laboratory MTP assays, and to fluorescence and chemiluminescence detection.
This article describes a new miniaturized platform for rapid, sensitive and inexpensive quantitation of multiple human cytokines based on a multiplexed fluoropolymer microcapillary film (MCF). The benefits of the new sandwich ELISA platform are demonstrated by integrating 10-bore test strips manufactured from a continuous melt-extruded MCF18 (Fig. S1b†) and operated with a portable, power free, 8-sample Multi-Syringe Aspirator (MSA) device (Fig. S1a and S1c†) capable of running up to 80 simultaneous ELISA tests.19 The small internal diameter of the capillaries (206 ± 12.2 μm) result in shortened diffusion distances and reduced volumes of reagents required to run an assay, consequently the total assay time is reduced by at least one order of magnitude when compared to standard methods.18 The flat measurement surfaces of the MCF and low refractive index of fluoropolymer material18 provides unique signal-to-noise ratios suitable for measuring low concentrations of chromogenic substrates. This is demonstrated by measuring a range of human cytokines, IL-1β, IL-6, IL-12p70 and TNFα with a flatbed scanner using commercial sandwich ELISA chemistry with total assay times between 15 and 50 min.
Experimental section
Immunoassay reagents
All the following human cytokines reagents were purchased from eBiosciences (Hatfield, UK): IL-1β (cat no: human recombinant protein #14-8018; Anti-Human IL-1β biotin #13-7016; Anti-Human IL-1β purified #14-7018); IL-12p70 (cat no: human recombinant protein #14-8129; Anti-Human IL-12p70 biotin #13-7129; Anti-Human IL-12p70 purified #14-7128); IL-6 (cat no: human recombinant protein #14-8069; Anti-Human IL-6 biotin #13-7068; Anti-Human IL-6 purified #14-7069); and Tumor Necrosis Factor-α (TNFα) (cat no: human recombinant protein #14-8329; Anti-Human TNFα biotin #13-7349; Anti-Human TNFα purified #14-7348).ExtrAvidin-Peroxidase (cat. no E2886), enzymatic substrate SIGMAFASTTM OPD (o-phenylenediamine dihydrochloride) tablets (cat. no P9187), Phosphate Buffered Solution (PBS, cat. no P5368-10PAK), pH 7.4, 0.01 M, Tween 20 diluted in PBS 0.05% v/v (PBS-T, P9416 – 50 ML) and Protease-free Albumin from Bovine Serum (BSA, cat no. A3858) were sourced from Sigma Aldrich Ltd (Dorset, SP8 4XT, UK). SuperBlock blocking buffer in PBS (cat. no PN37515), High Sensitivity Streptavidin–HRP (cat. no 21130) and High Sensitivity NeutrAvidin – HRP (cat. no 31030) were supplied by Thermo Scientific (Northumberland, UK). 3,3′,5,5′ Tetramethylbenzidine (TMB, cat. no DY999) was sourced from R&D Systems (Minneapolis, MN, USA). Human serum (cat no. #CR200) was sourced from TCS Biosciences (Buckingham, UK).
Fluoropolymer MCF
This consisted of a flat plastic ribbon containing 10 parallel capillaries (Fig. S1b†) with mean internal diameter 206 ± 12.2 μm, manufactured by Lamina Dielectrics Ltd (Billingshurst, West Sussex, UK) by a continuous melt-extrusion process from fluorinated ethylene propylene co-polymer (FEP-Teflon®). The external dimensions of the MCF were 4.5 ± 0.10 mm wide and 0.6 ± 0.05 mm thick.Sandwich ELISA protocol
Purified monoclonal anti-human antibodies (capAb) were immobilized by passive adsorption on the internal walls of the microcapillaries in 25–50 cm long strips of fluoropolymer MCF, which takes advantage of the hydrophobic surface of FEP microcapillaries. For singleplex cytokines detection, capAb diluted in PBS buffer was aspirated through the set of 10 parallel microcapillaries using a 1 ml syringe connected to the MCF with a 2 cm long silicone tube (ID 2 mm). For multiplex detection, each individual capillary was injected using a 30G × ½′′ (0.3 mm × 13 mm) gauge needle until the whole length of the capillary was visually filled with the solution. The capAb solution was then incubated for 2 hours after which all remaining binding sites were blocked with a suitable blocking solution and incubated for another 1 hour. All immunoassay steps were carried out at room temperature (20 °C). Subsequently, the MCF strip was washed with PBS-T and trimmed into a set of eight, 30 mm long test strips that were attached using a push-fit seals onto a prototype 8-channel semi-disposable multi-syringe aspirator (MSA) (Fig. S1a†), as described in Barbosa et al.19 Samples, wash buffer and reagents were sequentially aspirated from the custom multi-well plate into MCF test strips by turning the MSA control dial at the top of the device anti-clockwise, which simultaneously moves a set of 8 plungers in the array of 1 ml disposable syringes held within the MSA. All reagents and wash buffer could be pre-loaded on the custom multi-well plate and then sequentially aspirated with the device to complete the full sandwich assay, without requiring any further fluid handling devices.A 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 dilution series of human recombinant protein were prepared in the custom multi-well plate (150 μl per sample), and aspirated into the MCF test strips by turning the manual knob 6×, which corresponds to approximately 78 μl of solution, and incubated for 30 min. After washing with 150 μl of PBS-T, 150 μl of biotinylated anti-human cytokine antibody (detAb) diluted in PBS was aspirated with the MSA, and incubated for 10 min and washed with further 150 μl of PBS-T. Subsequently, high sensitivity streptavidin–HRP (HSS-HRP) conjugated enzyme diluted in PBS was aspirated and incubated for 10 min in the capillaries, followed by three washing steps with PBS-T. The enzyme conjugated was selected based on the optimization study described in ESI.† Overall, for the range of biotin-binding enzymes tested, avidin, streptavidin and neutravidin each were reported by the manufacturer to bind four biotins per molecule with high affinity and selectivity.20 Finally, 150 μl of enzymatic substrate consisting of OPD or TMB were aspirated and the whole MSA device placed in a HP ScanJet G4050 Scanner and the MCF strips scanned at 2400 dpi resolution in transmittance mode. The strips were scanned in intervals of 2–3 min over 30 minutes of incubation. The RGB images were analysed using ImageJ software, from which the absorbance on each individual capillary was determined from the grey scale peak height in the split blue channel as described in previous studies.18,19 Assay variability was determined based on sensitivity, precision and recovery, following the method described by Ederveen.21 Overall, CVs obtained for ELISA in the MCF were well within those reported for the 96-well MTP, which is in the range of 10–20%.21,22
:2 dilution series of human recombinant protein were prepared in the custom multi-well plate (150 μl per sample), and aspirated into the MCF test strips by turning the manual knob 6×, which corresponds to approximately 78 μl of solution, and incubated for 30 min. After washing with 150 μl of PBS-T, 150 μl of biotinylated anti-human cytokine antibody (detAb) diluted in PBS was aspirated with the MSA, and incubated for 10 min and washed with further 150 μl of PBS-T. Subsequently, high sensitivity streptavidin–HRP (HSS-HRP) conjugated enzyme diluted in PBS was aspirated and incubated for 10 min in the capillaries, followed by three washing steps with PBS-T. The enzyme conjugated was selected based on the optimization study described in ESI.† Overall, for the range of biotin-binding enzymes tested, avidin, streptavidin and neutravidin each were reported by the manufacturer to bind four biotins per molecule with high affinity and selectivity.20 Finally, 150 μl of enzymatic substrate consisting of OPD or TMB were aspirated and the whole MSA device placed in a HP ScanJet G4050 Scanner and the MCF strips scanned at 2400 dpi resolution in transmittance mode. The strips were scanned in intervals of 2–3 min over 30 minutes of incubation. The RGB images were analysed using ImageJ software, from which the absorbance on each individual capillary was determined from the grey scale peak height in the split blue channel as described in previous studies.18,19 Assay variability was determined based on sensitivity, precision and recovery, following the method described by Ederveen.21 Overall, CVs obtained for ELISA in the MCF were well within those reported for the 96-well MTP, which is in the range of 10–20%.21,22
Cytokine response curves
Unless otherwise stated, full response curves were built based on the absorbance data for 2 min enzymatic substrate incubation and fitted with a 4 Parameters Logistic (4PL) model using solver tool in Microsoft Excel and performed using the following conditions. Different immunoassay formats were tested in this study, (i) singleplex IL-1β, IL-12p70, IL-6 or TNFα detection; (ii) duplex qualitative detection of IL-1β and IL-6, and (iii) triplex quantitation of IL-1β, IL-12p70 and TNFα. Immunoassays were performed based on the assay conditions optimised for IL-1β as fully described in ESI,† for a concentration of recombinant protein in the range of 1 ng ml−1–0 pg ml−1 on a 1:2 dilution series for singleplex format or 1:3 dilution for triplex format. The standard concentration of capAb used in the assays was 20 μg ml−1 for IL-1β, IL-12p70 and TNFα, and 40 μg ml−1 for IL-6; 10 μg ml−1 for detAb; and 4 μg ml−1 for HSS-HRP. Experimental details specific to the qualitative duplex assay and quantitative triple assays are given in ESI.†
Results and discussion
Colorimetric microfluidic sandwich ELISA suited to simple optoelectronic measurement was optimised to achieve sensitive cytokine measurement and then assay performance evaluated for a novel miniaturised fluoropolymer MCF platform that exploits the unique properties of the fluoropolymer microfluidic material in a portable manually operated multi-sample device. Initially, singleplex assays were developed, where 10 replicate microcapillaries were used to measure a single cytokine; subsequently duplex and triplex assays were developed where multiple cytokines were measured simultaneously from each sample.Limit of quantitation of colorimetric signal with a flatbed scanner
Prior to optimising the cytokine sandwich ELISA protocol in the fluoropolymer MCF, the optical detection of chromogenic substrates was first studied. A series of dilutions of 2,3-diaminophenazine (DAP), the final product of OPD chromogenic substrate after conversion by HRP, were scanned in fluoropolymer MCF strips with a flatbed scanner, and in parallel peak absorbance (450 nm) of the same dilutions was measured in a 96-well MTP using a microplate reader. The DAP absorbance in the blue channel of the scanned image was calculated by image analysis. The minimum concentration of DAP that could be detected using the flatbed scanner was 62.5 μg ml−1, which is about 250 times less sensitive than the minimum concentration detectable in a MTP (0.244 μg ml−1, Fig. S5†). This reduced sensitivity is likely to be linked to three combined factors, all of which are shared with any colorimetric microfluidic measurement technique used for signal quantitation and based on optical imaging. Firstly, the 15-fold reduction in light path distance within the 200 μm internal diameter capillaries, compared to 100 μl in a microtiter plate well (≈3 mm liquid path), reduces the absolute Abs values. Secondly, the sensitivity is also limited by the broad wavelengths measured by the blue channel of the RGB sensor, in contrast to the narrow band of wavelengths measured by a dedicated UV-VIS plate reader that can be selected to closely match the peak absorbance of enzyme substrate product. Finally, the consumer imaging equipment may have intrinsically reduced sensitivity to low amounts of light absorbance compared to a microtiter plate reader, which was engineered specifically for sensitive absorbance readings. Such lack of sensitivity has therefore to be overcome by increasing colorimetric signal intensity and improving signal-to-noise ratio in miniaturized ELISA system, which is not straightforward as indicated from the lack of published reports of colorimetric ELISA sensitivity below ng ml−1 in microfluidic devices, including work in our research group with the same ELISA platform.19Despite the relatively poor sensitivity of flatbed scanner in respect to quantitation of colorimetric signal in microcapillaries, the dynamic range observed for DAP quantitation in the fluoropolymer MCF was greater than that in the MTP reader, exceeding 2log10 units. This can be attributed to the very short light path within the plastic microcapillaries, allowing quantitative measurement of very high concentrations (around 2 mg ml−1) of DAP expanding the concentration range for the Lambert–Beer law. In contrast, a maximum concentration of DAP in the range of 0.02 mg ml−1 was detected in the linear range for the MTP (Fig. S5†). This suggested that in theory limited sensitivity of optical reading could potentially be overcome by speeding up enzymatic chromogenic signal amplification in the MCF microcapillaries, yielding higher concentrations of coloured DAP product for the same analyte concentration.
Optimisation of cytokine ELISA detection in microcapillaries
A number of parameters widely recognised to strongly affect sandwich ELISA signal and sensitivity were screened in the fluoropolymer MCF strips in order to achieve high levels of sensitivity. These included: capAb concentration and incubation time; sample incubation time; detAb concentration; enzyme conjugate type and concentration; substrate concentration; type and incubation time of blocking solution; and reagent diluent and the number and volume of wash steps. Full details of optimisation study are described in the ESI† provided, and assay conditions summarised in Table S1 (ESI†).Overall, reagent concentrations required for optimal ELISA signal in MCF were significantly greater than those used for MTP, typically 10 to 20 times higher. The small diameter of the microcapillaries and format of the MSA device based on sequential solution aspiration through microcapillaries permitted, with the possibility of skipping some washing steps, simplifying the overall sandwich ELISA protocol. This is linked to the very short diffusion distances in the microcapillaries which enable using an excess of reagents for replacing the solutions in the microcapillaries; about 78 μl of reagents were aspirated at each step through each 30 mm long MCF strip, having internal volume of 10 μl, such that, in effect, each reagent was used as a washing solution. Nevertheless, it was noticed that a washing step before adding the antigen is very important to reduce the variability and improve signal strength. As an example see Fig. S2 in ESI.†
The performance of the colorimetric ELISA in the fluoropolymer MCF was found to be extremely sensitive to concentration of both capAb and detAb. A maximum signal-to-noise ratio was obtained with 20 μg ml−1 of capAb (Fig. S4d†), which is 10 to 20-fold higher than the concentration typically used for coating plastic surface in MTPs. This is ultimately linked to the larger surface-area-to-volume ratio of the 10-bore MCF compared to a MTP; a 200 μm internal diameter capillary has a surface-area-to-volume ratio about 16 times higher than a 96 well MTP with 100 μl of solution. It is worth noticing that a 9 mm long MCF strip has the same total surface area (i.e. 1.1 cm2) of a microwell in a 96 well MTP with 100 μl of solution. It can be shown that about 100 μg ml−1 of capAb is required to achieve theoretical full surface coverage in a 200 μm internal diameter microcapillary. This suggests antibody orientation in FEP is favoured at capAb concentrations lower than that required for full surface coverage, which agrees with antibody adsorption studies in other plastic surfaces.23
Given that colourimetric measurements of converted substrate with the flatbed scanner were far less sensitive than microtiter plate, is it perhaps surprising that the full ELISA is as sensitive as MTP. Again, this can be justified by the larger surface-area-to-volume ratio of MCF, which is completely coated with the capAb, yielding a higher surface monolayer concentration in a circular microcapillary, which exceeds the concentrations that can be obtained by immobilizing the capAb in just a fraction of the inner surface of a microchannel, or printing the capAb in spots within a microfluidic device. The surface-area-to-volume ratio in a circular or elliptical microcapillary entirely coated with capAb is 4 times larger than the one obtained by coating just one surface in a microchannel with the same internal dimensions. The kinetics of antibody–antigen binding are favoured by the increased concentration of free binding sites of capAb on the microcapillaries surface, which results in higher assay sensitivity. The binding of an immobilised capAb and recombinant protein (Ag) can be represented by the simple equilibrium reaction:
| capAb + Ag ↔ capAb–Ag |
According to Scatchard's model24 the equilibrium can be described by the expression:
| B/F = Ka(N − B) | (1) |
Singleplex cytokine quantitation
Full response curves were performed with four different human cytokines, IL-1β, IL-12p70, IL-6 and TNFα using assay conditions optimised for the IL-1β cytokine in the fluoropolymer MCF strips. A 4PL model was fitted to the experimental data based on minimum square differences and plotted as continuous lines (Fig. 1a). Each cytokine presented a unique response curve related to differences in antigen molecular weight, affinity and on/off binding rates for each antibody pair that ideally would need to be optimised individually. Response curves for all cytokines revealed a low background and higher signal compared to the absorbance values typically expected for MTP ELISA and those provided by the antibody supplier. | ||
| Fig. 1 (a) Full response curves for singleplex cytokines sandwich ELISA in fluoropolymer MCF strips using the multi-syringe aspirator device and HSS-HRP detection with a flatbed scanner. Response curves for IL-1β, IL-6, IL-12p70 and TNFα are represented after 2 min of OPD incubation. (b) Initial rate of colour development of each cytokine, in a singleplex assay. (c) Performance summary for each cytokine. Assay conditions as described in the Experimental section. | ||
A LoD in the range of 2.0–15 pg ml−1 was obtained for all cytokines measured for a total assay time of 50 min, plus 2 min of OPD conversion (Fig. 1c), in spite of the limited detection sensitivity for colorimetric detection of converted OPD substrate in microcapillaries in a flatbed scanner identified above. This corresponds to concentrations of between 3.5 × 10−14 to 7.1 × 10−13 molar. Recent work in our research group, involving sandwich ELISA measurement of a cancer biomarker Prostate Specific Antigen (PSA, a 26 kDa MW protein) in the same miniaturized ELISA platform,19 revealed a LoD around 1 ng m−1 (i.e. about 38 picomolar), which is 2 to 3 orders of magnitude larger than the LoD found for cytokines. This improvement in LoD is believed to be related to the higher affinity and quality of commercially available cytokine reagents, which favours the formation of capAb–Ag and subsequent Ag complexes on the surface of the plastic microcapillaries but also to the effectiveness of antibody adsorption in the FEP microcapillaries. Furthermore, the reported PSA assay system was optimised for faster assay times, rather than high sensitivity, since the clinically important range for PSA biomarker measurement is far higher than that of cytokines (i.e. >1 ng ml−1 rather than ∼1–10 pg ml−1).
Measuring initial reaction rates can in theory give a better measurement of enzyme concentration or immunoassay signal than endpoint measurement. By tracking the Abs development in each individual strip, it was possible to plot the initial rate against concentration for the whole set of cytokines measured. The linear colour development for each cytokine observed at early time points were used to measure initial velocity (Fig. 1b) the (note only data points in the linear range were plotted). However measuring initial rates did not result in improved LoD over endpoint absorbance, possibly because of the low scanning speed of the flatbed scanner, allowing just one scan every 2 or 3 minutes, which is related to the minimum 2400 dpi resolution required for image analysis. Rapid imaging using a digital camera might allow more accurate determination of initial rates and further improve assay performance.
Overall, IL-12 showed the lowest LoD among all cytokines measured, followed by TNFα, IL-1β and IL-6, with LoDs in molar basis of 35, 114, 426 and 713 femtomolar, respectively; there LoDs were calculated based on the MW of 57240 Da for IL-12, 57000 Da for TNFα, 17380 Da for IL-1β and 21041 Da for IL-6 provided by the reagent manufacturers. The CV values were all below 15% and the dynamic range for each cytokine was at least 1.5 to 2.0 log units.
Assay variability
A detailed variability study was performed for singleplex IL-1β detection as detailed in ESI.† From Fig. 1c the sensitivity of the assay herein defined by the LoD was 7.4 pg ml−1 for cytokine IL-1β. It is important to mention that LoD is referred to the level above which samples were considered positives, but this is not a very reliable measure of quantification; for that purpose LoQ is of better use. Consequently, the linear range obtained for IL-1β was 60 pg ml−1–250 pg ml−1, and from the data a calibration curve could be fitted: Abs = 0.8153 × [IL-1β] + 0.04 (R2 = 0.9973).The intra-assay precision was initially calculated across the strips where different capillaries in a given strip were compared (for each concentration: sample = 6; n = 10). The % CV was lower than 15% for all recombinant protein concentrations tested (LLoQ, MR and HLoQ) in all strips (Fig. 2a(i)).
 | ||
| Fig. 2 Variability study. (a) Shows the % CV and % recovery values of the assay across the strips, where different capillaries on the same strip are compared (for each concentration: sample = 6; n = 10). (b) Presents the % CV and % recovery values of the assay between strips (for each concentration: sample = 1; n = 6), which compares the mean values for each strip. (c) Shows the % CV values of the assay when same capillary is analysed and compared in different strips. The % CV values were revealed between 5% and 14% in (a) and (b) and between 3% and 33% in (c). 60%–160% is the range observed for the Recovery values. (d) Inter-assay precision: (i) CV values that are between 3% and 10% and (ii) % recovery values between 110% and 130%. The study was performed as described in supplementary experimental design section provided in ESI,† and used cytokine IL-1β as recombinant protein. All values are represented in percentage (%) basis. | ||
Some variability from strip to strip was noticeable and could be explained by small variation in the dimensions of the microcapillaries, which is intrinsic to the continuous melt-extrusion process used for manufacturing the MCF material used in this study. Microscope images of the cross section of the MCF taken 10 mm apart from the same batch (data not shown) revealed a 5% variation on internal diameter of the microcapillaries, which will improved in the future by optimising the melt-extrusion process and utilising in-line laser measurement to record film dimensions during production. Alternatively, we have recently shown19 that assay variability can be reduced in MCF by normalizing the absolute Abs signal for each capillary by the light path distance, which can be done by scanning a reference strip alongside the assay strips.
The assay accuracy is directly related with % recovery and consequently another parameter giving information about reliability of the assay. In terms of % recovery (Fig. 2a(ii)), the normal accepted range is between 80% and 120% that was in general not met for MR concentration (150 pg ml−1) of recombinant protein (% recovery higher than 150%), which might be related to suboptimal washing. For both LLoQ and HLoQ the % recovery was, in general, inside that range. The intra-assay precision between strips (for each concentration: sample = 1; n = 6), which compares the mean values for each strip revealed % CV lower than 10% for all concentrations tested (Fig. 2b(i)), though the % recovery remained higher than 120% at the higher recombinant protein concentrations (Fig. 2b(ii)), that can possibly be explained by the reasons previously highlighted. This data shows that the assay is precise, but for some concentrations the signal observed was sometimes higher than expected.
Each MCF strip contains 10 microcapillaries, allowing up to 10 replicate measurements for each small sample volume. Assay variability was therefore compared for each capillary position between different strips. This is particularly relevant for performance of duplex or multiplex assays in the fluoropolymer MCF strips. As expected, variability increased as the concentration of analyte decreased, but CVs remained well below 10% for all concentrations above LoQ, and close to the LoQ CVs of up to 32% were observed (Fig. 2c).
In order to understand reproducibility of the assay in MCF strips and MSA device, the Inter-assay precision was also studied. It was observed variability lower than 15% for MR concentration and lower than 10% for the LLoQ and HLoQ. The higher % recovery was obtained at the LLoQ which was 133%, followed by MR with an average % recovery of 123% (Fig. 2d). The Intra-assay analysis is mainly affected by internal variables such as fluid handling, washing steps and variation on the internal diameter of the capillaries, whereas the inter-assay is mainly depend on external variables such as operator, environment, device, etc., meaning that the assay internal variables are more critical in terms of accuracy of the method.
Quantitative triplex measurements
To demonstrate the multiplex capability of this platform, quantitative detection of three cytokines was performed (IL-1β, IL-12 and TNFα) in each fluoropolymer MCF strip using assay conditions optimised for singleplex IL-1β cytokine. This ultimately demonstrates the ability of this new miniaturized platform to perform portable and sensitive multiplex immunoassay measurements in a very cost effective manner.An important observation from Fig. 3 is that capillaries coated with 3% BSA showed Abs values close to 0.05, higher than the Abs values observed in the capillaries that were not coated with a capAb or protein (Fig. 3a). Note that all microcapilaries were subsequently coated with Superblock. This could be explained either by BSA coating interacting with one or more detAb to produce background, or less efficient blocking with BSA followed by Superblock. This highlights the importance of selecting a good blocking agent to achieve sub nanogram/milliliter detection.
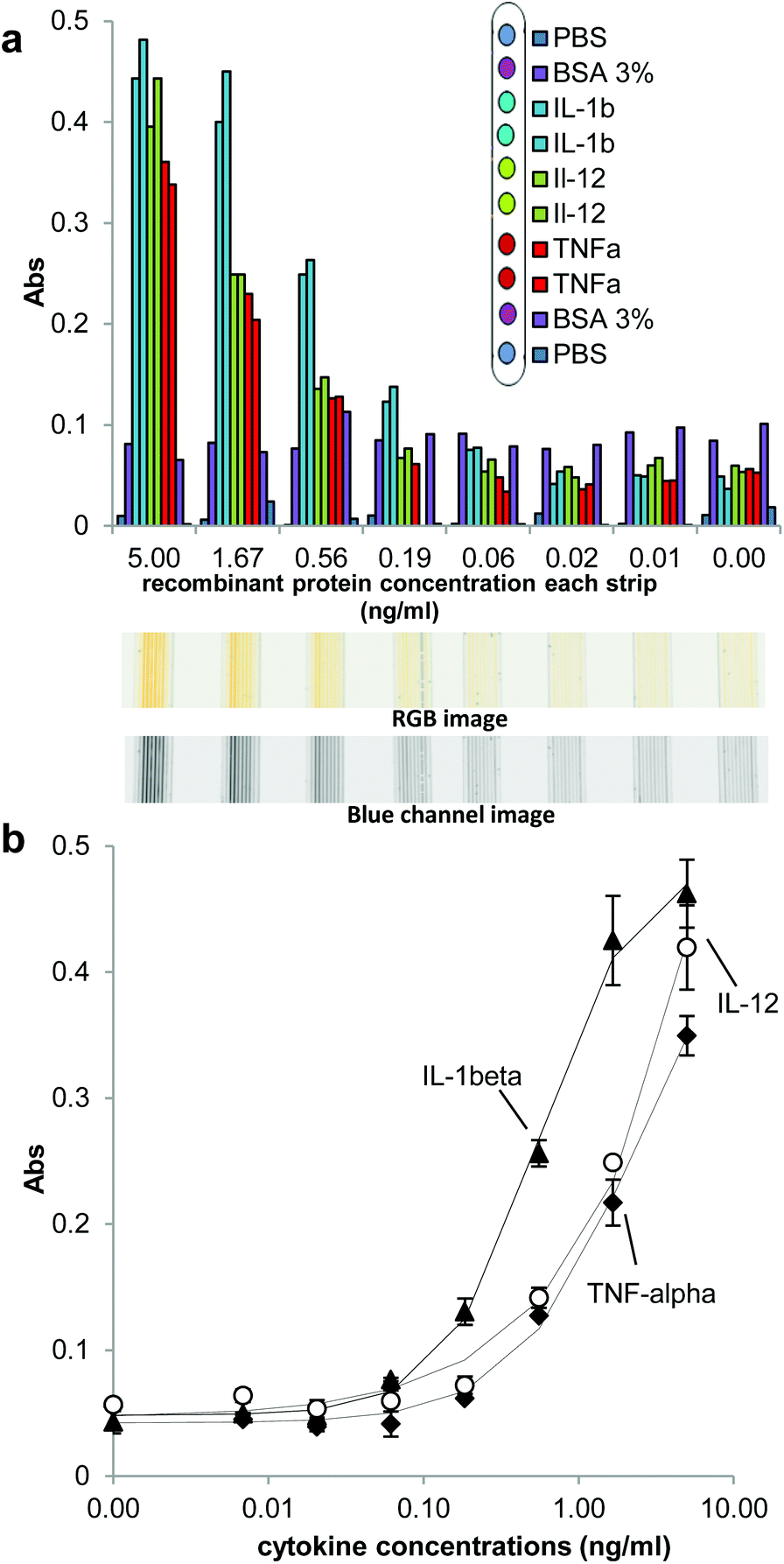 | ||
| Fig. 3 Triplex measurement of IL-1β, IL-12p70 and TNFα. (a) Capillaries 1 and 10 were coated with PBS; capillaries 2 and 8 coated with BSA 3%; the capillaries 3 and 4, 5 and 6 and 7 and 8 coated with anti-human IL-1β purified, anti-human IL-12 purified and anti-human TNFα purified, respectively. The assay was performed using 1:3 serial dilution starting from the stocking solution with 5 ng ml−1 of concentration as described in ESI.† (b) Cytokines full response curves for multiplex sandwich ELISA in MCF platform using the multi-syringe device and HSS-HRP detection using a flatbed scanner. Cytokines IL-1β, IL-12p70 and TNFα are represented for 2 min of OPD incubation. | ||
The full response curves for each cytokine measured in the multiplex strips were plotted by averaging Abs in the two capillaries coated with the same capAb on a given strip (Fig. 3b). The best-fitting of the 4PL model to the experimental data returned a cross-correlation coefficient, R2 = 0.998 for IL-1β, 0.996 for IL-12 and 0.998 for TNFα. Compared to the singleplex cytokines detection, the response curves showed similar profile in terms of absolute Abs values but also a small increase in the background. That was believed to be linked to the great excess of biotinylated detection antibodies resulting from mixing all different detAb. Consequently, the multiplex assay was further optimised in respect of detAb and HRP concentration in order to reduce this raised background and thus improve the multiplex assay to the same LoDs and LoQs reported by singleplex detection. The best combination was found to be 5 μg ml−1 of detAb and 2 μg ml−1 of Enzyme and, although the signal intensity dropped, it resulted in a clear improvement to the signal-to-noise ratio (Fig. S7†). In this instance, the Abs values for the control strips were determined from the average of three runs using the same conditions.
The full response curves and LoDs and LoQs values of the assay using the optimised conditions were then finally determined with a significant improvement on the LoDs and LoQs (Fig. 4) compared to previous conditions in Fig. 3. In addition to the conventional end-point Abs measurement, it was possible to read Abs values in MCF real-time and therefore plot response curves based on velocity data, as shown in Fig. 4b, with clear benefits in respect to LoD and LoQ for IL-1β and TNFα cytokines. In addition to the eventual problems of cross-reactivity for multi-analyte detection (that were not detectable in this study), it is important to consider the effect of cumulative increase in total biotinylated detection antibody concentration when developing a specific multiplex test panel.
 | ||
| Fig. 4 Optimised triplex measurement of IL-1β, IL-12p70 and TNFα (a) full response curve of each cytokine using the optimised conditions as described in the text. (b) Initial velocities of colour development of each cytokine in a triplex assay. (c) Typical performance of each cytokine using the optimised concentrations of detAb and Enzyme on the same conditions as the triplex assays shown in Fig. 3. | ||
Rapid cytokine detection
The robust and consistent cytokine measurements in the fluoropolymer MCF strips after optimization for analytical sensitivity allowed further assay time reductions. A rapid, 17 min IL-6 assay was performed using 40 μg ml−1 of capAb concentration by reducing the times of incubation of sample, detAb and HSS-HRP to only 5 minutes, plus 2 minutes for substrate conversion. It was noticed that not only that the signal was not significantly affected but also the LoD was slightly improved to 31 pg ml−1 when the rapid assay was performed (Fig. 5a). The high concentration of capAb immobilized on the surface of the fluoropolymer MCF strips appears to be a major factor for rapid assays by favouring the rapid binding of Ag molecules to the capAb without increasing the non-specific (background) signal. On the other hand, the enzyme concentration was decreased to 1 μg ml−1 in order to balance the high capAb concentration and keep the background values low. | ||
| Fig. 5 (a) Comparison of rapid (17 min total assay time) vs. long (52 min) IL-6 assay based on colorimetric signal for 2 min OPD conversion. The Abs values for each assay are similar and a small improvement in sensitivity was noticed for the rapid assay conditions. (b) Full response curve for 30 min IL-1β detection in undiluted human serum spiked with recombinant IL-1β protein; serum was incubated for 5 min, followed by 10 min incubation for detAb, 10 min enzyme and 5 min OPD conversion. On (a) and (b) the concentration of capAb used was 40 μg ml−1, and all other assay conditions remained as stated in Experimental section. | ||
Assay performance in undiluted human serum
Additional experiments demonstrated that MCF cytokine detection is also possible in undiluted human serum. Using a total assay time of 30 min and a concentration of capAb of 40 μg ml−1, the LoD obtained for IL-1β was 14 pg ml−1 (Fig. 5b), which compares well to the assay optimised in buffer based on 50 min total assay time (Fig. 1). Compared to full response curve in buffer (data not shown), the signal dropped by 18% for the same total assay time. We believe this is linked to higher viscosity of the matrix, which will be subject of future studies, and can be controlled by comparing measurements in human serum samples to a reference curve in a diluent that closely replicates human serum giving more appropriate reference values.Conclusions
A rapid, low cost, sensitive and precise microfluidic platform for multiple cytokine detection and quantitation was developed based on a miniaturized fluoropolymer MCF. The full response curves for singleplex detection of IL-1β, IL-12, IL-6 and TNFα confirmed an unprecedented LoD between 2.0–15.0 pg ml−1 for the whole set of cytokines tested, and CV values lower than 10% using colorimetric enzymatic amplification and detection using a flatbed scanner. The qualitative duplex and quantitative triplex assays provided evidence that the miniaturized MCF platform can efficiently measure multiple cytokines and deliver LoDs in the range of 60–150 pg ml−1 with CVs lower than 10%. The short diffusion distances in the microcapillaries offers also the possibility of performing full sandwich assay for cytokine measurement in less than 20 minutes or from undiluted human serum. The simple and inexpensive optical detection system combined with the capability of delivering 80 data points simultaneously on the same MSA device and cutting total assay time of sandwich ELISAs shows unique advantages suited to POC testing, with the possibility of being operated by a trained lab technician or an unskilled person on the field. Further work is focussed on further improving the analytical sensitivity and accuracy of multiplex assays, on measurements in clinical samples, and on automation of this manually operated device. The results presented for cytokine measurements are believed to be relevant for the development of a new generation of portable and inexpensive miniaturized diagnostics tools for early and accurate diagnostic of acute conditions such as sepsis, and assist clinicians on the POC detection of bacterial infections, reducing overuse of antibiotics.Acknowledgements
The authors are grateful to Patrick Hester from Lamina Dielectrics Ltd for supplying the fluoropolymer MCF to specifications. APC is also grateful to ERASMUS program for financial support.Notes and references
- J. Quinn, D. Gratalo, K. Haden and J. Moon, Accurate multiplex cytokine assay developed with VeraCode® technology, by Illumina, 2008.
- J. Rysz, M. Banach, A. Cialkowska-rysz, R. Stolarek, M. Barylski, J. Drozdz and P. Okonski, Cell. Mol. Immunol. Immunol., 2006, 3, 151–154 CAS.
- S. P. Fitzgerald, R. I. McConnell and A. Huxley, J. Proteome Res., 2008, 7, 450–455 CrossRef CAS PubMed.
- F. A. Bozza, J. I. Salluh, A. M. Japiassu, M. Soares, E. F. Assis, R. N. Gomes, M. T. Bozza, H. C. Castro-Faria-Neto and P. T. Bozza, Crit. Care, 2007, 11, R49 CrossRef PubMed.
- U. Sauer, P. Domnanich and C. Preininger, Anal. Biochem., 2011, 419, 46–52 CrossRef CAS PubMed.
- A. J. A. Lambeck, A. P. G. Crijns, N. Leffers, W. J. Sluiter, K. A. ten Hoor, M. Braid, A. G. J. van der Zee, T. Daemen, H. W. Nijman and W. M. Kast, Clin. Cancer Res., 2007, 13, 2385–2391 CrossRef CAS PubMed.
- M. Elsalhy, F. Azizieh and R. Raghupathy, Int. Endod. J., 2013, 46, 573–580 CrossRef CAS PubMed.
- A. Vahedi, I. Lotfinia and A. Alizadeh, Afr. J. Biotechnol., 2012, 11, 9869–9872 CAS.
- A. Prashant, P. Vishwanath, P. Kulkarni, P. Sathya Narayana, V. Gowdara, S. M. Nataraj and R. Nagaraj, PLoS One, 2013, 8, e68426 CAS.
- L. T. Osnes, B. Nakken, E. Bodolay and P. Szodoray, Autoimmun. Rev., 2013, 12, 967–971 CrossRef CAS PubMed.
- M. Holub, D. a. Lawrence, N. Andersen, A. Davidová, O. Beran, V. Marešová and P. Chalupa, Mediators Inflammation, 2013, 2013, 190145 Search PubMed.
- S.-I. Cho, S. Kuang, D.-K. Choo, J. deMello and A. J. Chang, Recent advances in microfluidic technologies for biochemistry and molecular biology, 2011 Search PubMed.
- T. B. Martins, B. M. Pasi, J. W. Pickering, T. D. Jaskowski, C. M. Litwin and H. R. Hill, Am. J. Clin. Pathol., 2002, 118, 346–353 CrossRef CAS PubMed.
- C. C. Wang, R. Huang, M. Sommer, H. Lisoukov, R. Huang, Y. Lin, T. Miller and J. Burke, J. Proteome Res. 2002337–343 Search PubMed.
- I-Micronews, Microfluid Substrates Mark Process trends, 2012 Search PubMed.
- X. Li, J. Tian and W. Shen, Anal. Bioanal. Chem., 2010, 396, 495–501 CrossRef CAS PubMed.
- L. Yu, C. M. Li, Y. Liu, J. Gao, W. Wang and Y. Gan, Lab Chip, 2009, 9, 1243–1247 RSC.
- A. D. Edwards, N. M. Reis, N. K. H. Slater and M. R. Mackley, Lab Chip, 2011, 11, 4267–4273 RSC.
- A. I. Barbosa, A. P. Castanheira, A. D. Edwards and N. M. Reis, Lab Chip, 2014, 2918–2928 RSC.
- U. Piran and W. Riordan, J. Immunol. Methods, 1990, 133, 141–143 CrossRef CAS.
- J. Ederveen, A Practical Approach to Biological Assay Validation, Hoofddorp, 2010 Search PubMed.
- J. R. Crowther, Methods in Molecular Biology: The ELISA guidebook, Humana Press, Totowa, New Jersey, 2000, vol. 149 Search PubMed.
- M. E. Wiseman and C. W. Frank, Langmuir, 2012, 28, 1765–1774 CrossRef CAS PubMed.
- T. Christopoulos and E. Diamandis, in Immunoassay, ed. T. Christopoulos and E. Diamandis, 1996, pp. 25–26 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary experimental design and supplementary results. See DOI: 10.1039/c5an00238a |
| This journal is © The Royal Society of Chemistry 2015 |