
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solvent-induced structural transitions of lysozyme in an electrospray ionization source†
Jong Wha
Lee
a and
Hugh I.
Kim
*ab
aDepartment of Chemistry, Pohang University of Science and Technology (POSTECH), Pohang, 790-784, South Korea
bDivision of Advanced Materials Science, Pohang University of Science and Technology (POSTECH), Pohang, 790-784, South Korea. E-mail: hughkim@postech.edu
First published on 24th March 2015
Abstract
The structural characterization of proteins using electrospray ionization mass spectrometry (ESI-MS) has become an important method for understanding protein structural dynamics. The correlation between the structures of proteins in solution and gas phase needs to be understood for the application of ESI-MS to protein structural studies. Hen egg white lysozyme (Lyz) is a small protein with a stable compact structure in solution. Although it was known that denatured Lyz in solution undergoes compaction during transfer into the gas phase via ESI, detailed characterization of the process was not available. In the present study, we show that the organic cosolvent, which denatures Lyz in solution, induces the collapse of the extended Lyz structure into compact structures during ESI. This process is further facilitated by the presence of acids, whose conjugate bases can interact with Lyz to reduce its charge state and the electrostatic repulsion between its charged residues (Analyst, 2015, 140, 661–669). Exposure of ESI droplets to acid and solvent vapors confirms that the overall process most probably occurs in the charged droplets from ESI. This study provides a detailed understanding of the possible influence of the solvent environment on protein structure during transfer into the gas phase.
Introduction
The recent development of electrospray ionization (ESI)1 has enabled structural investigations of various proteins using mass spectrometry (MS).2 During ESI, charged droplets are initially formed at the tip of a high-voltage capillary, and the droplets then undergo cycles of evaporation and fission until molecular ions are produced.3 This process applies significantly small energy to the analyte proteins allowing solution-phase proteins to be transferred intact into the gas phase. Many studies have supported positive correlations between protein structures in solution and the gas phase.4–12 For example, unfolding of a protein in solution is reflected by a shift in its charge state distribution (CSD) into higher charge states,4,5 and good agreement was observed between protein structural distributions in solution and protein CSDs in ESI-MS spectra.6,7 Moreover, the collision cross section (ΩD) values of proteins, obtained from ion mobility (IM) measurements, are similar to the theoretical ΩD values of protein crystal structures.9,10 A recent study showed a close relationship between ΩD and the Stokes radii of proteins and protein complexes.11 Supporting evidence for the preservation of salt bridge structures in the gas phase has also been reported.12 These reports suggest that the structural aspects of proteins in solution can be maintained after transfer into the gas phase. Utilizing the soft feature of ESI, numerous studies have successfully investigated protein conformations13–18 and noncovalent protein assemblies.19,20The transfer of solution-phase structural information into the gas phase is an important prerequisite for the application of ESI-MS to characterize protein structures. Generally, gentle parameters such as low capillary voltage and low source temperature could aid in preventing protein unfolding during ESI.21,22 However, recent reports show that structural transitions may occur during ESI even under gentle conditions. The unfolding of proteins during ESI can be facilitated by weak acids,23 low ionic strength,24 buffer decomposition,25 and enrichment of other additives.26 In addition, protein structures can collapse into smaller structures during or after transfer into the gas phase.27–29 For example, denatured myoglobin28 and lysozyme27 can assume compact structures after transfer into the gas phase. These examples show that it is important to identify factors that can influence protein structure during ESI for more reliable application of ESI-MS to investigate protein structures in solution.
Douglas and coworkers have previously shown that unfolded lysozyme (Lyz; 14.3 kDa) assumes compact structures after transfer into the gas phase.28 Although Lyz is known to be present in a helically unfolded conformation in 80% methanol solution at pH 2,30 they were unable to observe a shift of the CSD into higher charge states.28 Furthermore, the ΩD values and gas-phase hydrogen-deuterium exchange rates of Lyz ions from the native and denaturing solutions were indistinguishable.28 Thus, it was concluded that the Lyz structure collapses into compact structures while being transferred into the gas phase, and this was recently confirmed by Barran and coworkers.31 They reported that Lyz ions with extended conformations were not formed from low-pH organic cosolvents.31 The intriguing behavior of Lyz was interpreted as the refolding of Lyz driven by the four disulfide bonds within the protein,28,31 but further mechanistic characterization of this process has yet to be performed. While the disulfide bonds seem to be an important factor for the compaction of Lyz during ESI, we hypothesize that other factors also contribute to the process. In particular, acids and organic solvents used to denature Lyz in solution may actively facilitate the structural transition of Lyz during ESI. A more detailed investigation of this process would provide a better understanding of how proteins are transferred into the gas phase.
We have recently shown that acids can induce the structural transition of Lyz from aqueous solutions during ESI.23 It was demonstrated that weak acids, such as formic acid, induce the unfolding of Lyz during ESI, whereas the strong acid HCl suppressed unfolding. We inferred that formic acid provided protons to disrupt the salt bridges of Lyz, while the addition of HCl reduced electrostatic repulsion between charged residues of Lyz because of ion-pairing of chloride anions to Lyz.23 According to this explanation, organic cosolvents should be able to facilitate the compaction of proteins during ESI, as their intrinsic properties, such as low surface tension,32,33 high gas-phase basicity,34 and promotion of ion-pairing,35 would reduce the charge state and the electrostatic repulsion between the charged residues of Lyz. In this study, we aimed to reveal the influence of organic solvents on the structure of Lyz ions produced using ESI. We performed detailed structural characterization of Lyz in solution through circular dichroism (CD) spectroscopy and small-angle X-ray scattering (SAXS) experiments. Subsequently, the gas-phase structures of Lyz were investigated based on CSDs and IM measurements.
Experimental
Sample preparation
Hen egg white Lyz, ammonium acetate, HCl, formic acid, acetic acid, ethanol, and 1-propanol were purchased from Sigma-Aldrich (Saint Louis, MO, USA). HPLC-grade water, methanol, and acetonitrile were purchased from Avantor Performance Materials, Inc. (Center Valley, PA, USA) and used as solvents. An Orion 3 Star pH meter (Thermo Scientific, San Jose, CA, USA) was used to measure the pH of sample solutions.Circular dichroism (CD) spectroscopy
A Jasco J-815 polarimeter (Easton, MD, USA) was used at a speed of 50 nm min−1 and a Lyz concentration of 10 μM. All spectra were averaged from 15 spectra. The formic acid concentration was fixed to 1% by volume, because higher formic acid concentrations diminished the CD signals. The pH values of formic acid solutions are listed in the caption of Fig. 1. The samples at pH 4.5 and 7 were prepared by adding HCl or ammonia solution.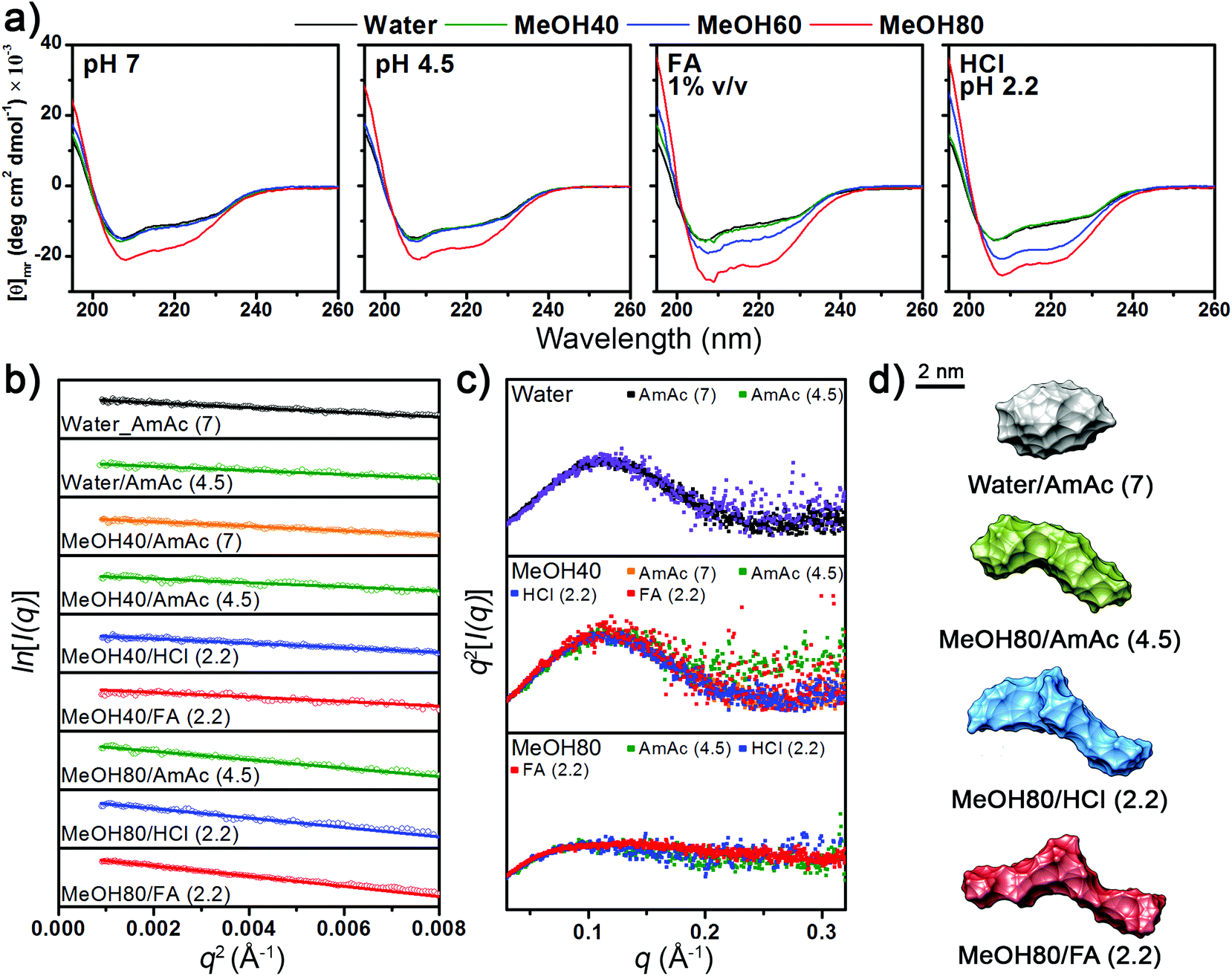 | ||
| Fig. 1 (a) CD spectra of Lyz at different pH and methanol (MeOH) concentrations. The pH values of formic acid solutions are 2.2, 2.3, 2.8, and 2.8 for 0%, 40%, 60%, and 80% methanol solutions, respectively. (b) Guinier plots, (c) Kratky plots, and (d) ab initio envelopes of Lyz from SAXS experiments. | ||
Small-angle X-ray scattering (SAXS)
SAXS experiments were performed at the 4C SAXS II beamline in Pohang Accelerator Laboratory (PAL), with experimental protocols identical to those used in our previous study.23 Ammonium acetate solutions were prepared by adding acetic acid to 200 mM ammonium acetate solution until the desired pH was obtained. Solvent scattering was subtracted from the sample scattering, and the data were analyzed using ATSAS 2.5.2.36–39 The radius of gyration (Rg) values of Lyz were estimated using the Guinier relationship (eqn (1)).40| ln[I(q)] = ln[I(0)] − Rg2q2/3 | (1) |
GNOM37 and GASBOR38 were used to obtain ab initio structures of Lyz in solution from SAXS curves. The structures were cross-checked with DAMMIN,41 and good agreement was observed.
Electrospray ionization ion mobility mass spectrometry (ESI-IM-MS)
Experiments were performed with a Waters Synapt G2 HDMS instrument with travelling wave ion mobility spectrometry (TWIMS) capability. Experimental parameters were optimized to ensure that gentle conditions were used. Overall, the parameters were similar to those used in our previous study,23 with the exception of desolvation gas flow, wave velocity, and wave height, which were 600 L h−1, 250 m s−1, and 18.0 V, respectively. The calibration of experimental arrival times into ΩD was performed using previously reported ΩD values for denatured ubiquitin, cytochrome c, and apomyoglobin.41,42 The IM spectra were smoothed once using the Savitzky–Golay algorithm with a one-unit window, using MassLynx 4.1 software (Waters, Milford, MA).Results and discussion
Structure of Lyz in solution
The CD spectra in Fig. 1a show that the secondary structural change of Lyz is negligible in up to 60% methanol at pH 4.5 and 7. Lowering the pH to below 3 by adding formic acid and HCl unfolds Lyz in 60% methanol solutions. However, Lyz still maintains its secondary structures in up to 40% methanol under highly acidic conditions, demonstrating its exceptional structural stability. The experimental results are consistent with those of Akasaka and coworkers, who showed that Lyz unfolds in solutions between 40% to 80% methanol depending on the pH.30The tertiary structural information of Lyz provided by the solution SAXS experiments generally agreed with the structural information from the CD spectra (see Fig. S1a in the ESI† for raw SAXS profiles). The Rg values of Lyz from Guinier plots of SAXS curves (Fig. 1b) are summarized in Table 1. Lyz in water and 40% methanol generally show Rg values of ∼15.3 Å, which are consistent with the Rg values of native Lyz reported previously.23,30,39 Slightly smaller Rg values were observed for Lyz in ammonium acetate solutions at pH 4.5, which is inferred to be due to the influence of acetate anions on Lyz (see Discussions in the ESI†).43 Kratky plots (Fig. 1c) of SAXS curves, which provide overall compactness of the protein, exhibit bell-shaped curves for up to 40% methanol, also demonstrating that Lyz is present as globular structures. At 80% methanol, Lyz undergoes unfolding even under weakly acidic conditions (pH 4.5) and is further unfolded under strongly acidic conditions (pH 2.2), in agreement with the structural information from the CD spectra (Fig. 1a). The Rg values of Lyz in these solutions are ∼22 Å, which are approximately 50% greater than the Rg value of native Lyz (Table 1). The ab initio SAXS envelopes38 for Lyz in Fig. 1d (see Fig. S1b in the ESI† for theoretical fitting curves) also illustrate that Lyz in 80% methanol solutions has more extended structures than native Lyz. SAXS experiments were not possible in 80% methanol solution at pH 7 due to the limited solubility of Lyz, but the CD spectra in Fig. 1a suggest that Lyz is likely to be unfolded under this condition.
| Solution (pH) | R g/Å | R 2 | Folding state |
|---|---|---|---|
| a Obtained from linear fitting curves of Guinier plots (Fig. 1b). b Obtained from Kratky plots (Fig. 1c). c AmAc: ammonium acetate. d Formic acid. | |||
| Water–AmAcc (7) | 15.3 | 0.97 | Globular |
| Water–AmAc (4.5) | 14.3 | 0.92 | Globular |
| MeOH40–AmAc (7) | 15.4 | 0.96 | Globular |
| MeOH40–AmAc (4.5) | 14.5 | 0.87 | Globular |
| MeOH40–HCl (2.2) | 15.2 | 0.96 | Globular |
| MeOH40–FAd (2.2) | 15.3 | 0.87 | Globular |
| MeOH80–AmAc (4.5) | 20.7 | 0.87 | Unfolded |
| MeOH80–HCl (2.2) | 21.9 | 0.91 | Unfolded |
| MeOH80–FA (2.2) | 22.9 | 0.98 | Unfolded |
The overall experimental results show that the structure of Lyz in solution is greatly dependent on the methanol concentration and pH. No significant dependency of the Lyz structure on the use of different acids was observed. Methanol concentrations required to denature Lyz were similar in both HCl and formic acid solutions, and the Rg values of Lyz after unfolding in HCl and formic acid solutions differed by less than 5%. Therefore, it is inferred that dominant factors determining the structural state of Lyz in solution are the electrostatic repulsion between its charged residues (low pH) and hydrophobic interactions (solvent composition).
Structure of Lyz after transfer into the gas phase
The MS spectrum of Lyz in water in Fig. 2 is centered at the +10 charge state, in agreement with the previously reported CSDs of Lyz.8,23,28,44 The CSD of Lyz from 80% methanol solution is highly similar to that from water, which indicates that CSDs are unable to describe the structural transition of Lyz in solution. Furthermore, while addition of acids further unfolds Lyz in 80% methanol solutions, the CSDs at pH 2.2 are shifted to lower charge states rather than to higher charge states. Unfolding of a protein generally enlarges its solvent accessible surface area and increases its charge states.5,45 An ∼50% increase in the Rg value of Lyz in 80% methanol solutions (Table 1) approximately corresponds to an ∼125% increase in its surface area. Therefore, the absence of charge shifts suggests that unfolded Lyz contracts into compact structures during transfer into the gas phase, before charging occurs.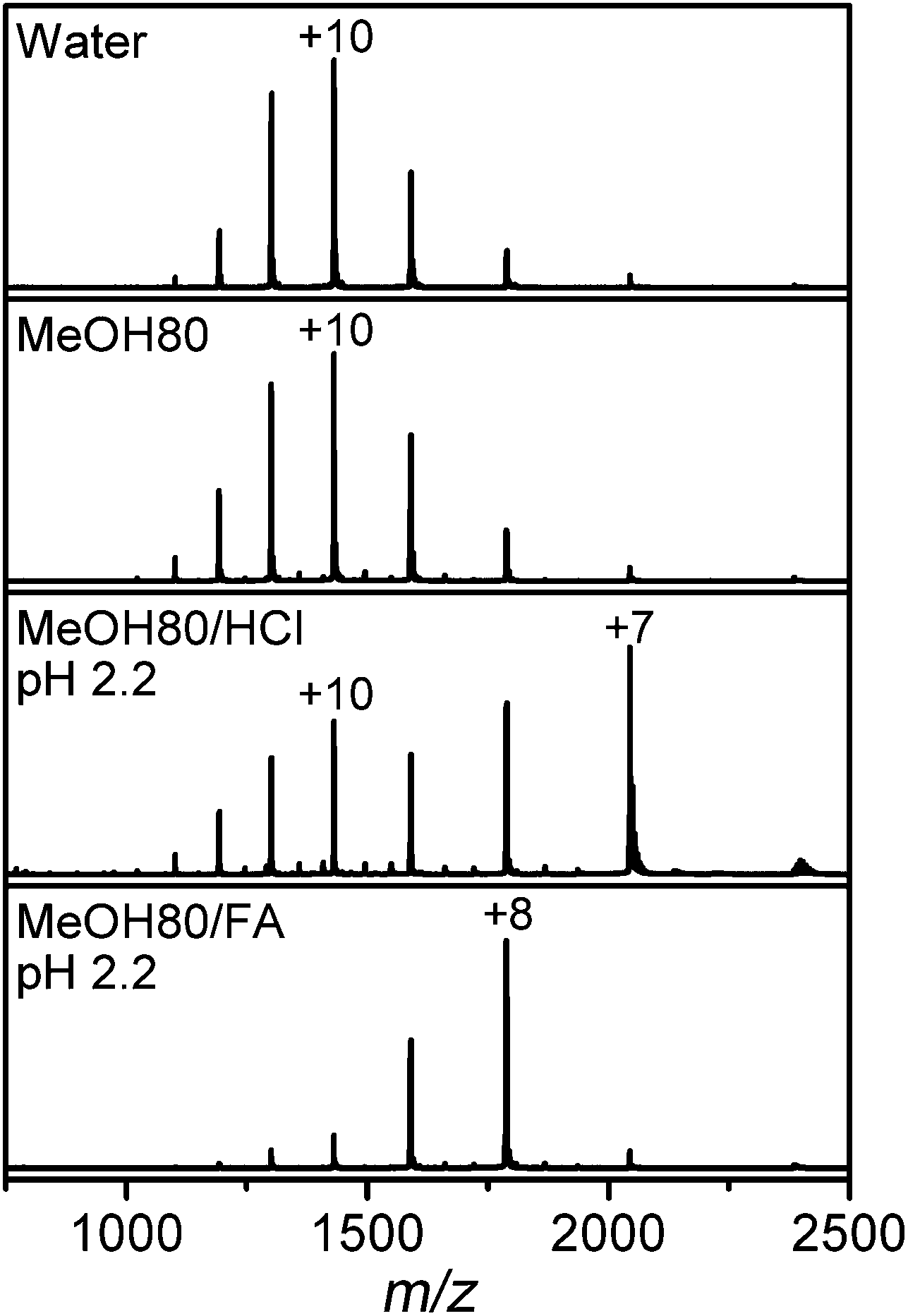 | ||
| Fig. 2 ESI-MS spectra of Lyz in pure water and 80% methanol (MeOH) solutions without acid, or containing HCl or formic acid (FA). | ||
In order to understand the correlation between structures of Lyz in solution and the gas phase, ΩD distributions of Lyz from different solutions were compared. A detailed structural characterization of Lyz ions in the gas phase was performed in our previous study.23 Briefly, we classified structures of Lyz ions in the gas phase into three distinct classes (A, B, and C) depending on their trends in charge-ΩD correlations (see Fig. 4a in ref. 23). We found that +6 and +7 charged Lyz ions have compact conformations that resemble the compact native state structure and are classified as A class ions (ΩD = 1320–1390 Å2). B class ions with greater ΩD are observed at higher charge states (+8 to +12), and their ΩD increases with increasing charge states (ΩD = 1750–2150 Å2). Addition of weak acids unfolds Lyz during ESI to facilitate the formation of B class ions at low charge states (+6 and +7) with ΩD values greater than those of the A class ions and C class ions with very large ΩD at high charge states (+10 to +12; ΩD = 2370–2510 Å2). Fig. 3 compares the ΩD distributions of Lyz from 80% methanol solutions (black solid lines) and pure water (red dots). For Lyz ions from the two different solutions, A class ions dominate at low charge states (+6 and +7) and B class ions dominate at higher charge states (+8 to +12) with no signature of C class ions. Analogously to the CSDs, the ΩD distributions of Lyz ions from 80% methanol solutions are similar to those of Lyz ions from pure aqueous solution. These observations suggest that Lyz does not maintain its solution-phase structural information after transfer into the gas phase.
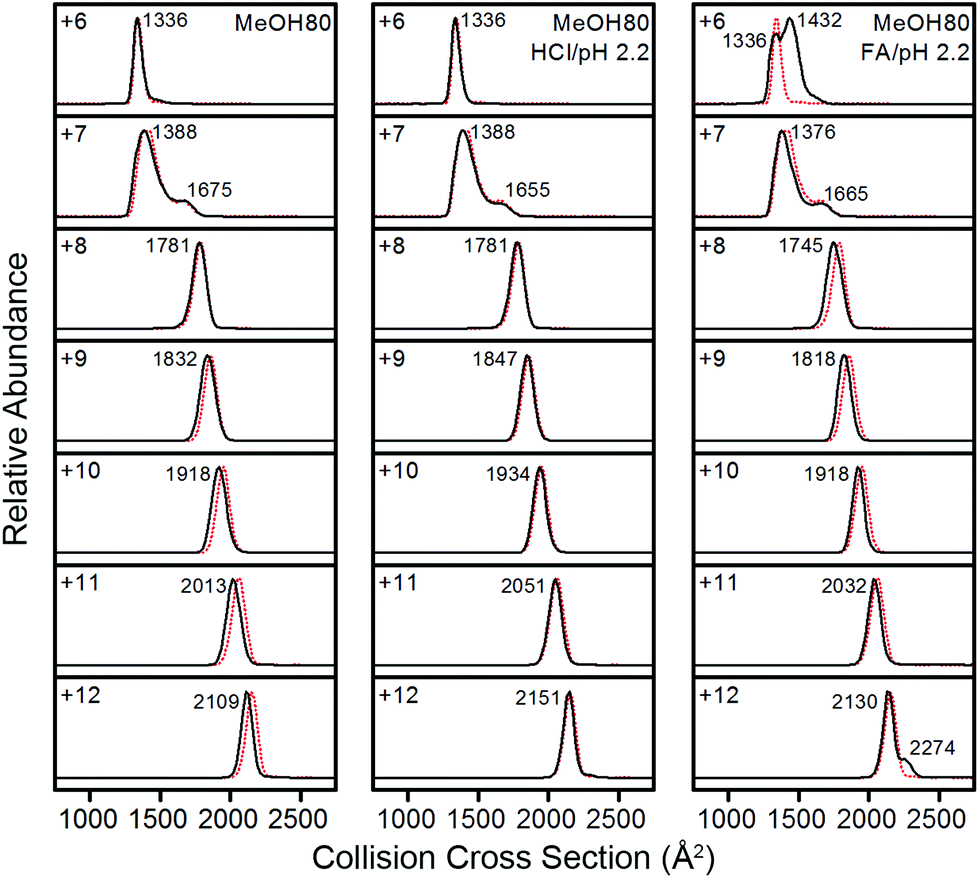 | ||
| Fig. 3 IM spectra of +6 to +12 charged Lyz ions from 80% methanol solutions (black lines). IM spectra of Lyz ions from pure water are given as red dotted lines for comparison. | ||
Fig. 3 shows that Lyz ions from the formic acid solution exhibit extra distributions with greater ΩD at +6 and +12 charge states in comparison with those from pure water. However, this result should be compared with the ΩD distributions of Lyz from aqueous formic acid solution (see Fig. 3 in ref. 23 for IM spectra), because the addition of formic acid causes more unfolded Lyz ions to be formed during ESI.23Fig. 4 illustrates that ions from 80% methanol–formic acid solution are relatively smaller in size than ions formed from aqueous formic acid solution by formic acid-induced unfolding.23 In particular, the ΩD of B class ions observed at +6 charge state is reduced (from 1495 Å2 to 1434 Å2), and the C class ions observed at +10 to +12 charge states disappear. Although not all different peaks can be completely resolved due to the low resolution of the IMS technique, these data were reproducible over three experiments and a time span of 9 months.
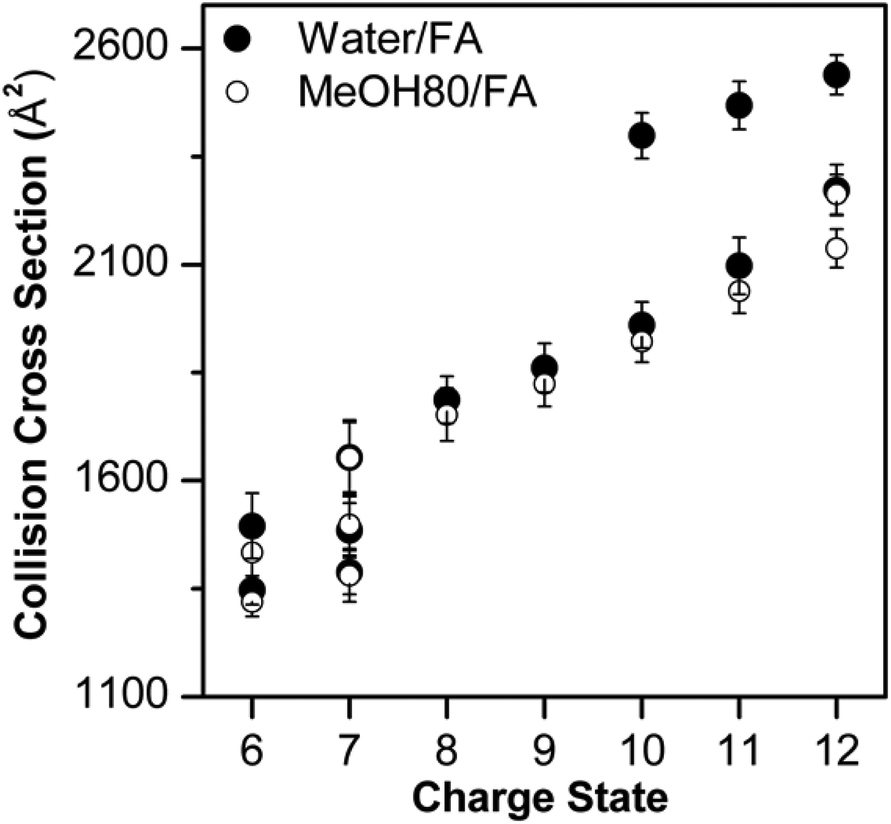 | ||
| Fig. 4 Comparison between Lyz conformers observed in water–formic acid solution and 80% methanol–formic acid solution at pH 2.2. The error bars represent half width at half maximum. | ||
The observation of smaller ions from methanol cosolvent is notable as it suggests that methanol is suppressing the formation of extended Lyz ions, which is a reversed effect of methanol in solution. Therefore, compaction of unfolded Lyz during transfer into the gas phase is not only due to the intrinsic properties of Lyz but is also facilitated by the presence of the organic solvent used to denature the protein in solution. Formic acid-induced unfolding is also suppressed in 40% methanol solution (Fig. S2 and S3 in the ESI†), but the ions formed are generally greater in size than those formed from 80% methanol solutions. As methanol unfolds Lyz in solution, this observation also supports the contention that methanol suppresses the formation of unfolded Lyz ions. Further evidence for the effect of methanol is given by the suppression of acetic acid-induced unfolding23 in both 40% and 80% methanol solutions (see Fig. S5 and Discussions in the ESI†).
Vapor exposure of Lyz during ESI and other organic solvent effects
For more detailed characterization of structural transitions of Lyz during ESI, vapor exposure experiments46–48 were employed. Vapor exposure of ESI droplets can induce folding or refolding of proteins within,46,47 and this experimental procedure would decouple possible solution-phase conformational effects on gas-phase Lyz structures. For the following series of experiments, the vapor was produced by placing a glass vial containing formic acid or methanol in the ESI chamber,48 and Lyz was subjected to ESI in the presence of the vapor.Regardless of solution compositions, exposure to formic acid or methanol vapor during ESI shifted the CSD of Lyz to lower states centered at the +8 charge state (Fig. S6 in the ESI†). This again suggests that the structural information of Lyz in solution does not remain in the CSDs. The IM distributions of Lyz ions formed in the presence (black lines) and the absence (red dots) of vapor show how structural transitions of Lyz occur during ESI (Fig. 5). First, exposure of Lyz in water to formic acid vapor caused unfolded Lyz ions (B class ions; ΩD = 1466 Å2) to be observed at the +6 charge state, showing that formic acid-induced unfolding occurs during ESI (Fig. 5a). When Lyz is in 80% methanol solution, the abundance of the unfolded Lyz ion is reduced, demonstrating that methanol can reduce unfolding of Lyz during ESI (Fig. 5b). It is further observed that methanol exposure also decreases abundance of the unfolded conformers formed by formic acid-induced unfolding (ΩD = 1491 and 1432 Å2, Fig. 5c and 5d, respectively). These series of experiments show that the structural transition of Lyz indeed occurs during ESI, and that methanol can actively participate in the contraction of the Lyz structure during the ionization process.
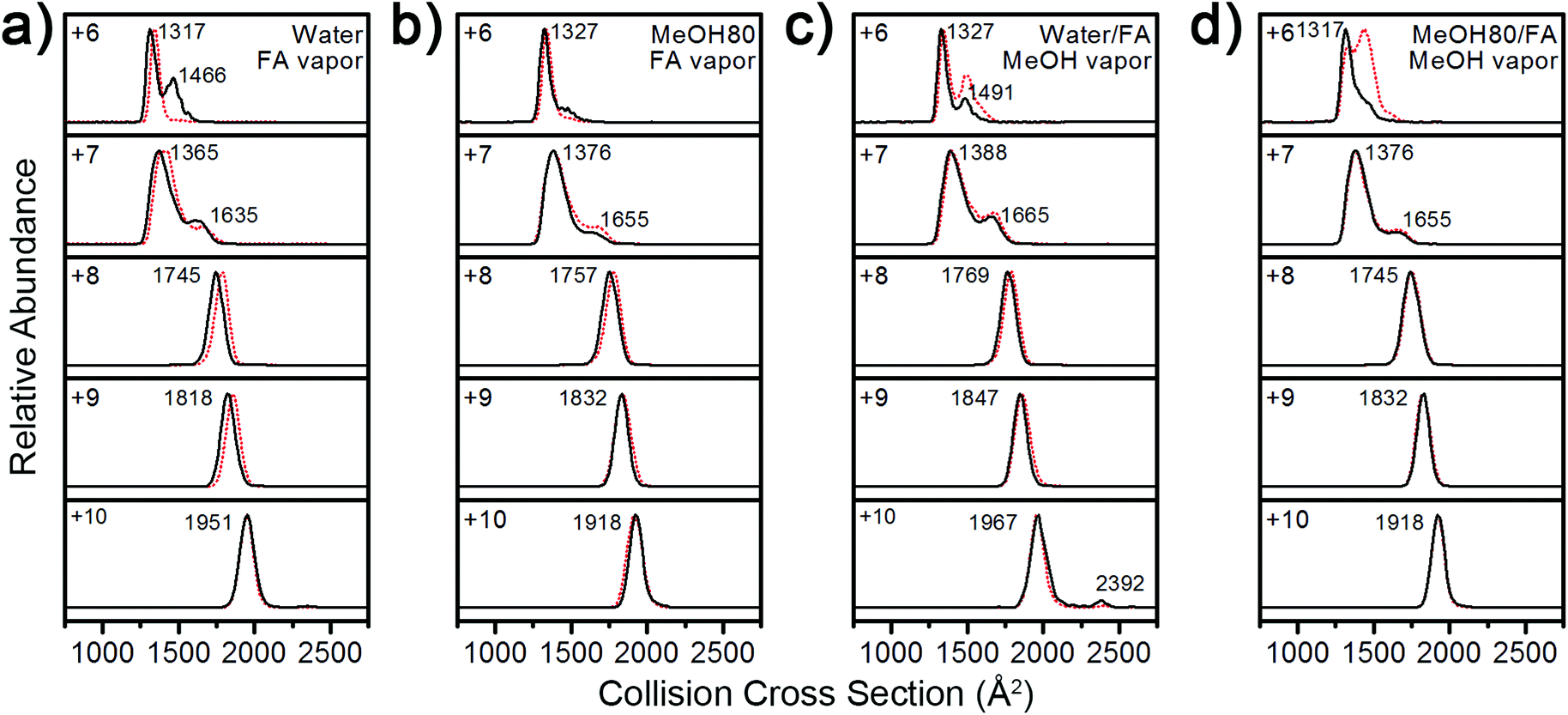 | ||
| Fig. 5 IM spectra of +6 to +10 charged Lyz ions from (a) water, exposed to formic acid vapor, (b) 80% methanol, exposed to formic acid vapor, (c) water with formic acid, exposed to methanol vapor, and (d) 80% methanol with formic acid, exposed to methanol vapor during ESI (black lines). IM spectra in the absence of vapor are given as red dotted lines for comparison. | ||
To test the generality of the solvent effect on the Lyz structure during ESI, further experiments were performed with other organic cosolvents (80% acetonitrile, ethanol, and 1-propanol) containing formic acid. It was observed that addition of these solvents also shifted the CSDs into lower charge states (Fig. 6a) and reduced the size or abundance of B class ions at the +6 charge state (Fig. 6b). These results indicate that suppression of Lyz unfolding is facilitated by properties that are commonly shared by different organic solvents.
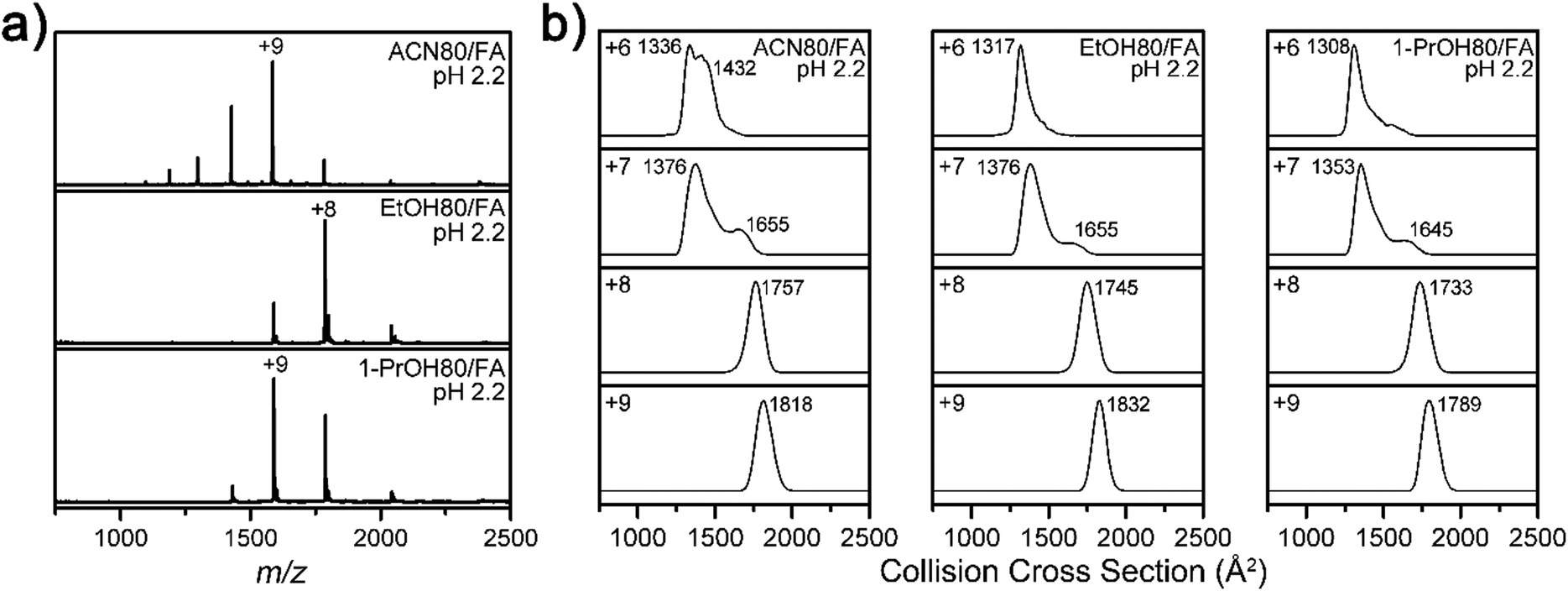 | ||
| Fig. 6 (a) ESI-MS spectra of Lyz in 80% acetonitrile (ACN), ethanol (EtOH), and 1-propanol (1-PrOH) solutions with formic acid, at pH 2.2. (b) IM spectra of Lyz ions from 80% ACN, EtOH, and 1-PrOH solutions with formic acid at pH 2.2. | ||
Influence of organic solvents on Lyz during transfer into the gas phase
Our experimental results show that both the addition of methanol to the ESI solution and exposure of aqueous ESI droplets to methanol vapor promoted compaction of Lyz during ESI. As organic solvents may operate in a different manner during these two processes, they require separate discussion. We first discuss the case where methanol was added directly to the ESI solution. The charged residue model (CRM)3,49 predicts that low surface tension of organic solvents reduces the charge density of ESI droplets, and thus reduces the charge state of protein ions.32 On the contrary, others discuss that protein charge states do not strictly follow the surface tension of the solvent used.8,50,51 Some explanation for conflicting interpretations of the role of solvent properties during ESI may come from the preferential evaporation of more volatile components.33 Our experimental results show that both CSDs and IM distributions of Lyz from 40% (Fig. S2 and S3 in the ESI†) and 80% methanol solution (Fig. 2 and 3) are highly similar to those from pure aqueous solution (Fig. 2 and 3). These results suggest that methanol is depleted preferentially from charged droplets generated with ESI and has a limited influence at the later stages of ESI when desolvated protein ions are formed.However, additional explanation is required, because the influence of methanol is significant in acidic solutions. The CSDs of Lyz from methanol solutions at low pH (Fig. 2) are generally centered at lower charge states in comparison to those we reported from aqueous solutions at low pH.23 It was also observed that methanol reduces the size or abundance of more unfolded conformers in a given charge state (Fig. 3), which indicates that the solvent effect can persist in charged droplets containing acids. We have previously reported that the acid effect persists to the later stages of ionization because acids protonate protein residues and induce protein unfolding or reduce protein charge states by adduction of acid anions.23 This explains why the effect of organic solvents is particularly significant under acidic conditions. Although methanol evaporates preferentially over water, the low dielectric constant of methanol could facilitate acid anion binding to Lyz during ESI.35 This effect may later be reflected in the CSDs as acid anions can abstract protein charges.52 Stabilization of compact conformers (A class ions at low charge states and B class ions at higher charge states) by methanol can be interpreted with reduction in the electrostatic repulsion between charged residues of Lyz following acid anion binding.23 Additionally, a decrease in the methanol content during ESI would also facilitate the structural collapse of Lyz, as water would relatively be enriched. The explanation based on ion-pairing also rationalizes the observation that the MS and IM spectra of Lyz in acetonitrile solution (Fig. S6 and Fig. 6) are most similar with those of Lyz in methanol solution (Fig. 2 and 3). Notably, the dielectric constants of methanol (33.0) and acetonitrile (36.64) are highly similar, whereas other properties such as gas-phase basicity, vapor pressure, surface tension, boiling point, and viscosity are either significantly different or also similar with those of other organic solvents (see Table S1 in the ESI†). This supports the notion that the ion association and dissociation phenomenon has significant effects on the formation of Lyz ions.
A very recent report by DeMuth and McLuckey has shown that exposure of ESI droplets to organic vapors can reduce metal adduction to protein ions.53 They discussed that organic solvents inside charged droplets can lower the barrier for ion evaporation of metal cations, and this argument was supported by intense signals of metal–organic solvent clusters observed at low m/z.53 The proposed mechanism can rationalize why methanol vapor prevented the formation of unfolded ions during ESI, because ion evaporation of protons facilitated by methanol vapor would reduce the proton density within the ESI droplets. Consequently, the proton concentration available for Lyz would decrease, and anions would be relatively enriched to decrease the overall charge state of Lyz during ESI. It is also possible that further charge reduction by methanol, which is more basic than water, affects the charge state of Lyz after desolvation in the gas phase. However, our vapor exposure experiments (Fig. 5) show that the solvent effect is mostly operative in the charged droplet. Overall, the structural transition of Lyz from an unfolded state in solution to a compact structure in the gas phase during ESI is facilitated by charge reduction of Lyz by organic solvents.
Conclusion
Although the peculiar properties of Lyz during transfer into the gas phase have long been known, a detailed investigation of the process was not available. We have shown that methanol and acids cooperate during ESI to facilitate the structural transition of Lyz from an unfolded state in solution to compact structures in the gas phase. This study provides additional insight into the influence of organic solvents on the charge state and structural distribution of protein ions formed using ESI, which is currently not completely understood. Although the low surface tension of organic solvents has received the most attention, our study shows that the low dielectric properties of organic solvents could exert additional effects on proteins during transfer into the gas phase. In addition, our detailed characterization of Lyz indicates that its structure in the gas phase is most dependent on the ionization process; therefore, Lyz may be used to reveal other potential factors influencing protein structures during ESI.Acknowledgements
This work was supported by a Basic Research Program (grant no. 2013R1A1A2008974) through the National Research Foundation (NRF) of Korea funded by the Ministry of Science, ICT, and Future Planning (MSIP) and a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare of Korea (grant no. HT13C-0011-040013).Notes and references
- J. Fenn, M. Mann, C. Meng, S. Wong and C. Whitehouse, Science, 1989, 246, 64–71 CAS.
- R. H. H. v. d. Heuvel and A. J. R. Heck, Curr. Opin. Chem. Biol., 2004, 8, 519–526 CrossRef PubMed.
- L. Konermann, E. Ahadi, A. D. Rodriguez and S. Vahidi, Anal. Chem., 2013, 85, 2–9 CrossRef CAS PubMed.
- Z. Hall and C. V. Robinson, J. Am. Soc. Mass Spectrom., 2012, 23, 1161–1168 CrossRef CAS PubMed.
- L. Testa, S. Brocca and R. Grandori, Anal. Chem., 2011, 83, 6459–6463 CrossRef CAS PubMed.
- L. Konermann, B. A. Collings and D. J. Douglas, Biochemistry, 1997, 36, 5554–5559 CrossRef CAS PubMed.
- A. Dobo and I. A. Kaltashov, Anal. Chem., 2001, 73, 4763–4773 CrossRef CAS.
- M. Šamalikova, I. Matecko, N. Muller and R. Grandori, Anal. Bioanal. Chem., 2004, 378, 1112–1123 CrossRef PubMed.
- T. Wyttenbach and M. T. Bowers, J. Phys. Chem. B, 2011, 115, 12266–12275 CrossRef CAS PubMed.
- D. E. Clemmer, R. R. Hudgins and M. F. Jarrold, J. Am. Chem. Soc., 1995, 117, 10141–10142 CrossRef CAS.
- D. Hewitt, E. Marklund, D. J. Scott, C. V. Robinson and A. J. Borysik, J. Phys. Chem. B, 2014, 118, 8489–8495 CrossRef CAS PubMed.
- Z. Zhang, S. Browne and R. Vachet, J. Am. Soc. Mass Spectrom., 2014, 25, 604–613 CrossRef CAS PubMed.
- S. W. Heo, T. S. Choi, K. M. Park, Y. H. Ko, S. B. Kim, K. Kim and H. I. Kim, Anal. Chem., 2011, 83, 7916–7923 CrossRef CAS PubMed.
- J. A. Loo, R. R. O. Loo, H. R. Udseth, C. G. Edmonds and R. D. Smith, Rapid Commun. Mass Spectrom., 1991, 5, 101–105 CrossRef CAS PubMed.
- U. A. Mirza, S. L. Cohen and B. T. Chait, Anal. Chem., 1993, 65, 1–6 CrossRef CAS.
- J. W. Lee, S. W. Heo, S. J. C. Lee, J. Y. Ko, H. Kim and H. I. Kim, J. Am. Soc. Mass Spectrom., 2013, 24, 21–29 CrossRef CAS PubMed.
- S. J. C. Lee, J. W. Lee, T. S. Choi, K. S. Jin, S. Lee, C. Ban and H. I. Kim, Anal. Chem., 2014, 86, 1909–1916 CrossRef CAS PubMed.
- A. Litwińczuk, S. R. Ryu, L. A. Nafie, J. W. Lee, H. I. Kim, Y. M. Jung and B. Czarnik-Matusewicz, Biochim. Biophys. Acta, Proteins Proteomics., 2014, 1844, 593–606 CrossRef PubMed.
- M. Sharon and C. V. Robinson, Annu. Rev. Biochem., 2007, 76, 167–193 CrossRef CAS PubMed.
- A. J. R. Heck, Nat. Methods, 2008, 5, 927–933 CrossRef CAS PubMed.
- H. J. Sterling, C. A. Cassou, A. C. Susa and E. R. Williams, Anal. Chem., 2012, 84, 3795–3801 CrossRef CAS PubMed.
- U. A. Mirza and B. T. Chait, Int. J. Mass Spectrom. Ion Processes, 1997, 162, 173–181 CrossRef CAS.
- J. W. Lee and H. I. Kim, Analyst, 2015, 140, 661–669 RSC.
- H. Lin, E. N. Kitova, M. A. Johnson, L. Eugenio, K. K. S. Ng and J. S. Klassen, J. Am. Soc. Mass Spectrom., 2012, 23, 2122–2131 CrossRef CAS PubMed.
- J. B. Hedges, S. Vahidi, X. Yue and L. Konermann, Anal. Chem., 2013, 85, 6469–6476 CrossRef CAS PubMed.
- H. J. Sterling, J. S. Prell, C. A. Cassou and E. R. Williams, J. Am. Soc. Mass Spectrom., 2011, 22, 1178–1186 CrossRef CAS PubMed.
- H. Sterling, M. Daly, G. Feld, K. Thoren, A. Kintzer, B. Krantz and E. Williams, J. Am. Soc. Mass Spectrom., 2010, 21, 1762–1774 CrossRef CAS PubMed.
- D. M. Mao, K. R. Babu, Y. L. Chen and D. J. Douglas, Anal. Chem., 2003, 75, 1325–1330 CrossRef CAS.
- K. Pagel, E. Natan, Z. Hall, A. R. Fersht and C. V. Robinson, Angew. Chem., Int. Ed., 2013, 52, 361–365 CrossRef CAS PubMed.
- Y. O. Kamatari, T. Konno, M. Kataoka and K. Akasaka, Protein Sci., 1998, 7, 681–688 CrossRef CAS PubMed.
- R. Beveridge, S. Covill, K. J. Pacholarz, J. M. D. Kalapothakis, C. E. MacPhee and P. E. Barran, Anal. Chem., 2014, 86, 10979–10991 CrossRef CAS PubMed.
- A. T. Iavarone and E. R. Williams, J. Am. Chem. Soc., 2003, 125, 2319–2327 CrossRef CAS PubMed.
- R. L. Grimm and J. L. Beauchamp, J. Phys. Chem. A, 2010, 114, 1411–1419 CrossRef CAS PubMed.
- A. Iavarone, J. Jurchen and E. Williams, J. Am. Soc. Mass Spectrom., 2000, 11, 976–985 CrossRef CAS.
- G. Wang and R. Colecor, J. Am. Soc. Mass Spectrom., 1996, 7, 1050–1058 CrossRef CAS.
- P. V. Konarev, V. V. Volkov, A. V. Sokolova, M. H. J. Koch and D. I. Svergun, J. Appl. Crystallogr., 2003, 36, 1277–1282 CrossRef CAS.
- D. Svergun, J. Appl. Crystallogr., 1992, 25, 495–503 CrossRef.
- D. I. Svergun, M. V. Petoukhov and M. H. J. Koch, Biophys. J., 2001, 80, 2946–2953 CrossRef CAS.
- D. Svergun, C. Barberato and M. H. J. Koch, J. Appl. Crystallogr., 1995, 28, 768–773 CrossRef CAS.
- C. D. Putnam, M. Hammel, G. L. Hura and J. A. Tainer, Q. Rev. Biophys., 2007, 40, 191–285 CrossRef CAS PubMed.
- B. T. Ruotolo, J. L. P. Benesch, A. M. Sandercock, S.-J. Hyung and C. V. Robinson, Nat. Protoc., 2008, 3, 1139–1152 CrossRef CAS PubMed.
- M. F. Bush, Z. Hall, K. Giles, J. Hoyes, C. V. Robinson and B. T. Ruotolo, Anal. Chem., 2010, 82, 9557–9565 CrossRef CAS PubMed.
- A. Ducruix, J. P. Guilloteau, M. Riès-Kautt and A. Tardieu, J. Cryst. Growth, 1996, 168, 28–39 CrossRef CAS.
- M. I. Catalina, R. H. H. van den Heuvel, E. van Duijn and A. J. R. Heck, Chem. – Eur. J., 2005, 11, 960–968 CrossRef CAS PubMed.
- L. Konermann, J. Phys. Chem. B, 2007, 111, 6534–6543 CrossRef CAS PubMed.
- A. Kharlamova, B. M. Prentice, T.-Y. Huang and S. A. McLuckey, Anal. Chem., 2010, 82, 7422–7429 CrossRef CAS PubMed.
- A. Kharlamova, J. C. DeMuth and S. McLuckey, J. Am. Soc. Mass Spectrom., 2012, 23, 88–101 CrossRef CAS PubMed.
- K. Sokratous, L. V. Roach, D. Channing, J. Strachan, J. Long, M. S. Searle, R. Layfield and N. J. Oldham, J. Am. Chem. Soc., 2012, 134, 6416–6424 CrossRef CAS PubMed.
- J. Fernandez de la Mora, Anal. Chim. Acta, 2000, 406, 93–104 CrossRef CAS.
- M. Šamalikova and R. Grandori, J. Am. Chem. Soc., 2003, 125, 13352–13353 CrossRef PubMed.
- M. Šamalikova and R. Grandori, J. Mass Spectrom., 2005, 40, 503–510 CrossRef PubMed.
- U. A. Mirza and B. T. Chait, Anal. Chem., 1994, 66, 2898–2904 CrossRef CAS.
- J. C. DeMuth and S. A. McLuckey, Anal. Chem., 2015, 87, 1210–1218 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional discussions and additional data (Fig. S1–S6 and Table S1). See DOI: 10.1039/c5an00235d |
| This journal is © The Royal Society of Chemistry 2015 |