
Design and performance of a high-flux electrospray ionization source for ion soft landing
K. Don D.
Gunaratne†
a,
Venkateshkumar
Prabhakaran†
a,
Yehia M.
Ibrahim
b,
Randolph V.
Norheim
b,
Grant E.
Johnson
a and
Julia
Laskin
*a
aPacific Northwest National Laboratory, Physical Sciences Division, P.O. Box 999, MSIN K8-88, Richland, Washington 99352, USA. E-mail: Julia.Laskin@pnnl.gov
bBiological Sciences Division and Environmental Molecular Sciences Laboratory, Pacific Northwest National Laboratory, 3335 Innovation Avenue (K8-98), P.O. Box 999, Richland, Washington 99352, USA
First published on 23rd March 2015
Abstract
We report the design and evaluation of a new high-intensity electrospray ionization source for ion soft-landing experiments. The source incorporates a dual ion funnel, which enables operation with a higher gas load through an expanded diameter heated inlet into the additional first region of differential pumping. This capability allowed us to examine the effect of the inner diameter (ID) of the heated stainless steel inlet on the total ion current transmitted through the dual funnel interface and, more importantly, the mass-selected ion current delivered to the deposition target. The ion transmission of the dual funnel is similar to the transmission of the single funnel used in our previous soft landing studies. However, substantially higher ion currents were obtained using larger ID heated inlets and an orthogonal inlet geometry, in which the heated inlet was positioned perpendicular to the direction of ion propagation through the instrument. The highest ion currents were obtained using the orthogonal geometry and a 1.4 mm ID heated inlet. The corresponding stable deposition rate of ∼1 μg of mass-selected ions per day will facilitate future studies focused on the controlled deposition of complex molecules on substrates for studies in catalysis, energy storage, and self-assembly.
Introduction
Soft and reactive landing of mass-selected ions onto surfaces is ideally suited for the highly controlled preparation of novel materials and purification of compounds present in complex mixtures. This approach allows the isolation of selected species on substrates and subsequent characterization of their structure and reactivity.1–11 In addition, precise control of the composition, ionic charge state, kinetic energy, and shape of the ion beam facilitates fundamental studies of the chemistry and physics of ion–surface interactions that are not complicated by the presence of residual reactants, counterions, and solvent molecules. Applications of mass-selected ion deposition include the preparation of protein arrays12–14 and carbon-based surfaces,15–17 studies of how surfaces impact protein conformations,18 preparation of conformationally-selected peptide layers on surfaces,19,20 chiral enrichment of enantiomers,21,22 deposition of redox-active molecules23,24 and molecular magnets,25 covalent modification of polymer films,26,27 covalent immobilization of organic and biological molecules on surfaces,28–32 deposition of catalytic organometallic complexes,33–35 molecular clusters,36,37 as well as metal and metal alloy clusters and larger nanoparticles.2,38–45 These diverse applications of ion soft landing will all benefit directly from the development of instrumentation capable of delivering intense focused beams of mass-selected ions to surfaces.Historically, soft and reactive landing of mass-selected ions have been used primarily for the preparation of relatively low coverage (∼10% of a monolayer) samples dispersed over small areas (deposition area ∼5 mm2) for fundamental studies in surface science and catalysis.3,10,46–49 The main objective in many of these experiments has been to isolate well-defined molecules and clusters on supports at low enough coverages that they do not interact with their neighbors or agglomerate into larger poorly-defined aggregates.8,50 Similar low surface coverages are also desirable for high resolution microscopy studies of the structure and magnetic properties of individual proteins and molecular clusters soft landed from solution using electrospray deposition.18,25 It is, therefore, extremely fortunate that the high sensitivity of many surface analytical techniques such as scanning tunneling microscopy (STM),18,51 atomic force microscopy (AFM),52 electron microscopy,53,54 and secondary ion mass spectrometry (SIMS)55,56 has enabled soft landed materials to be characterized even at these relatively low coverages (∼1011–1012 total ions on <1 cm2 area). While microscopy techniques often boast single molecule detection limits, infrared reflection absorption spectroscopy (IRRAS)32 that provides important structural information about soft landed ions requires substantially higher surface coverages (≥1013 ions) to obtain sufficient signal-to-noise ratios. This typically requires several hours of continuous ion deposition. Other powerful structure-specific analytical techniques such as nuclear magnetic resonance spectroscopy (NMR)57 require at least 1016 ions, which is currently out of limits for existing soft-landing instrumentation. It follows that a high flux ion source will make it possible to routinely deliver enough mass-selected ions to surfaces that structurally insightful but less sensitive spectroscopic techniques such as these may be applied to well-defined samples free of contaminants. Furthermore, it will enable soft landing and characterization of a wider range of ions including species that are present with low abundance instead of only the most intense ions in beams. Perhaps most intriguing, however, is the fact that substantially larger ion currents will allow ion soft landing to transition from preparing low coverage samples with small dimensions for studies in fundamental science to the fabrication of high coverage two- and three-dimensional materials61 (>1014 ions) over larger (∼1 cm2) areas for advanced applications in specialty devices such as chemical sensors,58 electrochemical capacitors,59 and molecular cluster batteries,60 as well as preparation of high density microarrays of proteins on surfaces for applications in high-throughput biological screening.12 It is expected that numerous unforeseen applications will also emerge as the high-flux ionization source technology matures.
The ion current delivered to surfaces is the key factor determining the rate of ion deposition. Electron impact (EI) ionization typically generates intense ion beams making it possible to deliver up to 20 nA of mass-selected ions to a deposition target.62,63 High ion currents have also been obtained using high frequency laser vaporization sources.64,65 The source, reported by Heiz and co-workers, generated currents up to 1 nA of mass-selected Nbn+ (n = 1–18) cluster ions.66 Ion currents in the range of 5–8 nA were also reported by Judai et al.67 for vanadium-benzene cluster ions produced using a similar cluster ion source. Similar ion currents have been reported for deposition instruments equipped with magnetron sputter gas aggregation and pulsed arc discharge cluster ion sources.68 For example, notable ion currents of 6.3 nA and 2 nA were recently obtained by Vajda and co-workers for mass-selected Ag3+ and Cu4+ ions, respectively, produced using this technique.69 Similarly, high ion currents of 3–6 nA were reported for small Sin+ (n = 4–10) clusters produced using pulsed arc discharge68 and 4–8 nA for mass-selected 3–4 nm Cu nanoparticles generated using magnetron sputtering.70 Electrospray ionization (ESI) is also a widely used method of ionization for ion soft-landing experiments. ESI provides access to a broad range of precursor ions with the capability to generate stable continuous ion beams over extended time periods with relatively little source maintenance required from the operator. Ion currents of 1–5 nA without mass selection56,71–73 and up to 600 pA with mass selection56,74 have been previously reported for ion deposition systems equipped with ESI sources.
In this study, we describe the design and performance of a new high-intensity ESI source for ion soft-landing experiments that combines several key developments in instrumentation. Specifically, mass-selected ion currents of up to 2.5 nA were obtained for the triply charged Keggin polyoxometalate (POM) anion, PMo12O403−, corresponding to a deposition rate of ∼1 μg per day. This deposition rate is at least five times higher than what may be achieved using the single-funnel instrument described elsewhere.32,75 The higher ion currents were obtained by combining a wide-bore stainless steel heated inlet in an orthogonal geometry for desolvation and droplet transfer into the first region of differential pumping with a tandem ion funnel developed by Smith and co-workers for efficient transfer of ions through two stages of differential pumping to vacuum conditions for subsequent mass-selection and deposition.76
Experimental setup
A tandem ion funnel, shown schematically in Fig. 1, was installed on an existing ion deposition system described in detail elsewhere.32,75 Briefly, the instrument is equipped with an ESI source, a tandem ion funnel system (described in more detail later in the text), an RF-only collision quadrupole (CQ), a quadrupole mass filter (Extrel CMS, Pittsburgh, PA), and three einzel lenses which focus the ion beam onto a deposition target. Ions produced using ESI are transferred into vacuum through a 100 mm long resistively heated stainless steel (SS) inlet tube maintained at a temperature of 150 °C. The instrument is equipped with both a direct inlet76 and an orthogonal inlet geometry.77 It has been demonstrated that orthogonal ion injection efficiently decouples ion transport from the gas flow dynamics, eliminates neutral molecules and solvent droplets from the instrument axis, and reduces electrode contamination.77 The heated tube in the direct inlet is located ∼6 mm off the center instrument axis and positioned close to the entrance plate of the first ion funnel. The orthogonal inlet is introduced through a 20 mm × 20 mm cutout in the middle of the first section (F1) of the outer funnel, as shown in Fig. 1. The benefit of having the orthogonal inlet and a direct inlet with an offset from the ion beam is the reduced transfer of neutral droplets into the high vacuum region of the instrument, preventing neutral contaminants from reaching the surface during soft landing experiments.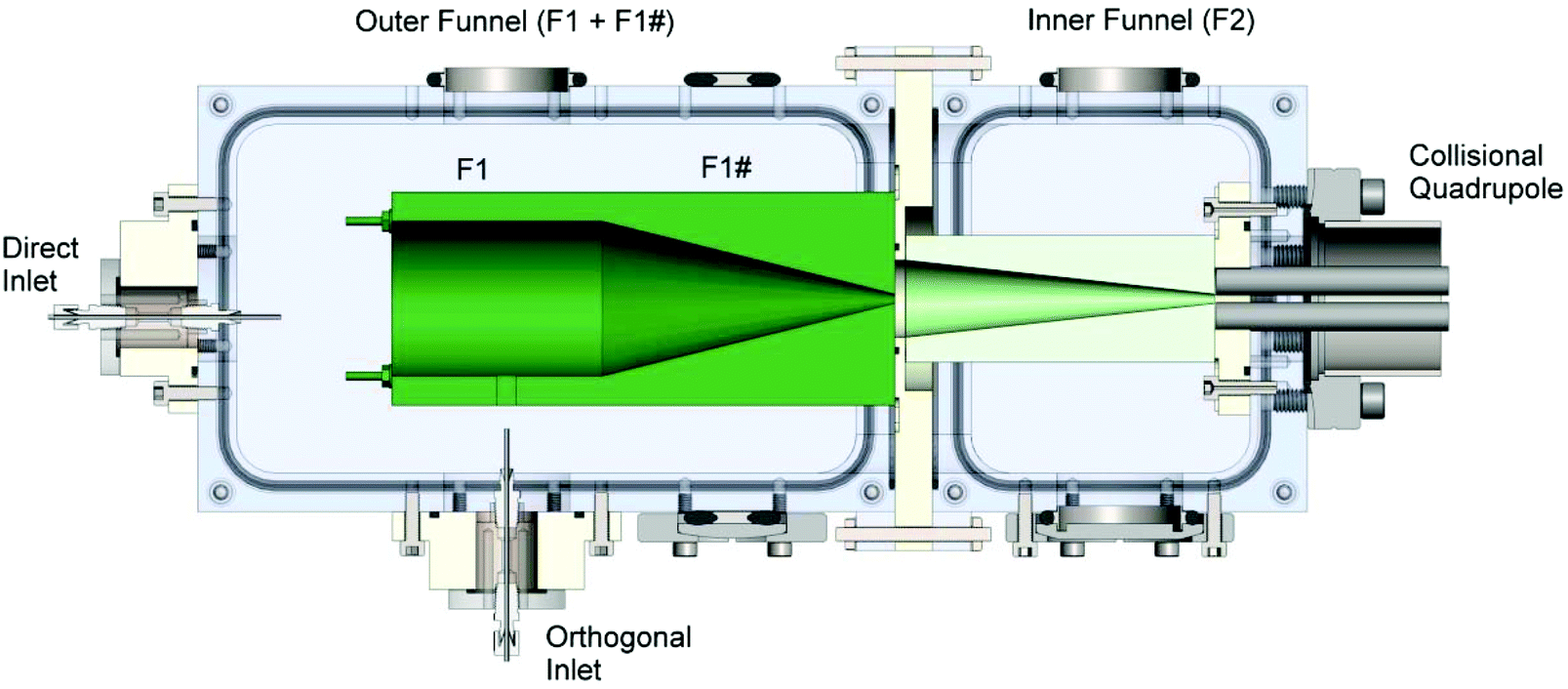 | ||
| Fig. 1 Schematic drawing of the tandem ion funnel interface showing the orthogonal and direct heated inlets, the first and second funnels, and the collisional quadrupole. The first funnel is separated into two regions denoted as F1 and F1#. Typical pressures and ion currents are listed in Tables 1 and 2, respectively. | ||
The tandem ion funnel consists of two RF ion funnels fabricated using printed circuit board technology described in detail elsewhere77,78 and housed in two differentially pumped vacuum regions. The first ion funnel, which is denoted as the outer funnel in Fig. 1, is comprised of a total of 128 ring electrodes. The first 64 electrodes have the same inner diameter (ID) of 50.8 mm (2 inches) while the ID of the last 64 electrodes decreases linearly from 50.8 mm to 2.5 mm. The second ion funnel, referred to hereafter as the inner funnel, is comprised of 68 ring electrodes. The first 34 electrodes have the same ID of 25.4 mm (1 inch) while the ID of the last 34 electrodes decreases linearly from 25.4 mm to 2.5 mm. Both ion funnels are powered by home-built high-Q heads; the outer funnel resonates at a frequency of ∼2080 kHz while the inner funnel resonates at ∼860 kHz. In each funnel, an RF voltage of the same amplitude and frequency is applied to all electrodes with a phase shift of 180 degrees between neighboring electrodes. Typical peak-to-peak voltages are ∼148 V and ∼122 V in the outer and inner funnel, respectively. A series of 0.5 MΩ resistors connected between all adjacent electrodes of the ion funnel generates a DC potential gradient when voltages are applied to the front and the back plates of the funnel. The outer funnel is separated into two DC regions, the first stack having 42 electrodes (F1) and the second stack (F1#) with 86 electrodes. This arrangement provides better control over the DC gradient applied to the funnel, which is particularly useful when ions are introduced orthogonally. The inner funnel has a single DC power supply which provides a gradient from the beginning to the final ring electrode. The following representative potentials were applied to generate DC gradients in the inner and outer funnel: F1 front plate, −394 V; F1 back plate, −367 V; F1# front plate, −366 V; F1# back plate, −126 V; F2 front plate, −175 V; F2 back plate, −44 V.
The chamber housing the outer funnel is differentially pumped by an Edwards 80 two stage rotary vane mechanical pump (56 cubic feet per minute (cfm)) to an operating pressure of 1–10 Torr, while the vacuum chamber housing the inner funnel is differentially pumped by a Leybold TriVac® D25B rotary vane mechanical pump (20 cfm) to an operating pressure of 0.4–1.5 Torr. The last plate of the outer funnel serves as the conductance limit between the first and second regions of differential pumping, while the last plate of the inner funnel serves as the conductance limit between the second and the third differentially pumped vacuum regions of the instrument. Typical operating pressure in the CQ located in the third differentially pumped region of the system is in a range of 10–50 mTorr. The quadrupole mass filter and the deposition target are located in the fourth differentially pumped region of the instrument maintained at 7 × 10−5–1 × 10−4 Torr during deposition by a Varian TV-301 Navigator turbomolecular pump (250 L s−1) backed by another Leybold TriVac® D25B rotary vane mechanical pump (20 cfm).
Sodium phosphomolybdate hydrate (Na3[PMo12O40]·xH2O CAS: 1313-30-0, Sigma-Aldrich, St. Louis, MO.) was dissolved in methanol to a final concentration of 150 μM and introduced into the ESI source using a syringe pump (Legato 180, KD Scientific, Holliston, MA) at a flow rate of 65–80 μL h−1. Charged microdroplets were produced by applying a ∼3 kV negative potential to the stainless steel union connected to the ESI emitter. Three types of ESI emitters were used in this study; (i) single bore (ii) double-bore (iii) four-bore emitters. Single bore emitters (Polymicro Technologies, Phoenix, AZ) are polyimide-coated fused silica capillaries with an ID of 100 μm and an outer diameter (OD) of 360 μm. Double bore and four bore emitters were prepared by pulling 500 μm OD boro-silicate glass tubes (VitroCom, NJ, USA) having two and four bores (ID of each bore: 125 μm), respectively, to a final OD of 130 μm using a micropipette puller (P-2000, Sutter Instrument Company, Novato, CA). Typical pulling parameters (in relative units used by the instrument software) were as follows: heat, 350; filament, 4; velocity, 30; delay, 200; pull strength, 0. PMo12O403− ions, the dominant species observed after droplet desolvation, were used for the optimization and characterization of the high-intensity ESI interface. Ion current measurements were performed using a picoammeter (model 9103, RBD Instruments, Bend, OR).
Results and discussion
The ability to operate at higher pressure in the outer funnel region while maintaining <10−4 Torr pressure in the deposition region is one of the major advantages of this dual funnel system. Our previous studies used a single-funnel interface,32,75,79 which allowed us to work with a heated inlet with an ID of up to 0.76 mm. However, further increases in the heated inlet ID were not possible due to pumping limitations. Specifically, it was not possible to maintain the required pressure in the fourth chamber housing the quadrupole mass filter and deposition target due to the increased ID of the inlet, which prevented mass-selection of ions prior to deposition. The same inlet was used in this study for comparison between the dual- and single-funnel interfaces. Using the dual ion funnel interface, we examined the effect of the heated inlet ID on the total ion current transferred through the dual funnel system measured at the CQ rods as well as the mass-selected ion current at the deposition target. The parameters of the four 100 mm long stainless steel heated inlets evaluated in this study and the corresponding pressures in the different regions of the instrument are summarized in Table 1. For the smaller ID heated inlets, an auxiliary gas flow through a vacuum valve in the first ion funnel chamber was required to obtain the pressure necessary for optimum ion currents. Ion current exiting the dual funnel interface was measured on the CQ rods. Ion currents on the CQ rods and on the deposition target obtained for each heated inlet using both the direct and orthogonal geometry and three different types of ESI emitters are summarized in Table 2 and in Fig. 2.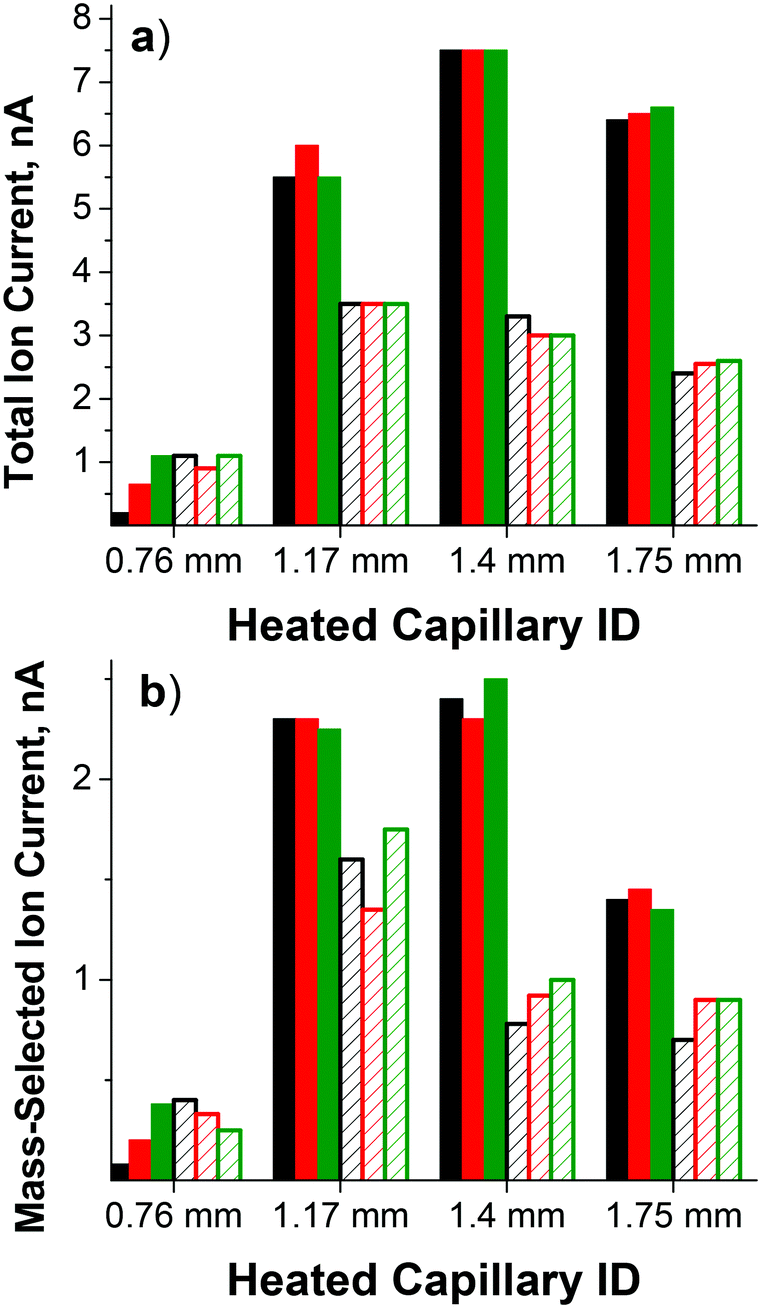 | ||
| Fig. 2 (a) Total ion current measured on the CQ rods and (b) mass-selected ion current of PMo12O403− on the deposition target obtained using the single-bore ESI emitter (black), double-bore ESI emitter (red), and four-bore ESI emitter (green). The heated inlet was introduced either in the orthogonal geometry (filled bars) or in the direct geometry (hatched bars) as described in the text. | ||
| Pressure regions | Pressure (Torr) | |||
|---|---|---|---|---|
| OD: 1/16′′ | OD: 1/16′′ | OD: 1/8′′ | OD: 1/8′′ | |
| ID: 0.030′′ | ID: 0.046′′ | ID: 0.055′′ | ID: 0.069′′ | |
| (0.76 mm) | (1.17 mm) | (1.40 mm) | (1.75 mm) | |
| Funnel 1 | 2.8 | 3.3 | 3.1 | 4.5 |
| Funnel 2 | 0.70 | 0.79 | 0.81 | 1.0 |
| CQ | 0.035 | 0.020 | 0.023 | 0.036 |
| Deposition target | 1 × 10−4 | 9 × 10−5 | 9 × 10−5 | 1 × 10−4 |
| Additional gas flow | Yes | Yes | No | No |
| ESI configuration | Ion currents (nA) | |||||||
|---|---|---|---|---|---|---|---|---|
| OD: 1/16′′ | OD: 1/16′′ | OD: 1/8′′ | OD: 1/8′′ | |||||
| ID: 0.030′′ | ID: 0.046′′ | ID: 0.055′′ | ID: 0.069′′ | |||||
| (0.76 mm) | (1.17 mm) | (1.40 mm) | (1.75 mm) | |||||
| Direct inlet | CQ | Sample | CQ | Sample | CQ | Sample | CQ | Sample |
| Single bore | 1.1 | 0.40 | 3.5 | 1.6 | 3.3 | 0.8 | 2.4 | 0.7 |
| Double bore | 0.9 | 0.33 | 3.5 | 1.4 | 3.0 | 0.9 | 2.6 | 0.9 |
| Four bore | 1.1 | 0.40 | 3.5 | 1.8 | 3.0 | 1.0 | 2.6 | 0.9 |
| Orthogonal inlet | CQ | Sample | CQ | Sample | CQ | Sample | CQ | Sample |
|---|---|---|---|---|---|---|---|---|
| a ESI of 150 μM Na3PMo12O40 in methanol; syringe pump flow rates: 65 μL h−1 for 0.57 mm and 0.76 mm ID tubes and 80 μL h−1 for 1.40 mm and 1.75 mm ID tubes. | ||||||||
| Single bore | 0.2 | 0.08 | 5.5 | 2.3 | 7.5 | 2.4 | 6.4 | 1.4 |
| Double bore | 0.7 | 0.20 | 6.0 | 2.3 | 7.5 | 2.3 | 6.5 | 1.5 |
| Four bore | 1.1 | 0.38 | 5.5 | 2.2 | 7.5 | 2.5 | 6.6 | 1.4 |
Ion currents obtained using the single-bore ESI emitter with a flared 0.76 mm ID heated inlet introduced in the direct geometry (1 nA on the CQ rods and 0.3 nA on the sample) are similar to the values obtained using the same heated inlet installed on the single-funnel interface. This result indicates similar transmission through both the single- and dual-funnel interfaces. For this size heated inlet, the highest ion current was obtained using the direct geometry and the four-bore ESI emitter. A substantial increase in the ion current was observed with larger ID heated inlets. Specifically, the ion current obtained using a 1.17 mm ID heated inlet introduced in the direct geometry is at least a factor of 3 higher than the value obtained using the 0.76 mm ID heated inlet. Even higher ion currents were obtained using the larger ID heated inlets introduced in the orthogonal geometry. Specifically, the ion current obtained using the orthogonal geometry is higher than the value obtained using the direct geometry by a factor of 1.6, 2.3, and 2.6 for 1.17 mm, 1.40 mm, and 1.75 mm ID heated inlets, respectively.
When combined with the 0.76 mm ID heated inlet, multi-bore ESI emitters produce higher ion currents than the single-bore emitter. In contrast, when larger ID heated inlets are used for transferring the charged droplets from atmospheric pressure into the first differentially pumped vacuum region of the ion deposition instrument, ion currents are similar for all ESI emitters. Differences in the size distribution of the charged droplets produced by different ESI emitters as well as the droplet desolvation efficiency and gas flow dynamics in different ID heated inlets, may contribute to the observed trends.
The highest ion current through the 1.17 mm heated inlet was obtained in the presence of an auxiliary gas flow perpendicular to the gas flow from the orthogonal inlet and to the instrument axis. The auxiliary gas flow could not be used with the larger ID heated inlets (i.e. 1.40 mm and 1.75 mm) due to pumping limitations. The high ion currents obtained using the orthogonal geometry along with the observed ion current enhancement in the presence of the auxiliary gas flow indicate that, in addition to voltage gradients in the ion funnel, gas dynamics play an important role in determining the ion transfer efficiency in the dual-funnel interface. Specifically, the introduction of auxiliary gas flow through the front inlet may increase the degree of mass transfer across the different vacuum regions of the dual ion funnel interface. This may induce the stream of charged droplets from the orthogonal inlet to take a perpendicular turn and align along the instrument axis. This combined effect may result in the higher ion transfer rate through the dual-funnel by entrainment of ions in the larger gas flow.
Similar ion transmission from the CQ to the deposition target is observed for both direct and orthogonal inlets and for different ESI emitters. The average ion transmission values of 39 ± 23%, 43 ± 5%, 31 ± 4%, and 27 ± 7% were obtained for 0.76 mm, 1.17 mm, 1.40 mm, and 1.75 mm ID heated inlets, respectively. The drop in ion transmission from 43% with the 1.17 mm ID inlet to 31% for 1.40 mm heated inlet is likely attributed to the higher pressure in the CQ which reduces the ion transfer efficiency through this radial focusing element.
The highest flux of mass-selected PMo12O403− ions measured on the deposition target of 2.5 nA was obtained using the four-bore ESI emitter and the 1.40 mm heated inlet introduced through the orthogonal geometry. For comparison, under the same conditions we obtained 0.8 nA of mass-selected current for the singly charged Mn(salen)+ [salen = N,N′-ethylenebis(salicylaldiminato)] complex and 1.8 nA of mass-selected doubly charged ruthenium tris(bipyridine) cations indicating that high ion currents may be obtained for both singly and multiply charged ions of both polarities that span a considerable mass range. As expected, the ion current is proportional to the charge of the ion. Stable ion current was also obtained over an extended period of time as shown in Fig. 3 indicating that the increased ion current does not result in more rapid degradation of the instrument and increased maintenance. Assuming that a majority of ions are retained on the deposition target, this ion current corresponds to a deposition rate of ∼1 μg of mass-selected ions per day, which is a substantial improvement over previously reported values.
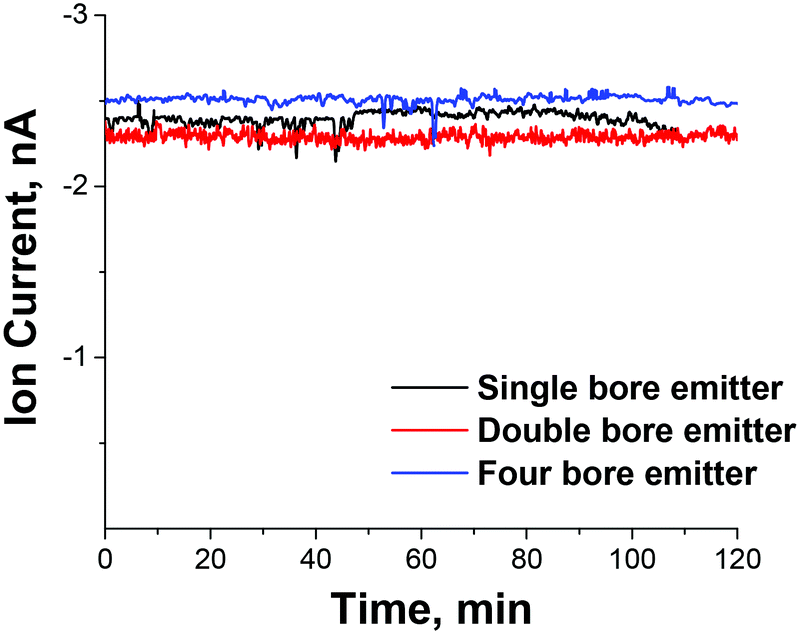 | ||
| Fig. 3 The stability of the mass-selected ion current of PMo12O403− on the deposition target as a function of time obtained using the 1.4 mm ID heated inlet introduced in the orthogonal geometry and three different ESI emitters. | ||
The total efficiency of ion formation and delivery to the surface can be estimated based on the solution concentration of analyte ions and the flow rate of the syringe pump. For PMo12O403− ions, we estimate that ∼350 μg of material is consumed for deposition of ∼1 μg of mass-selected ions, which corresponds to ∼0.3% efficiency. A substantially higher efficiency of ∼5–10% is estimated for ion transfer from the ESI source to the surface assuming typical ion currents of 20–40 nA generated by the ESI emitters used in this study. Therefore, the bottleneck limiting the efficiency of deposition is that only a very small fraction of the material present in solution is ionized. It follows that improved understanding of ESI mechanisms and better control of the ionization efficiency may result in a significant further increase in ion currents for ion soft-landing experiments.
Conclusions
The high-intensity electrospray ionization source described in this study will enable faster accumulation of mass- and charge-selected cations and anions on liquid and solid supports for subsequent characterization. By combining the dual ion funnel interface with a larger ID heated inlet in an orthogonal geometry for efficient droplet desolvation and ion transfer into the vacuum region of the instrument, we were able to increase the deposition rate by a factor of 5–6 corresponding to ∼1 μg of mass-selected ions per day. To the best of our knowledge, this is the highest stable deposition rate achieved for mass-selected ions produced using ESI, which is important for many applications ranging from the controlled preparation of organometallics, metal oxide and ligated metal clusters to the deposition of peptides, proteins, and large non-covalent complexes on surfaces.Acknowledgements
This work was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences & Biosciences and performed in EMSL, a national scientific user facility sponsored by the DOE's Office of Biological and Environmental Research and located at the Pacific Northwest National Laboratory (PNNL). PNNL is operated by Battelle for the U.S. DOE.References
- V. Grill, J. Shen, C. Evans and R. G. Cooks, Rev. Sci. Instrum., 2001, 72, 3149–3179 CrossRef CAS PubMed.
- U. Heiz and E. L. Bullock, J. Mater. Chem., 2004, 14, 564–577 RSC.
- B. Gologan, J. R. Green, J. Alvarez, J. Laskin and R. G. Cooks, Phys. Chem. Chem. Phys., 2005, 7, 1490–1500 RSC.
- A. M. Bittner, Surf. Sci. Rep., 2006, 61, 383–428 CrossRef CAS PubMed.
- B. Gologan, J. M. Wiseman and R. G. Cooks, in Principles of Mass Spectrometry Applied to Biomolecules, ed. J. Laskin and C. Lifshitz, John Wiley & Sons., Inc., Hoboken, NJ, 2006 Search PubMed.
- P. Wang and J. Laskin, in Ion Beams in Nanoscience and Technology, ed. R. Hellborg, H. J. Whitlow and Y. Zhang, Springer, London, New York, 2009 Search PubMed.
- G. E. Johnson, Q. C. Hu and J. Laskin, Annu. Rev. Anal. Chem., 2011, 4, 83–104 CrossRef CAS PubMed.
- V. N. Popok, I. Barke, E. E. B. Campbell and K. H. Meiwes-Broer, Surf. Sci. Rep., 2011, 66, 347–377 CrossRef CAS PubMed.
- J. Cyriac, T. Pradeep, H. Kang, R. Souda and R. G. Cooks, Chem. Rev., 2012, 112, 5356–5411 CrossRef CAS PubMed.
- G. Verbeck, W. Hoffmann and B. Walton, Analyst, 2012, 137, 4393–4407 RSC.
- L. Hanley and S. B. Sinnott, Surf. Sci., 2002, 500, 500–522 CrossRef CAS.
- Z. Ouyang, Z. Takats, T. A. Blake, B. Gologan, A. J. Guymon, J. M. Wiseman, J. C. Oliver, V. J. Davisson and R. G. Cooks, Science, 2003, 301, 1351–1354 CrossRef CAS PubMed.
- T. A. Blake, Z. Ouyang, J. M. Wiseman, Z. Takáts, A. J. Guymon, S. Kothari and R. G. Cooks, Anal. Chem., 2004, 76, 6293–6305 CrossRef CAS PubMed.
- M. Volny, W. T. Elam, A. Branca, B. D. Ratner and F. Turecek, Anal. Chem., 2005, 77, 4890–4896 CrossRef CAS PubMed.
- A. Bottcher, P. Weis, S. S. Jester, D. Loffler, A. Bihlmeier, W. Klopper and M. M. Kappes, Phys. Chem. Chem. Phys., 2005, 7, 2816–2820 RSC.
- D. Loffler, S. Ulas, S. S. Jester, P. Weis, A. Bottcher and M. M. Kappes, Phys. Chem. Chem. Phys., 2010, 12, 10671–10684 RSC.
- S. Ulas, D. Strelnikov, P. Weis, A. Bottcher and M. M. Kappes, J. Chem. Phys., 2012, 136 Search PubMed.
- Z. T. Deng, N. Thontasen, N. Malinowski, G. Rinke, L. Harnau, S. Rauschenbach and K. Kern, Nano Lett., 2012, 12, 2452–2458 CrossRef CAS PubMed.
- P. Wang and J. Laskin, Angew. Chem., Int. Ed., 2008, 47, 6678–6680 CrossRef CAS PubMed.
- Q. C. Hu, P. Wang and J. Laskin, Phys. Chem. Chem. Phys., 2010, 12, 12802–12810 RSC.
- K. J. Koch, F. C. Gozzo, S. C. Nanita, Z. Takats, M. N. Eberlin and R. G. Cooks, Angew. Chem., Int. Ed., 2002, 41, 1721–1724 CrossRef CAS.
- S. C. Nanita, Z. Takats, R. G. Cooks, S. Myung and D. E. Clemmer, J. Am. Soc. Mass Spectrom., 2004, 15, 1360–1365 CrossRef CAS PubMed.
- F. Mazzei, G. Favero, M. Frasconi, A. Tata, N. Tuccitto, A. Licciardello and F. Pepi, Anal. Chem., 2008, 80, 5937–5944 CrossRef CAS PubMed.
- F. Pepi, A. Ricci, A. Tata, G. Favero, M. Frasconi, S. D. Noci and F. Mazzei, Chem. Commun., 2007, 3494–3496 RSC.
- S. Kahle, Z. T. Deng, N. Malinowski, C. Tonnoir, A. Forment-Aliaga, N. Thontasen, G. Rinke, D. Le, V. Turkowski, T. S. Rahman, S. Rauschenbach, M. Ternes and K. Kern, Nano Lett., 2012, 12, 518–521 CrossRef CAS PubMed.
- E. T. Ada, O. Kornienko and L. Hanley, J. Phys. Chem. B, 1998, 102, 3959–3966 CrossRef CAS.
- M. B. J. Wijesundara, Y. Ji, B. Ni, S. B. Sinnott and L. Hanley, J. Appl. Phys., 2000, 88, 5004–5016 CrossRef CAS PubMed.
- T. Pradeep, B. Feng, T. Ast, J. S. Patrick, R. G. Cooks and S. J. Pachuta, J. Am. Soc. Mass Spectrom., 1995, 6, 187–194 CrossRef CAS.
- N. Wade, C. Evans, S. C. Jo and R. G. Cooks, J. Mass Spectrom., 2002, 37, 591–602 CrossRef CAS PubMed.
- M. Volny, W. T. Elam, B. D. Ratner and F. Turecek, Anal. Chem., 2005, 77, 4846–4853 CrossRef CAS PubMed.
- P. Wang, O. Hadjar, P. L. Gassman and J. Laskin, Phys. Chem. Chem. Phys., 2008, 10, 1512–1522 RSC.
- Q. C. Hu, P. Wang, P. L. Gassman and J. Laskin, Anal. Chem., 2009, 81, 7302–7308 CrossRef CAS PubMed.
- G. E. Johnson and J. Laskin, Chem. – Eur. J., 2010, 16, 14433–14438 CrossRef CAS PubMed.
- W. P. Peng, G. E. Johnson, I. C. Fortmeyer, P. Wang, O. Hadjar, R. G. Cooks and J. Laskin, Phys. Chem. Chem. Phys., 2011, 13, 267–275 RSC.
- N. Hauptmann, C. Hamann, H. Tang and R. Berndt, J. Phys. Chem. C, 2013, 117, 9734–9738 CAS.
- M. Mitsui, S. Nagaoka, T. Matsumoto and A. Nakajima, J. Phys. Chem. B, 2006, 110, 2968–2971 CrossRef CAS PubMed.
- S. Nagaoka, K. Ikemoto, K. Horiuchi and A. Nakajima, J. Am. Chem. Soc., 2011, 133, 18719–18727 CrossRef CAS PubMed.
- S. Vajda, R. E. Winans, J. W. Elam, B. D. Lee, M. J. Pellin, S. Seifert, G. Y. Tikhonov and N. A. Tomczyk, Top. Catal., 2006, 39, 161–166 CrossRef CAS PubMed.
- G. Kwon, G. A. Ferguson, C. J. Heard, E. C. Tyo, C. R. Yin, J. DeBartolo, S. Seifert, R. E. Winans, A. J. Kropf, J. Greeley, R. L. Johnston, L. A. Curtiss, M. J. Pellin and S. Vajda, ACS Nano, 2013, 7, 5808–5817 CrossRef CAS PubMed.
- R. E. Palmer, S. Pratontep and H. G. Boyen, Nat. Mater., 2003, 2, 443–448 CrossRef CAS PubMed.
- M. Di Vece, N. P. Young, Z. Y. Li, Y. Chen and R. E. Palmer, Small, 2006, 2, 1270–1272 CrossRef CAS PubMed.
- F. Yin, Z. W. Wang and R. E. Palmer, J. Am. Chem. Soc., 2011, 133, 10325–10327 CrossRef CAS PubMed.
- U. Heiz, F. Vanolli, A. Sanchez and W. D. Schneider, J. Am. Chem. Soc., 1998, 120, 9668–9671 CrossRef CAS.
- M. J. Berr, F. F. Schweinberger, M. Doblinger, K. E. Sanwald, C. Wolff, J. Breimeier, A. S. Crampton, C. J. Ridge, M. Tschurl, U. Heiz, F. Jackel and J. Feldmann, Nano Lett., 2012, 12, 5903–5906 CrossRef CAS PubMed.
- G. E. Johnson, R. Colby and J. Laskin, Nanoscale, 2015, 7, 3491–3503 RSC.
- U. Heiz and W. D. Schneider, Crit. Rev. Solid State Mater. Sci., 2001, 26, 251–290 CrossRef CAS.
- J. A. van Bokhoven and S. Vajda, Phys. Chem. Chem. Phys., 2014, 16, 26418–26420 RSC.
- V. Habibpour, M. Y. Song, Z. W. Wang, J. Cookson, C. M. Brown, P. T. Bishop and R. E. Palmer, J. Phys. Chem. C, 2012, 116, 26295–26299 CAS.
- G. E. Johnson, Q. C. Hu and J. Laskin, Annu. Rev. Anal. Chem., 2011, 4, 83–104 CrossRef CAS PubMed.
- F. Yin, S. S. Lee, A. Abdela, S. Vajda and R. E. Palmer, J. Chem. Phys., 2011, 134 Search PubMed.
- S. Bonanni, K. Ait-Mansour, M. Hugentobler, H. Brune and W. Harbich, Eur. Phys. J. D, 2011, 63, 241–249 CrossRef CAS.
- S. J. Davila, D. O. Birdwell and G. F. Verbeck, Rev. Sci. Instrum., 2010, 81 Search PubMed.
- G. E. Johnson, R. Colby and J. Laskin, Nanoscale, 2015, 7, 3491–3503 RSC.
- Z. Y. Li, N. P. Young, M. Di Vece, S. Palomba, R. E. Palmer, A. L. Bleloch, B. C. Curley, R. L. Johnston, J. Jiang and J. Yuan, Nature, 2008, 451, 46–U42 CrossRef CAS PubMed.
- G. E. Johnson, M. Lysonski and J. Laskin, Anal. Chem., 2010, 82, 5718–5727 CrossRef CAS PubMed.
- Z. X. Nie, G. T. Li, M. P. Goodwin, L. Gao, J. Cyriac and R. G. Cooks, J. Am. Soc. Mass Spectrom., 2009, 20, 949–956 CrossRef CAS PubMed.
- P. Y. Liu, R. G. Cooks and H. Chen, Angew. Chem., Int. Ed., 2015, 54, 1547–1550 CrossRef CAS PubMed.
- W. Y. Zhang, D. Du, D. Gunaratne, R. Colby, Y. H. Lin and J. Laskin, Electroanalysis, 2014, 26, 178–183 CrossRef CAS.
- V. Ruiz, J. Suarez-Guevara and P. Gomez-Romero, Electrochem. Commun., 2012, 24, 35–38 CrossRef CAS PubMed.
- H. Wang, S. Hamanaka, Y. Nishimoto, S. Irle, T. Yokoyama, H. Yoshikawa and K. Awaga, J. Am. Chem. Soc., 2012, 134, 4918–4924 CrossRef CAS PubMed.
- V. Singh, P. Grammatikopoulos, C. Cassidy, M. Benelmekki, M. Bohra, Z. Hawash, K. W. Baughman and M. Sowwan, J. Nanopart. Res., 2014, 16, 2373 CrossRef.
- J. P. Biesecker, G. B. Ellison, H. Wang, M. J. Iedema, A. A. Tsekouras and J. P. Cowin, Rev. Sci. Instrum., 1998, 69, 485–495 CrossRef CAS PubMed.
- A. Bottcher, P. Weis, A. Bihlmeier and M. M. Kappes, Phys. Chem. Chem. Phys., 2004, 6, 5213–5217 RSC.
- T. G. Dietz, M. A. Duncan, D. E. Powers and R. E. Smalley, J. Chem. Phys., 1981, 74, 6511–6512 CrossRef CAS PubMed.
- M. A. Duncan, Rev. Sci. Instrum., 2012, 83 CrossRef CAS PubMed , 041101.
- U. Heiz, F. Vanolli, L. Trento and W. D. Schneider, Rev. Sci. Instrum., 1997, 68, 1986–1994 CrossRef CAS PubMed.
- K. Judai, K. Sera, S. Amatsutsumi, K. Yagi, T. Yasuike, S. Yabushita, A. Nakajima and K. Kaya, Chem. Phys. Lett., 2001, 334, 277–284 CrossRef CAS.
- B. Klipp, M. Grass, J. Müller, D. Stolcic, U. Lutz, G. Ganteför, T. Schlenker, J. Boneberg and P. Leiderer, Appl. Phys. A, 2001, 73, 547–554 CrossRef CAS.
- C. Yin, E. Tyo, K. Kuchta, B. von Issendorff and S. Vajda, J. Chem. Phys., 2014, 140 Search PubMed.
- A. Majumdar, D. Köpp, M. Ganeva, D. Datta, S. Bhattacharyya and R. Hippler, Rev. Sci. Instrum., 2009, 80 CrossRef CAS PubMed , 095103.
- M. Volny and F. Turecek, J. Mass Spectrom., 2006, 41, 124–126 CrossRef CAS PubMed.
- M. Pauly, M. Sroka, J. Reiss, G. Rinke, A. Albarghash, R. Vogelgesang, H. Hahne, B. Kuster, J. Sesterhenn, K. Kern and S. Rauschenbach, Analyst, 2014, 139, 1856–1867 RSC.
- C. Hamann, R. Woltmann, I.-P. Hong, N. Hauptmann, S. Karan and R. Berndt, Rev. Sci. Instrum., 2011, 82, 033903 CrossRef PubMed.
- K. D. D. Gunaratne, G. E. Johnson, A. Andersen, D. Du, W. Zhang, V. Prabhakaran, Y. Lin and J. Laskin, J. Phys. Chem. C, 2014, 118, 27611–27622 CAS.
- O. Hadjar, P. Wang, J. H. Futrell, Y. Dessiaterik, Z. H. Zhu, J. P. Cowin, M. J. Iedema and J. Laskin, Anal. Chem., 2007, 79, 6566–6574 CrossRef CAS PubMed.
- Y. Ibrahim, K. Q. Tang, A. V. Tolmachev, A. A. Shvartsburg and R. D. Smith, J. Am. Soc. Mass Spectrom., 2006, 17, 1299–1305 CrossRef CAS PubMed.
- M. E. Belov, E. Damoc, E. Denisov, P. D. Compton, S. Horning, A. A. Makarov and N. L. Kelleher, Anal. Chem., 2013, 85, 11163–11173 CrossRef CAS PubMed.
- Y. M. Ibrahim, E. S. Baker, W. F. Danielson III, R. V. Norheim, D. C. Prior, G. A. Anderson, M. E. Belov and R. D. Smith, Int. J. Mass spectrom., 2015 DOI:10.1016/j.ijms.2014.1007.1034.
- G. E. Johnson, M. Lysonski and J. Laskin, Anal. Chem., 2010, 82, 5718–5727 CrossRef CAS PubMed.
Footnote |
| † These authors contributed to this work equally. |
| This journal is © The Royal Society of Chemistry 2015 |